Teil Chromatographische und Elektrophoretische Trennverfahren
Markus Kalberer HCI, E330 Tel. 632 29 29
kalberer@org.chem.ethz.ch
Selektivität einer Trennmethode Quantitative Methoden
Kapitel 2: Gas-Chromatographie (GC)
Trennung von eher flüchtigen Substanzen
Überblick über die für GC üblichen Trennsysteme
Aufbau und Funktion eines Gaschromatographen und der gebräuchlichsten Detektoren
Kapitel 3: Flüssig-Chromatographie (LC)
Trennung von eher polaren, weniger flüchtigen Substanzen
Aufbau und Funktion einer Flüssigchromatographie-Apparatur und der gebräuchlichen Detektoren
Überblick über verschiedene Varianten der LC
Kapitel 4: Elektrophoretische Trennverfahren
Kurzer theoretischer Überblick – Unterschiede zur Chromatographie Aufbau und Funktion einer Messapparatur
Kapitel 5: Probenaufarbeitung
Extraktions-, Aufkonzentrations-, Reinigungsmethoden
Bücher
D.A. Skoog, J.J. Leary,
Instrumentelle Analytik, Grundlagen, Geräte, Anwendungen, Springer, Berlin, 1996
K. Cammann,
Instrumentelle Analytische Chemie, Verfahren, Anwendungen, Qualitätssicherung,
Spektrum Akademischer Verlag, Heidelberg, 2001
R. Kellner, J.-M. Mermet, M. Otto, H.M. Widmer, Analytical Chemistry,
Wiley-VCH Verlag, Weinheim, 1998
K. Robards, P.R.Haddad, P.E. Jackson,
Principles and practice of modern chromatographic methods, Academic Press, London, 1994
... und viele mehr, siehe u.a. Chemie Bibliothek !
1. Theoretische Grundlagen 1.1 Typen von Trennmethoden
Trennung verschiedener Komponenten in der chemischen Analytik beruht im allgemeinen auf Unterschieden in der Verteilung zwischen zwei Phasen und/oder in der Beweglichkeit innerhalb einer Phase der zu trennenden Substanzen. Es werden bevorzugt eindimensionale Trennverfahren eingesetzt, bei denen eine Bewegung aller Komponenten in der gleichen Richtung erfolgt. Die folgenden Bewegungstypen sind relevant:
Typ 1 Typ 2 Typ 3
1 2
1
2 1
2
Elektrophorese (Kap. 4) Gas Chromatographie Membrantrenn-
Isoelektrische Fokussierung GC (Kap. 2) verfahren
Isotachophorese Flüssig Chromatographie Dialyse
Kapillarzonenelektrophorese HPLC (Kap. 3) Elektrodialyse (CZE)
Mischformen:
Gelpermeationschromatographie
Elektrokinetische Micellarchromatographie SDS-PAGE (Trägerelektrophorese)
Zweidimensionale Trennmethoden sind meist zwei zeitlich hintereinander geschaltete eindimensionale Trennungen. In der Polypeptidanalytik kommen z.B. häufig 2D- Elektrophoresemethoden zur Anwendung (Trennung nach Molmasse und isoelektrischem Punkt).
Trennkriterien:
a) Kinetische Eigenschaften
b) Gleichgewichtseigenschaften (Phasengleichgewichte, Verteilungsgleichgewichte) Oft liegt eine Kombination von a) und b) vor.
a) Trennmethoden unter Ausnutzung von kinetischen Eigenschaften:
Massenspektrometrie Elektrophorese Ultrazentrifuge Enzymatischer Abbau
Trennverfahren mit porösen Membranen (Dialyse, Elektrodialyse, Gasdiffusion)
b) Trennmethoden unter Ausnutzung von Gleichgewichtseigenschaften
- Phasengleichgewichte: Phasen gebildet durch zu trennende Komponenten (z.B. Destillation)
- Verteilungsgleichgewichte: Phasen enthalten zum grossen Teil Spezies, die nicht zum zu trennenden Gemisch gehören (z.B. GC, HPLC)
Übersicht über verschiedene Trennmethoden
zweite Phase
gasförmig fluid flüssig fest
gasförmig thermische Diffusion
Gas-Flüssigkeits- Chromatographie
Gas-Adsorptions- Chromatographie
fluid Fluid-Flüssigkeits-
Chromatographie
Fluid-Adsorptions- Chromatographie
flüssig Destillation
Flotation
Flüssigkeits- Flüssigkeits- Chromatographie Dialyse
Flüssig-Flüssig- Extraktion Ultrafiltration
Flüssigkeits- Adsorptions- Chromatographie Ionenaustausch Ausfällen Kristallisieren Abscheiden
(Zonenelektrophorese) Erste
Phase
fest Sublimation Fluid- Fest- Extraktion
Flüssig-Fest- Extraktion
Übersicht über Chromatographische Methoden
mobile Phase
flüssig fluid gasförmig
fest Flüssigkeits-Adsorptions- Chromatographie
Tswett, 1906
Kuhn, Winterstein, Lederer, 1931
Superkritische Fluid-
Adsorptions- Chromatographie Klesper, 1962 Rijnders, 1966 Novotny, 1981
Gas- Adsorptions- Chromatographie Ramsay, 1905 Cremer, 1951 Janak, 1953 stationäre
Phase
flüssig Flüssigkeits-Flüssigkeits- Chromatographie
Martin, Synge, 1941
Superkritische Fluid-
Flüssigkeits - Chromatographie Klesper, 1962
Gas- Flüssigkeits - Chromatographie James, Martin, 1952
1.2 Anschauliches Konzept für das Funktionsprinzips der Chromatographie:
Gedanklich kann man sich eine chromatographische Trennung als eine Reihe von diskreten Extraktionen vorstellen, bei denen sich die Analyten entsprechend ihrem Verteilungsgleichgewicht (Stoffkonstante) zwischen zwei Phasen verteilen. Vorraussetzung ist, dass sich die zwei Phasen nicht ineinander mischen.
Phase 1 (z.B. Wasser)
Phase 2 (z.B.
organisches Lösungsmittel)
Analyt zuerst 100% in dieser Phase
weniger Analyt
mehr Analyt
Zeit
Sind zwei Analyten A und B (mit unterschiedlichem Verteilungsgleichgewicht) in unserem System, haben wir eine Trennung der beiden Analyten erreicht.
Phase 1
Phase 2
A nd B zuerst 100% in dieser Phase
weniger A
,
mehr B
mehr A ,
weniger B
Zeit
Eine vollständige Trennung von A und B kann mit einer einmaligen Extraktion nicht erreicht werden. Um eine bessere Trennung zu erreichen, können mehrere Extraktionen hintereinander durchgeführt werden.
beliebige Wiederholungen neue Phase 2
neue Phase 1
Dieses Konzept kann man sich nun kontinuierlich vorstellen:
Phase 1 Phase 2
Die Phasen müssen gegen einander fliessen, sonst wird das System zu einer einzigen grossen Extraktionsstufestufe. Sie dürfen aber nicht zu schnell fliessen, sodass lokal jeweils Gleichgewichte etabliert werden können. Diese Methode heisst Gegenfluss-Extraktion (Countercurrent Extraction).
Da experimentell sehr aufwändig, wird die Gegenfluss-Extraktion jedoch nur selten angewendet.
Viel praktikabler ist es, wenn Phase 2 fest statt flüssig ist. In einer solchen Anordnung fliesst eine Flüssigkeit (Phase 1) über die feste Phase 2, was technisch sehr leicht zu machen ist.
Definition der Chromatographie:
• Physikalische Trennmethode, bei der die zu trennenden Komponenten zwischen einer feststehenden (stationären) und einer beweglichen (mobilen) Phase verteilt werden.
• Im Idealfall verteilen sich alle zu trennenden Substanzen unterschiedlich zwischen den zwei Phasen.
Hauptanwendungen für chromatographische Methoden:
• Analytische Anwendungen
• Präparative Anwendungen Grundarten der Chromatographie:
• Säulenchromatographie
• Planare Chromatographie
Fluss- richtung
Trennung eines Gemisches von A und B in einer Säulenchromatographie und das zeitabhängige Detektorsignal.
mobile Phase
A+B
A B
A B
Zeit
Zeit
t
At
B1.3 Einflussfaktoren der Wandergeschwindigkeit einer Verbindung in einem Chromatographischen System
1. Verteilungskoeffizient K beschreibt das Verhältnis der Konzentrationen der Substanz A in der mobilen und der stationären Phase im thermodynamischen Gleichgewicht.
!
Amobil thermodynamischesGleichgewicht
" $ $ $ $ $ $ $ $ # Astationär
!
K =
[ ]
A stationär[ ]
A mobil(1.1)
Falls verschiedene Substanzen in einem gegebenen chromatographischen System verschiedene K’s aufweisen, kann eine Trennung erreicht werden.
2. Retentionszeit tR beschreibt die Zeit die eine Substanz braucht um ein chromatographisches System zu durchlaufen.
Der erste Peak mit der Retentionszeit tM stellt eine Substanz dar, die von der stationären Phase nicht zurückgehalten wird. tM wird häufig als dead time oder solvent delay bezeichnet.
Zeit t R,1
t M
M 1
Detektorsignal
3. Kapazitätsfaktor k’ (auch Retentionsfaktor) beschreibt die Wanderungsgeschwindigkeit von einer Substanz in einer Säule.
!
kA' =KA VS
VM (1.2)
wobei KA der Verteilungskoeffizient von A, Vs der Volumen der stationären Phase und VM
das Volumen der mobilen Phase Durch Umformungen erhält man
!
kA' =tR "tM
tM (1.3)
4. Selektivitätsfaktor α beschreibt die Wanderungsgeschwindigkeit zweier Substanzen A und B relativ zueinander. α ist definiert als
!
"=KB
KA (1.4)
wobei per Definition A immer jene Substanz ist, die schwächer zurückgehalten wird, sodass α immer grösser 1 ist.
Ersetzen von KA und KB in Gleichung (1.4) durch k’ (Gleichung (1.2) und (1.3)) erhält man eine Gleichung, die den Selektivitätsfaktor als Funktion der Retentionszeiten ausdrückt,
!
"=tR,B #tM
tR,A #tM (1.5)
wobei tR,B die Retentionszeit der Substenz B ist und tR,A die Retentionszeit der Substanz A.
Später in diesem Kapitel sehen wir, dass man α und k’ benutzen kann um die Trennleistung einer chromatographischen Säule zu optimieren.
1.4 Effizienz einer chromatographischen Säule
Wir können das zu Beginn verwendete Bild der hintereinander geschalteten Extraktionen wieder verwenden, bei denen sich für jede Substanz jeweils das Verteilungsgleichgewicht zwischen den Phasen einstellt. Entsprechend diesem Konzept ist es erstrebenswert, wenn möglichst viele Extraktionen hintereinander durchgeführt werden, um eine möglichst vollständige Trennung zu erhalten. Diese theoretischen Extraktionen werden theoretische Böden N genannt, wobei die Effizienz einer Säule mit der Anzahl der theoretischen Böden N zunimmt.
Um N experimentell aus einem Chromatogramm zu bestimmen, müssen wir uns zuerst die Form eines typischen (idealen) Elutionspeaks in einem Chromatogramm ansehen.
tr ist die Retentionszeit, w die Basispeakbreite, und σ die Standardabweichung der Peakbreite.
Für einen idealen Peak gilt
!
w=4" (1.6)
!
"2 =w2
16 (1.7)
σ2 nennt man Varianz und stellt ein Mass für die Breite des Peaks dar.
Die Effizienz einer chromatographischen Säule, ausgedrückt als Anzahl theoretischen Böden N, kann man definieren als
!
N = tR2
"2 (1.8)
Zeit
Detektorsignal
t R
w σ
Da es oft nicht sehr praktisch ist σ2 aus einem Chromatogramm herauszulesen kann man Gleichung (1.7) in (1.8) einsetzten und erhält
!
N =16 tR w
"
#
$ $
%
&
' '
2
(1.9)
Die Anzahl der theoretischen Böden nimmt mit zunehmender Säulenlänge zu. Um unterschiedlich lange Säulen dennoch miteinander vergleichen zu können, wurde ein weiterer Parameter definiert, die Bodenhöhe H
!
H= L
N (1.10)
wobei L die Länge der Säule ist, die oft in mm angegeben wird.
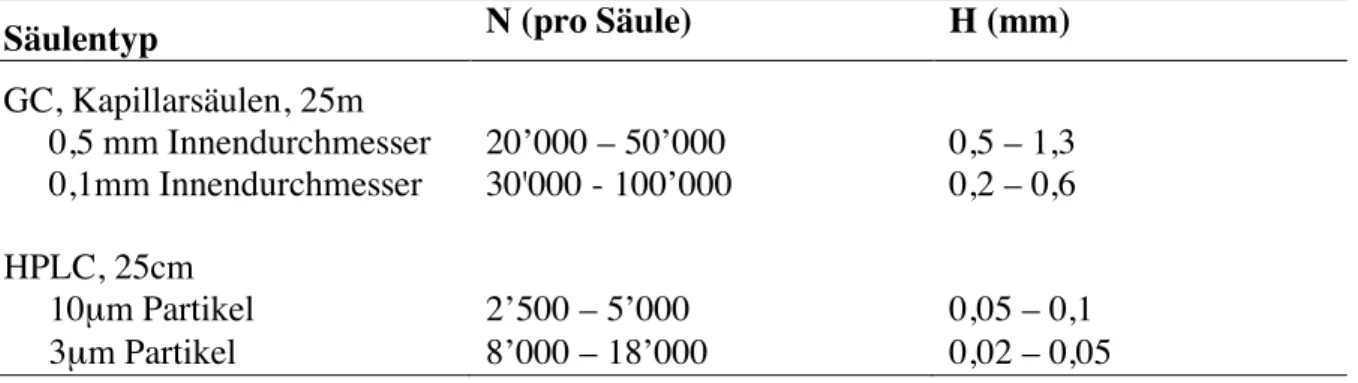
Typische Werte von H und N für verschiedene chromatographische Methoden sind in Tabelle 1.1 zusammengestellt.
Tabelle 1.1. Bodenzahl und Bodenhöhe für typische Gaschromatographie (GC)- und Hochdruck-Flüssigchromatographie (HPLC)-Säulen
Säulentyp N (pro Säule) H (mm)
GC, Kapillarsäulen, 25m
0,5 mm Innendurchmesser 20’000 – 50’000 0,5 – 1,3 0,1mm Innendurchmesser 30'000 - 100’000 0,2 – 0,6 HPLC, 25cm
10µm Partikel 2’500 – 5’000 0,05 – 0,1
3µm Partikel 8’000 – 18’000 0,02 – 0,05
1.5 Einfluss von kinetischen Säulenvariablen auf die Bodenhöhe H
In einem chromatographischen System fliesst die mobile Phase fortlaufend über die stationären Phase, wodurch die Analyten von der mobile Phase stets weiter getragen werden, bevor sich ein tatsächliches thermodynamisches Verteilungsgleichgewicht einstellen kann.
Die Flussgeschwindigkeit der mobilen Phase hat daher einen entscheidenden Einfluss auf die Trenneffizienz einer Säule.
Mit der folgenden Gleichung kann die Änderung der Effizienz (ausgedrückt als H) in Abhängigkeit der Fliessgeschwindigkeit beschrieben werden (Van-Deemter-Gleichung, hier die modifizierte Form nach Hawkes). Die Fliessgeschwindigkeit der mobilen Phase wird mit u bezeichnet.
!
H= B
u +csu+cmu (1.11)
!
B
u beschreibt die longitudinale Diffusion des Analyten von Bandenzentrum weg. Dieser Term ist proportional zur Zeit, welche die Substanzen in der Säule verbringen, also umgekehrt proportional zur Strömungsgeschwindigkeit in der Säule (vor allem wichtig in der Gaschromatographie).
Massentransfer-Effekte:
!
csu +
!
cmu
Die Einstellung des Gleichgewichts zwischen mobiler und stationärer Phase wird vom Massentransfer der Analytmoleküle zwischen den zwei Phasen bestimmt. Man unterscheidet dabei den Anteil in der stationären und in der mobilen Phase.
!
csu beschreibt die Verzögerung des Massentransfers aufgrund von Eigenschaften der stationären Phase. Die Dicke der stationären Phase hat den weitaus wichtigsten Einfluss auf diesen Term, aber auch der Diffusionskoeffizient des Analyten in der stationären Phase spielt eine Rolle.
Flussgeschwindigeit Diffusion
!
cmu beschreibt den Massentransfer der mobilen Phase, auch Wirbel-Diffusion genannt und beschreibt u. a. die unterschiedlichen Weglängen, welche die Analytmoleküle in der Säule zurückzulegen. Dieser Faktor ist weitgehend von der Strömungsgeschwindigkeit unabhängig (v.a. wichtig in der Flüssigchromatographie).
Graphische Darstellung der Van-Deemter-Gleichung
16 14 12 10 8 6 4 2 0
Bodenhöhe (H)
8 6
4 2
Flussgeschwindigkeit, cm/s
H
csu
cmu
B/u
1.6 Auflösung einer Säule
Die Auflösung einer Säule Rs beschreibt wie gut die Peaks von zwei Substanzen voneinander getrennt werden und kann direkt aus einem Chromatogramm berechnet werden. Eine hohe Auflösung erreicht man, wenn zwei Peaks möglichst gut voneinander getrennt sind und wenn sie eine scharfe Peakform aufweisen.
!
Rs= tR,B "tR,A wA
2 + wB
2
Durch weitere Umformungen erhält man eine Beziehung, die Rs beschreibt als eine Funktion der Bodenzahl N, des Selektivitätsfaktors α und des Kapazitätsfaktors k’B.
!
Rs= N 4
" #1
"
$
% & ' ( ) kB'
1+kB'
$
%
&
&
' (
) ) (1.12)
Detektorsignal
t R, B
wA wB
t R, A
Zeit
1.7 Optimieren einer chromatographischen Methode
Bei praktischen Anwendungen ist es häufig wünschenswert, die Retentionszeiten zu verringern. Dabei muss jedoch geachtet werden, dass die Auflösung nicht zu klein wird.
Unter Zuhilfenahme obiger Definitionen kann man die Retentionszeit darstellen als
!
tR =16Rs2H u
"
"#1
$
% & '
( )
(
1+kB')
3kB'
( )
2 (1.13)wobei u die Fliessgeschwindigkeit der mobilen Phase ist.
Aus den Gleichungen (1.12) und (1.13) ist ersichtlich, dass die beiden Parameter Rs und tR
von α, k’, N, und im Falle von tR auch noch von H abhängen. Alle diese Faktoren können optimiert werden.
α hängt von den Analyteigenschaften ab und kann u.a. verändert werden durch variieren der Säulentemperatur, der stationären Phase (z.B. Veränderung der Polarität) oder mobilen Phase (z.B. ändern des pH). Bei einer Veränderung der mobilen Phase kann oft eine bessere Selektivität erreicht werden, ohne dass die k’-Werte sich gross ändern.
k’ ist ein Parameter, der von der stationären Phase und dem Analyten abhängt. Oft erreicht man durch eine höhere Temperatur eine Erhöhung von k’ und damit eine bessere Auflösung. k’-Werte >10 erhöhen die Auflösung Rs meist kaum mehr und haben nur noch eine verlängerte Retentionszeit zur Folge.
10 8
6 4
2 0
Kapazitätsfaktor, k' Auflösung Rs
Retentionszeit tR
Oft ist eine Auflösung bei Kapazitätsfaktoren zwischen 5-10 ideal.
N kann durch die Länge der Säule oder durch die Bodenhöhe verändert werden (siehe unten und Diskussion der Van-Deemter-Gleichung)
H wird stark beeinflusst durch die Flussgeschwindigkeit der mobilen Phase, sowie den Parametern, die in die Van-Deemter-Gleichung einfliessen (Diffusionskoeffizient des Analyten in der mobilen und stationären Phase, Dicke der stationären Phase und Teilchengrösse der Packung (siehe Kap. 1.5)).
In der Praxis ist es oft üblich, die Bedingungen während eines chromatographischen Experiments zu ändern, da nicht alle Peaks bei den gleichen Bedingungen genügend getrennt werden können. So wird z.B. in der Gaschromatographie sehr häufig die Temperatur während eines Experiments laufend erhöht und in der Flüssigchromatographie die Zusammensetzung der mobilen Phase (sog. Gradientenelution).
Einfluss der verschiedenen Faktoren auf die Auflösung anhand eines Beispiels:
1.8 Nicht ideale Elutionspeaks
Symmetrische, gaussförmige Elutionspeaks werden in den praktischen Anwendungen leider oft genug nicht gemessen. Abweichungen von der Gleichgewichtsverteilung zwischen mobiler und stationärer Phase haben eine nicht ideale Peakform zur Folge, wie auf folgender Darstellung ersichtlich ist.
1 2 3
1 lineare Isotherme: Konzentrationsunabhängige Verteilung (Idealfall) zwischen mobiler und stationärer Phase.
Folgen: Symmetrisches Signal; Retentionszeit
nicht von injizierter Probenmenge beeinflusst
2 Tailing: Wegen Übersättigung der stationären Phase (häufig Normalfall) werden grosse Probenkonzentrationen
(überladene Säule zu wenig stark zurückgehalten.
mit Komponenten Folgen: Verschiebung des Peakmaximums in
relativ niedriger Retention) Richtung kleinerer Retention, asymmetrisches Signal und Verkürzung der Retentionszeit bei
hohen Probenkonzentrationen.
3 Fronting (leading): Tritt z.B. dann auf, wenn grosse Mengen von (überladene Säule mit Probekomponenten in der stationären Phase
Komponenten kondensieren.
relativ hoher Retention) Folgen: Asymmetrisches Signal und eine
Zunahme der Retentionszeit bei grossen
Probenkonzentrationen.
1.9 Quantitative Methoden
Chromatographische Methoden hätten in den letzten Jahrzehnten wohl nicht die Wichtigkeit erlangt, wenn nicht sehr präzise quantitative Aussagen möglich wären. Man unterscheidet im wesentlichen drei Quantifizierungsmethoden: die Quantifizierung mittels externem Standard und mittels internem Standard und die Standardaddition.
Externer Standard
In den Analyseproben werden die Peakhöhen oder Peakflächen der zu quantifizierenden Substanzen ermittelt, die dann anhand der Kalibrationsgerade (Konzentration- Signalintensitäts-Beziehung) in Konzentrationen umgerechnet werden.
Ein Nachteil dieser Methode ist, dass die aufgetragenen Probemengen für die Standardproben und die Analyseproben genau übereinstimmen müssen. Da oft sehr kleine Mengen (µl) eingesetzt werden, kann dies zu grossen Fehlern führen.
Konzentration der Analyten
In den eigentlichen Proben können unter Umständen Störungen auftreten, welche die Elution, oder Detektion des Analyten beeinträchtigen, die aber in den Standardproben nicht vorhanden waren und somit auch in der Kalibrationsgerade nicht berücksichtigt werden konnten.
Interner Standard
Bei dieser Methode werden den Standardproben eine oder mehrere zusätzliche Substanzen beigegeben, die man nicht in den Analyseproben erwartet (oft verwendet man markierte Verbindungen z.B. mit 13C- oder 2H-Atomen). Diese Substanzen nennt man Interne Standards.
Konzentration der Analyten Peakhöhe des AnalytenPeakhöhe des Analyten Peakhöhe des internen Standards
Für die Kalibrationsgerade wird nun das Verhältnis der Peakhöhen von Analyt und Internem Standard gegen die Konzentration des Analyten aufgetragen, wobei für alle Messungen der Kalibrationsgerade die Konzentration des Internen Standards gleich gross ist und die Konzentration der zu quantifizierenden Substanzen variiert wird.
Zu den Analyseproben wird nun dieselbe Menge an Internem Standards zugegeben. Aus dem Verhältnis von Analyt und Internem Standard kann die gesuchte Konzentration des Analyten mit Hilfe der Kalibrationsgerade ermittelt werden. Diese Methode ist viel weniger anfällig auf Änderungen der Analysemengen.
Standardaddition
Bei der Standardaddition werden mehrmals immer gleiche Probemengen von bekannten Konzentrationen des Analyten der Probe beigegeben. Durch Extrapolation der Messkurve auf den Schnittpunkt der Konzentrationsachse kann die Konzentration der Probelösung bestimmt werden.
Konzentration der Analyten
-10 -5 0 5 10 15 Konzentration
in der Probe
Blindwert
Zu jeder quantitativen Bestimmung gehört neben der Konzentrationsbestimmung der Probe auch eine Bestimmung des Blindwertes, womit Spuren des Analyten im Lösungsmittel und weiteren Reagenzien (z.B. Puffer) bezeichnet werden, sowie Verunreinigungen, die durch die Aufbereitung in die Probe gelangen. Zur Bestimmung des Blindwertes wird die Konzentration des Analyten in einer sogenannten Blindprobe bestimmt, die genau gleich zusammengesetzt ist (Lösungsmittel, Standards, ev. weiter Zusätze) und aufgearbeitet wird wie eine echte Probe, jedoch keine Probelösung enthält.
Die Analytkonzentration in der Blindprobe muss von der Konzentration der Probelösung abgezogen werden um ein korrektes Messergebnis zu erhalten.
Peakhöhe des Analyten