vieratomiger dianionisher
Trielidluster
Diplomarbeit
inTheoretisherChemie
Daniel S. Lambreht
Düsseldorf
im Juli 2003
vieratomiger dianionisher
Trielidluster
von
Daniel Sebastian Lambreht
Diplomarbeit
inTheoretisherChemie
angefertigtam
Institutfür Theoretishe Chemie undComputerhemie
vorgelegt der
Mathematish-Naturwissenshaftlihen Fakultät
derHeinrih-Heine-Universität Düsseldorf
Juli 2003
habe.
Düsseldorf,den 17.7.2003
Referentin: Frau Prof. Dr.Christel M. Marian
Korreferent: HerrProf. Dr. KarlKleinermanns
Antoine de Saint-Exupery
1 Einleitung und Motivation 1
I Theoretishe Grundlagen 5
2 Nihtrelativistishe Methoden 7
2.1 Die Shrödingergleihung . . . 7
2.2 Methoden dernihtrelativistishen Quantenhemie . . . 8
2.2.1 DieSCF-Näherung . . . 9
2.2.2 ExpansionimBasissatz . . . 10
2.2.3 Ab-Initio-Elektronenkorrelationsmethoden . . . 11
2.2.3.1 Kongurationswehselwirkung (CI) . . . 12
2.2.3.2 Multikongurations-SCF (MCSCF) . . . 13
2.2.3.3 CoupledCluster (CC) . . . 13
2.2.4 Dihtefunktionaltheorie (DFT) . . . 17
3 Relativistishe Methoden 19 3.1 Die Dira-Gleihung . . . 19
3.1.1 Symmetrieim relativistishen Fall . . . 21
3.2 Methoden derrelativistishen Quantenhemie . . . 23
3.2.1 DieVielteilhen-Diragleihung . . . 23
3.2.2 DieDira-Hartree-Fok SCF-Näherung . . . 24
3.2.3 ExpansionimBasissatz . . . 25
3.2.4 VierkomponentigesrelativistishesCoupled-Cluster . . . . 26
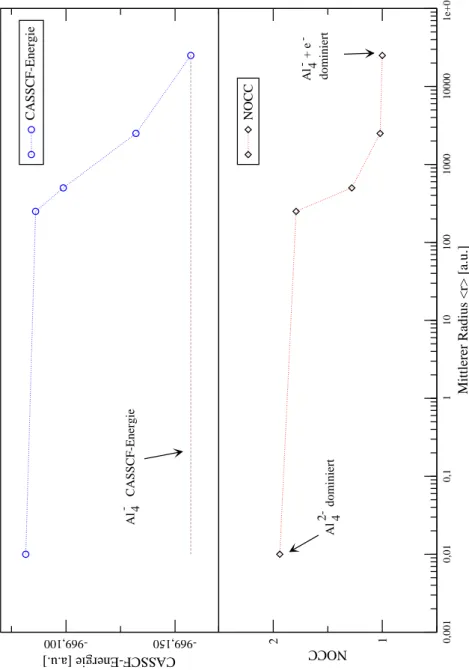
II Stabilität des isolierten Dianions in der Gasphase 31 4 Untersuhung des isolierten Al 2 4 33 4.1 Problemstellung . . . 33
4.2 Geometrieoptimierung . . . 36
4.3 Relative Stabilitäten derAl ( 0;1;2) 4 -Systeme . . . 42
4.4 Superposition mitderKontinuumswellenfunktion . . . 46
4.5 Fazit . . . 53
5 Vorarbeiten 61
5.1 Problemstellung. . . 61
5.2 WahlderBasissätze . . . 62
6 Eziente Geometrierelaxation 67 6.1 Motivation. . . 67
6.2 Wahldes Koordinatensystems . . . 68
6.3 Behandlung derMolekülsymmetrie . . . 69
6.4 DerOptimierungsalgorithmus . . . 69
6.4.1 Gitterbasierter Algorithmus . . . 70
6.4.2 Brent-Algorithmus . . . 72
6.5 Implementierung . . . 76
6.6 Evaluierung . . . 77
7 Geometrieoptimierungen 81 7.1 Vorüberlegung . . . 81
7.2 DFT-Geometrien . . . 81
7.3 CC-Geometrien . . . 83
8 Vertikale Detahment-Energien 87 8.1 Motivation. . . 87
8.2 Methode . . . 87
8.3 Ergebnisse und Diskussion . . . 88
9 Vierkomponentige relativistishe Berehnungen 93 9.1 Motivation. . . 93
9.2 Berehnungen . . . 95
9.3 Diskussionund Zusammenfassung. . . 103
10 Zusammenfassung 107
A Berehnung der atomaren Elektronenanitäten 117
B Basissätze 121
C Danksagung 129
Einleitung und Motivation
Aromaten gehören zu den wihtigsten und bestuntersuhten Verbindungsklas-
sen der Chemie [1℄. Dennoh ist es kürzlih überrashend gelungen, eine neue
ArtvonmetallishenAromatenherzustellen,waseinesovielversprehende Ent-
dekung darstellt,dasssie unlängst inSiene vorgestellt wurde [2℄.Die vorlie-
gendeArbeitbeshäftigt sih mitderquantenhemishen Untersuhung einiger
Prototypen dieserMetallluster.
Bei den neuartigen Aromaten handelt es sih um anionishe Cluster der
Triele, den Elementen der dritten Hauptgruppe. Diese Cluster zeihnen sih
durhplanareStruktureinheitenderKonstitutionM 2
4
(M=Al,Ga,In, Tl)aus;
dieM 2
4
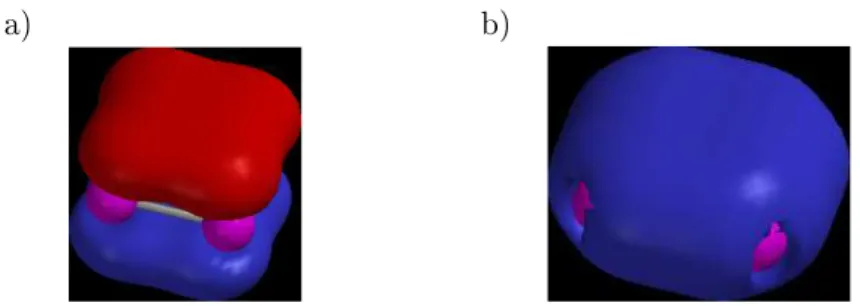
-Einheitenweisen jezweivollständigdelokalisierte-und-Elektronen
auf (Abb. 1.1 a) und b) und werden darum als (Hükel-)Aromaten angesehen
[2,3℄.
a) b)
Abbildung 1.1:Das(a)HOMO (-Orbital) (b)HOMO-5(-Orbital) derAl 2
4 -
Struktureinheit.
Berehnungen der NMR-hemishen Abshirmungskonstanten und magne-
tishinduziertenRingströme[4 6℄stützendieKlassikationalsHükelaromaten
wieauhdasVorliegeneinerdelokalisierten-Bindung.Demnahsheintessih
beidenTrielidlusternsowohlum-alsauhum-Aromatenzuhandeln[3,7℄.
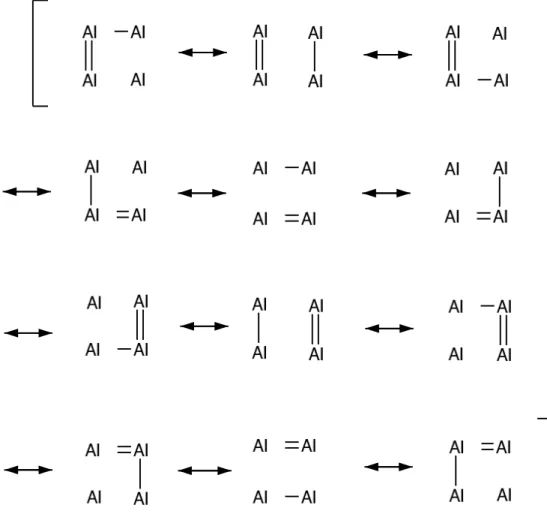
EineanshauliheDarstellungderBindungsdelokalisationindenM 2
4
-Einheiten
istinAbbildung 1.2 amBeispieldesAl 2
4
gegeben.
Abbildung 1.2:Valene-Bond-Modell derBindungsdelokalisationimAl 2
4
.Man
beahte, dass niht nur die Doppel-,sondern auh die Einfahbindungen delo-
kalisiertsind. Angelehnt an [3℄.
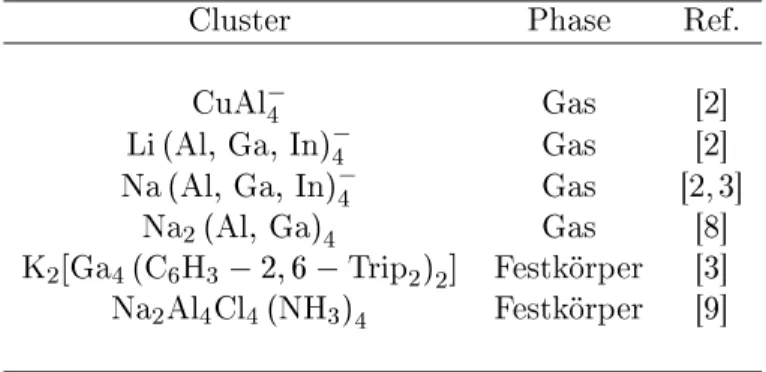
Wie der erste Trielidluster CuAl
4
(Abb. 1.3) wurde auh die Mehrzahl
der später entdekten aromatishen Cluster in der Gasphase hergestellt (Tab.
1.2).IsolierteM 2
4
-Einheiten sindthermodynamish instabil undwurdendaher
bislangniht dargestellt.
Cluster Phase Ref.
CuAl
4
Gas [2℄
Li(Al;Ga ;In)
4
Gas [2℄
Na( Al;Ga; In)
4
Gas [2,3℄
Na
2
(Al; Ga)
4
Gas [8℄
K
2 [Ga
4 ( C
6 H
3
2;6 Trip
2 )
2
℄ Festkörper [3℄
Na
2 Al
4 Cl
4 (NH
3 )
4
Festkörper [9℄
Tabelle 1.2: Bisher synthetisierte Trielidluster mit M 2
4
-Einheit. Cluster des
shweren HomologenTl wurden bislangniht dargestellt.
Al
Al Al
Cu Al
Abbildung 1.3: Der Trielidluster mit Summenformel CuAl
4
war der erste
Allmetall-Aromat, der entdekt wurde und hat in der Gasphase pyramidale
Struktur(C
4v
-Symmetrie).
Mittlerweile ist maneher an der Herstellung von Festkörpernmit aromati-
shen Struktureinheiten M 2
4
als an gasförmigen Molekülen interessiert [3℄. So
erhot man sih beispielsweise Einsatzmöglihkeiten in der Nanotehnologie,
z.B. für elektrish leitende Nanopartikel oder als Ersatz für organishe Aro-
maten [10℄. Von zusätzlihem Interesse ist die Synthese neuartiger Zintl-Ionen
jenseitsderZintl-Grenze,zudenendievieratomigenTrielidanionengehören[11℄.
VonexperimentellerSeiteistüberdieTrielidlusternohniht vielbekannt,
wenn man von der Existenz einiger Photoelektronenspektren absieht. Gründe
hierfürsindindernohshwierigenexperimentellenZugänglihkeitderSysteme
undderKonzentration aufdie Suhe nah neuen Festkörpernzu sehen[3℄.
Auh die bisherigen quantenhemishen Untersuhungen beshränken sih
hauptsählih auf die Bestimmung von Geometrie und vertikalen Detahment-
energienderleihterenCluster[25,7,9℄,wobeiMethodenwieDFToderCoupled
Cluster/Outer Valene Green's Funtions (OVGF) in Verbindung mit kleinen
Basissätzen verwendet wurden. Dabei wurden auh bei den shwereren Homo-
wohl bekannt ist, dass sie einen deutlihen Beitrag zur Korrelation leisten, der
für untershiedlihe Geometrien und Zustände untershiedlih starksein kann.
Auÿerdemwurden einige dieserBerehnungen amthermodynamish instabilen
M 2
4
durhgeführt, obgleih zu bezweifeln ist, dass die Verwendung quanten-
hemisher Programme, die zur Beshreibung gebundener Zustände entwikelt
wurden, auh bei metastabilen Systemen, d.h. lediglih quasigebundenen Zu-
ständen,anwendbar sind.
Damitergibt sihfür diese Arbeitdie folgendeZielsetzung:
1. Am Beispiel des Al 2
4
soll die Anwendbarkeit der Bound-State-Beshrei-
bung vonquasigebundenen Zuständen untersuht werden. DieseUntersu-
hungistfürdieFragederAromatizitätderTrielidlusterfundamental,da
mansihbeiderKlassizierung derTrielidlusterals Aromatenzueinem
Groÿteilauf solhe Untersuhungen stützt(siehez.B. [2℄).
2. GeometrienundvertikaleElektronenverlustenergien(VertialEletronDe-
tahment Energies,VDEs) werden fürCluster desTyps LiM
4
berehnet,
wobeiauhdieWihtigkeitderd-ElektronenfürdieKorrelationsbeshrei-
bung untersuht wird. Zudem sollen eventuelle Trends in geometrishen
und elektronishenStrukturen untersuht werden.
3. DieUntersuhung dershwerenIndium-und Thalliumhomologe maht es
nötig, zu einer relativistishen Beshreibung überzugehen. Dabei ergibt
sihdieMöglihkeit, Anwendung undMethodenerforshung zu kombinie-
ren:
(a) Es wird untersuht, welhe Trends sih unter dem Einuss relati-
vistisher Eekte innerhalb der shweren Homologen ergeben. Des
Weiteren soll untersuht werden, ob dasThalliumhomolog existiert,
da esbislangnoh niht hergestelltwerden konnte.
(b) Die tehnishen Voraussetzungen zurvierkomponentigen relativisti-
shen Beshreibung der shweren Homologe LiIn
4
und LiTl
4 wird
untersuht.BeiderImplementierung derrelativistishenElektronen-
korrelationsmethoden im integral-direkten Code DIRAC [12℄ ist die
Anwendung auf kleine Moleküle vorgesehen [13℄, wurde aber noh
niefür Systeme mit viershwerenZentrengetestet [14℄.Es wirdda-
her untersuht, unter welhen Bedingungen sih vierkomponentige
Coupled-Cluster-Berehnungen anSystemen mit mehrerenshweren
Kernen durhführen lassen und an welhen Stellen die momentane
Implementierung eventuellverändert werden muss.
Theoretishe Grundlagen
Nihtrelativistishe Methoden
2.1 Die Shrödingergleihung
InderQuantenhemie bedient mansih derQuantenmehanik,umdasVerhal-
tenvonElektronen inAtomen undMolekülen zu beshreiben.
Fundamentale Gleihung dernihtrelativistishen Quantenmehanik ist die
Shrödingergleihung
ih
t
jni=Hj ni: (2.1)
SiebeshreibtdiezeitliheEntwiklungdesZustandsvektorsjni,derdaszugehö-
rigephysikalishe Systemim Zustandnharakterisiert[15℄. DerZustandsraum
der Quantenmehanik ist der Hilbertraum H , welher als Abstraktion der in
derursprünglihen Formulierung der Theorie verwendeten isomorphen Räume
derquadratintegrablen Funktionen L
2
undder komplexwertigen Folgen`
2 ein-
geführtwurde. Indieserabstrakten Formulierungist einzigeBedingung fürdie
Zustandvektoren, dass sie Elemente eines Hilbertraums sind.
jni enthält sämtlihe Informationen, die mandurh Messungen amSystem
erhalten kann.
1
Zentrale Aufgabe der Quantenhemie ist es daher, durh ap-
proximative LösungderShrödingergleihung ZustandsvektorenfürAtomeund
MolekülezubestimmenunddarausInformationenüberderenEigenshaftenwie
beispielsweise Energie, GeometrieoderLadungsverteilung zu erhalten.
Im Allgemeinen verwendet man die Ortsraumprojektion von jni, die Wel-
lenfunktion
n
(x)hxjni: (2.2)
IhrBetragsquadrat ergibt nah Borneineräumlihe Wahrsheinlihkeitsdihte,
die nah Multiplikation mit dem Volumenelement dV die Wahrsheinlihkeit
ergibt,daszugehörige Teilhen ineiner -UmgebungU
dV
vonx zunden:
P[x2U
dV
(x)℄=j
n (x)j
2
dV hnjxihxj nidV; (2.3)
1
TatsählihentbrannteeinjahrzehntelangerDisputüberdieExistenzsog.verstekterVa-
riabler[16℄,dienihtgemessenwerdenkönnen,aberdenAusgangvonMessungenbeeinussen.
MitHilfederQuantenlogikkonntegezeigtwerden,dassdiequantenmehanisheBeshreibung
derRealität vollständigistundesverstekteVariablennihtgebenkann[17℄.
wobei die Wahrsheinlihkeit, ein Teilhen im gesamten Raum zu nden, bei
gebundenen Zuständenzu jedemzulässigenZeitpunkt auf1 normiert ist(on-
servationof probability),
hnjni =1 und
t
hnjni =0: (2.4)
MoleküleigenshaftenlassensihentwedermitHilfedesHellmann-Feynman-
Theorems,ausdemEnergiefunktionaldesSystemsoderdurhAnwendungquan-
tenmehanisherOperatorenaufdieWellenfunktionberehnen.Vonbesonderer
Bedeutung ist dabeiderErwartungswert
hAi
hnjAj ni
hnjni
; (2.5)
der den Mittelwert vieler Messungen einer Eigenshaft A mit korrespondieren-
demOperatorAan gleihpräpariertenSystemen desZustands nrepräsentiert.
Diese Mittelwertbildung weist die QMals statistishe Theorie aus eskönnen
keinedeterministishenVorhersagengetroenwerden,sonderndieAussagender
QMhaben probabilistishen Charakter.
Nahdemih dieGrundlagen derQuantenhemie angedeutet habe,folgt in
dennähstenAbshnittendieBeshreibungder wihtigstenquantenhemishen
Methoden, dieindieserArbeitzum Einsatzkommen.
2.2 Methoden der nihtrelativistishen Quantenhe-
mie
Zur Beshreibung von stationären Zuständen von Atomen und Molekülen ver-
wendet man indernihtrelativistishen Theorie die zeitunabhängige Shrödin-
gergleihung inOrtsdarstellung,
H (k)
( r;R)=E
k
(k)
(r;R) (2.6)
mitElektronenkoordinatenr(r1;r2;:::;rn),KernkoordinatenR(R1;R2;:::;RN)
und demmolekularen Hamiltonoperator inatomarenEinheiten
H= X
A 1
2M
A r
2
A X
i 1
2 r
2
i X
i;A 1
r
iA +
X
A<B Z
A Z
B
R
AB +
X
i<j 1
r
ij
: (2.7)
Umdiekomplizierte molekulareShrödingergleihung lösenzu können,müssen
einige Näherungen eingeführtwerden. Meist wirddieShrödingergleihung zu-
nähstdurhVerwendung derBorn-Oppenheimer-Näherung [18℄ineinenKern-
und einenElektronenteilsepariert.
Dabei geht man davon aus, dass die Kerne ortsfest sind (lamped nulei
approah) und man betrahtet nur die Bewegung der Elektronen. Diese Vor-
gehensweise ist dadurh gerehtfertigt,dass Kerne wesentlih shwerer sind als
Man setzt die Gesamtwellenfunktion als Produkt einer Kern- und Elektro-
nenwellenfunktion an,
(n;k)
( r;R)= (n)
el r;R
(k)
nu
(R): (2.8)
Rdeutetdabeian,dassdieKernkoordinaten festgehaltenwerdenundnurpara-
metrishindieelektronisheWellenfunktioneingehen.NahEinsetzenvon(2.8)
indie Shrödingergleihung kann diese separiert werden, da dieKopplungster-
mezwishen elektronisher und Kernwellenfunktion unter o.g. Annahmen ver-
shwinden. So erhält man in der BO-Näherung die elektronishe Shrödinger-
gleihung
H
el ( n)
el r;R
=E
n (n)
el r;R
(2.9)
mitdem elektronishen Hamiltonoperator
H
el
= 1
2 X
i r
2
i X
i;A Z
A
r
iA +
X
i<j 1
r
ij
: (2.10)
Die BO-Näherung liefert in den meisten Fällen gute Ergebnisse und so ist
sie einedermeistverwendeten fundamentalen Näherungen derQuantenhemie.
SieführtzumwihtigenKonzeptderPotentialhyperähen,dasinderMolekül-
spektroskopie und Aufklärung von Reaktionsmehanismen Verwendung ndet.
So wird auh in dieser Arbeit vollständig im Rahmen der BO-Näherung gear-
beitet.
2.2.1 Die SCF-Näherung
Nah Separation der Shrödingergleihung in einen Kern- und Elektronenan-
teilmuss daselektronishe Problemgelöst werden, was dasHauptproblem der
Quantenhemie ist.Ausgangspunktfür dieLösungderelektronishenShrödin-
gergleihungstellt dieimfolgenden vorgestellteSelf-Consistent-Field-Näherung
(SCF-Näherung) dar.
Der Operator der interelektronishen Wehselwirkung r 1
ij
verhindert die
Separationderelektronishen Shrödingergleihung. InderSCF-Näherungver-
nahlässigt man die instantane Korrelation der Elektronen und verwendet ein
gemitteltesPotential,dasnurnohvoneinerElektronenkoordinateabhängt.So
lässtsihdasn-Elektronenproblem derShrödingergleihung näherungsweise in
nEinelektronenprobleme separieren.
DazusetztmandieelektronisheWellenfunktionalsSlaterdeterminantevon
Spinorbitalen
i an:
j 1;2;:::;ni= 1
p
n!
1 (x
1
)
2 ( x
1
)
n ( x
1 )
1 (x
2
)
2 ( x
2
)
n ( x
2 )
.
.
.
.
.
. .
.
. .
.
.
1 (x
n
)
2 ( x
n
)
n (x
n )
: (2.11)
Die Koordinaten x
i
sind dabei als Vereinigung von Raumkoordinate r
i und
Spinkoordinate ! deniert: x fr;!g.
Als Bestimmungsgleihungen für dieSpinorbitale ergeben sih ausder For-
derungÆE=0unterVariation!+ÆmitNebenbedingunghij ji =Æ
ij die
Hartree-Fok-SCF-Gleihungen
^
f( 1)jii=
i
jii (2.12)
mit demFok-Operator
^
f(1)=
^
h(1)+ X
j6=i
^
J
j (1)
^
K
j (1)
: (2.13)
Hierbeiist
^
h ein Einelektronen-Hamiltonoperator, und dieOperatoren
^
J
j bzw.
^
K
j
beshreiben die gemittelte Coulomb- bzw. Austaushwehselwirkung des
Elektronsim Orbital
i
mit denen inden
j .
DerFehler beim SCF-Verfahren ist,dass dieElektronen nur dasgemittelte
Feld der anderen Elektronen spüren, niht aber die instantane Coulombab-
stoÿung. Dadurh kommen sih die Elektronen im Mittelzu nahe, sodass das
Verfahren beispielsweise zu hoheEnergien und niht ganz korrekte Elektronen-
verteilungen liefert.SCF-Berehnungen stellenausdiesemGrundmeistnurden
Ausgangspunkt für aufwändigere Berehnungen dar, in denen die Elektronen-
korrelation explizit berüksihtigt wird.
2.2.2 Expansion im Basissatz
DieSCF-GleihungenkönnenfürkleineSystemenumerishgelöstwerden.Meist
abertransformiert mandie HF-Integrodierentialgleihungen durh Expansion
der Molekülorbitale
i
in einer Basis von atomaren Funktionen
(LCAO 2
-
Ansatz)
i ( x)=
X
C
i
( x); (2.14)
ineinalgebraishes Gleihungssystem,die Roothaan-Gleihungen
FC=SC: (2.15)
Die Matrix der optimalen MO-Koezienten C wird dann in einer iterativen
Prozedur bestimmt.
Als Basisfunktionenverwendet manaus rehentehnishen Gründen 3
über-
wiegend Linearkombinationen (Kontraktionen) von Gauÿfunktionen g(x;),
(x)=
X
a p
a g
a ( x;
a
); (2.16)
wobei dieExponenten und Kontraktionskoezienten p
a
meist aus Energie-
optimierungen aufatomarem Niveau bestimmt werden.
Bei der Konstruktion von Basissätzen ist darauf zu ahten, dass vor allem
die Valenzregion gut beshrieben wird, da sie von groÿer Bedeutung für das
2
LinearCombinationofAtomiOrbitals
3
hemishe Verhalten von Systemen ist. Dies erzielt man beispielsweise durh
zusätzliheBasisfunktioneninderValenzregion unddurhVerwendungvonPo-
larisationsfunktionen.ZurBeshreibunganionisher SystememüssenBasissätze
umdiuseFunktionenaugmentiertwerden,umdieausgedehnteLadungsvertei-
lungimAnion angemessenbeshreiben zukönnen.
FürkorrelierteBerehnungensolltenauÿerdemsowohlradialealsauhWin-
kelkorrelation möglihst gut beshrieben werden, d.h. Elektronen sollten sih
sowohlinihrerradialenalsauhWinkelverteilung gutausdemWeggehenkön-
nen.Daserfordert(1.)einehinreihend groÿeAnzahlanFunktionen derselben
DrehimpulsquantenzahlmitvershiedenenExponentenund(2.)höhereDrehim-
pulse.Wihtig istdabeiauh einebestimmteBalaniertheitderBasis,d.h. das
Verhältnis derAnzahl vonFunktionen fürWinkel-und Radialkorrelation sollte
ausgeglihen sein.
ZurKlassizierungvonBasissätzenwurdederBegridersog.n-Tupel-Zeta-
Qualität eingeführt: Ein Single-Zeta-Basissatz enthält je besetztem AO genau
eine kontrahierte Basisfunktion pro Atom, während eine Double-Zeta-Basis je
besetztemAOgenauzweikontrahierteBasisfunktionenenthält.GemäÿVariati-
onsprinzipwirddieBeshreibung einesSystemsmitzunehmenderBasissatzgrö-
ÿe besser, da dievariationelle Freiheit zunimmt. Oft wirdauh dieAnzahl von
unkontrahierten FunktioneninrundenKlammernangegebenunddieAnzahlan
kontrahierten Funktioneninekigen Klammern, z.B.(4s3p1d)/[3s2p1d℄.
Mittlerweile existieren ganze Bibliotheken von Basissätzen für SCF- und
korrelierte Berehnungen. Drei Beispiele für populäre Familien solher Basis-
sätze sind Ahlrihs' TZV-Basen [19℄, Dunnings korrelationskonsistente Basis-
sätze [20,21℄ und die Popleshen Basen [22,23℄. Welher Basissatz für die je-
weilige Berehnung verwendetwird, hängt stark vonangestrebter Genauigkeit,
RehenmethodeundSystemgröÿeab.AlsFaustregelistjedohbekannt,dassfür
korrelierteBerehnungen mindestensBasissätze vonDouble-, ehernoh Triple-
Zeta-Qualität verwendetwerden sollten[24℄.
2.2.3 Ab-Initio-Elektronenkorrelationsmethoden
Das Dezit der SCF-Methode liegt in der falshen Beshreibung der Elektro-
nenkorrelation, 4
beiderzweiArten untershieden werden:
Als dynamishe (oder kurzreihweitige Korrelation) bezeihnet man die
Coulomb-Wehselwirkung zweier Elektronen,diedurhden Operatorr 1
ij
beshrieben wird.
UnterdemBegriderstatishen(oderlangreihweitigenKorrelation)fasst
man dasPhänomen der Nahe-Entartung bzw. dasVorkommen von meh-
rerenKongurationen/DeterminantenmitrelativgroÿemGewihtzusam-
men.
4
Hier ist die Coulomb-Korrelation gemeint. Die Fermi-Korrelation wird durh SCF be-
Der durh Verwendung eines gemittelten Elektron-Elektron-Wehselwirkungs-
potentials verursahte Fehler kann nahträglih durh Verwendung von Korre-
lationsmethoden ausgeglihen werden.
2.2.3.1 Kongurationswehselwirkung (CI)
Kongurationswehselwirkung (CI) ist die konzeptuell einfahste Möglihkeit,
das HF-Ergebnis zu verbessern. Darum soll sie hier kurz beshrieben werden,
obwohlCI-Methoden indieserArbeit eineuntergeordnete Rolle spielen.
IdeebeiCIistes,dieGesamtwellenfunktion alsLinearkombination von Sla-
terdeterminanten bzw. Kongurationszustandsfunktionen (Conguration State
Funtions, CSFs)vershiedener AnregungsstufenvonderReferenzwellenfunkti-
on
0
auszudrüken [25℄:
=
0 +
X
I;A
A
I
A
I +
X
I<J X
A<B
AB
IJ
AB
IJ
+::: (2.17)
Werdenalle AnregungenindieExpansioneinbezogen, kommtmanzumGrenz-
falldesFullCI(FCI),dasinnerhalbdesvonderEinelektronenbasisaufgespann-
tenRaumsexakteErgebnisseliefert.LeiderskaliertFCIfaktoriellmitderBasis-
satzgröÿe, sodassbeidenmeistenpraktishen Anwendungenbesserskalierende
Methoden verwendetwerden müssen.
Manuntersheidet prinzipiellzwishenzweiMöglihkeiten,dasSkalierungs-
verhalten vonCI-Verfahren zu beinussen [26℄:
Preseletion,d.h.Vorauswahlderin(2.17)eingehendenKongurationen
überden GradderAnregung (z.B.CISD,CISDTQ)undAusnutzungvon
Symmetrie und Spinerhaltung
Sreening,d.h. Vernahlässigen vonTermen in(2.17)z.B.durh pertur-
bativeAbshätzungdesEinusseseinzelnerKongurationen.Ein Beispiel
für einsolhesVerfahren istdassog. nth-order interating spae[27℄.
Abgebrohene Single-Referenz-CI-Verfahren wie CISD haben heute kaum Be-
deutung mehr [26℄, da durh Vernahlässigung höherer Anregungen meist nur
ein wesentlih geringerer Anteil an Korrelationsenergie erhalten wird als bei
anderen Korrelationsmethoden. Ein weiterer Nahteildieser Methoden ist ihre
fehlende Gröÿenkonsistenz.
Meist ist es wünshenswert, niht nur Valenz-, sondern auh kernnähere
Elektronenzukorrelieren.DabeiführtdieEinbeziehunghöhererAnregungenim
Outer-Core- oder Core-Elektronen-Bereih zu extrem hohem Rehenaufwand.
Mit dem Generalized Ative Spae-CI-Verfahren (GASCI) können Räume von
Orbitalenbeliebig deniertwerden, innerhalbderer bzw. ausdenen heraus nur
bestimmteAnregungstypenerlaubtsind.BeispielsweisekannimValenzraumein
FCI und für die kernnäheren Elektronen (outer Core) ein CISD durhgeführt
werden. Dieses Verfahren liefert oft Ergebnisse von nahezu FCI-Qualität bei
2.2.3.2 Multikongurations-SCF (MCSCF)
Beim MCSCF-Verfahren wird die Wellenfunktion in mehreren Determinanten
expandiert,
j
MCSCF i=e
^
k
e
^ s
j
0
i; (2.18)
wobei
^
k Orbitalrotationen durhführt und s^ Kongurationen erzeugt. Beim
MCSCF wird die Form der Orbitale unter Berüksihtigung der Valenzkorre-
lationoptimiert.
Eine populäre Variante ist CASSCF, beidem die MOs in drei Räume auf-
geteilt werden, den inaktiven, aktiven undden virtuellen Raum. Innerhalb des
aktiven RaumswerdensämtliheKongurationen erzeugt,diedurhBesetzung
deraktiven MOs mit den aktiven Elektronen erhalten werden können, es wird
also ein FCI im aktiven Raum durhgeführt. Die inaktiven Orbitale bleiben
stetsdoppelt besetztund dievirtuellenOrbitale bleiben unbesetzt.Häug gibt
mandie Anzahl an aktiven Elektronen n und aktiven Orbitalen m mit (n,m)-
CASSCFan.
LeiderwähstdieAnzahlanmöglihenDeterminantenshnellmitderGröÿe
desaktiven Raums an:
N
Det:
=
2m
n
: (2.19)
Beimheutigen Stand der Tehnik gehört ein (16;16)-CAS mit 601080390 De-
terminantenzu den gröÿtenpraktish durhführbaren Berehnungen.
Die Auswahldesaktiven Raums erfolgtnahden folgenden Regeln[28℄:
Für jedesbesetztebindende Orbitalsollte daskorrespondierende antibin-
dende Orbital einbezogen werden.
Je kleinerdieEnergiedierenz zwisheneinem besetztenundeinem virtu-
ellen Orbital der selben Symmetrie ist, desto gröÿer ist ihre Wehselwir-
kung.
DieHF-Orbitalenergien können beiSystemen mit ausgeprägtem Multireferenz-
harakter qualitativfalsh sein.Insolhen Fällen sollteauf natürlihe Orbitale
zurükgegrienwerden, diez.B.durh einekleine CI-Berehnung erhaltenwer-
denkönnen.
MCSCF-Wellenfunktionen vermögen die Eekte der statishen Korrelati-
onbeigeshikter WahlderReferenzfunktionen qualitativkorrekt zu beshrei-
ben, so beispielsweise die Bindungsdissoziation. Dynamishe Korrelation wird
aber nur unzureihend beshrieben, und so werden im Anshluss an MCSCF-
Berehnungenmeistweitere(Multireferenz-)Korrelationsmethodenangewendet.
2.2.3.3 Coupled Cluster (CC)
Zentrale Idee bei der Methode des Coupled Cluster (CC) [29℄ ist die Berük-
Dies erreiht man durh Anwendung einer Exponentialfunktion des Cluster-
Operators Tauf dieWellenfunktion [31℄
CC
= exp(T)
0
=
1+T+ T
2
2 +
T 3
6 +:::
0
: (2.20)
Der Cluster-Operator istdabeieine SummevonAnregungsoperatoren
T=T
1 +T
2
+::: (2.21)
mit derWirkung
T
1
0
= o:
X
a virt:
X
r t
r
a
r
a
(2.22)
T
2
0
= o:
X
a<b virt:
X
r<s t
rs
ab
rs
ab
(2.23)
usw.
Durh denAnsatz als Exponentialfunktion ergeben sih sowohlverbundene
als auh niht verbundene Anregungen (onneted/disonneted exitations),
beispielsweise T
2
und T 2
1
als verbundene bzw. niht verbundene Doppelanre-
gungen(onneted/disonneteddoubles).Mankannzeigen,dassdasAuftreten
höherer Anregungenals ProduktniedrigererAnregungen zurGröÿenkonsistenz
führt.
Formulierung durh Projektion auf angeregte Zustandsfunktionen.
Die Clusteramplituden t bestimmt man aus den Amplitudengleihungen, die
durhProjektionvon Hexp(T)j
0
imitangeregten Kongurationenvonlinks,
z.B. mith u
m
j, erhaltenwerden können:
h u
m
jHexp( T)j
0 i =E
CC h
u
m
jexp(T)j
0
i : (2.24)
Die Amplitudengleihungen sind nihtlinear inden Amplituden und auÿerdem
ist dieAnzahlan Amplitudenextrem groÿ.Darum verwendetmanzurBestim-
mung derti.A.ein Projektionsverfahren [32℄. AlsCC-Energie ergibt sih
E
CC
=E
HF +
o:
X
a<b virt:
X
r<s (t
rs
ab +t
r
a t
s
b t
s
a t
r
b
)(h abjrsi habj sri); (2.25)
was auh für abgebrohene CC-Methoden gültig ist. Die Terme habjrsi stehen
dabeikurz fürdie Zweielektronenintegrale
ab
r 1
12
jrsi = Z
dx
1 Z
dx
2
a (x
1 )
b (x
2 )r
1
12
r ( x
1 )
s (x
2
): (2.26)
Bei der CCSD-Methode wird der Clusteroperator durh T
1 +T
2
appro-
erzielbaren Ergebnisse sind i.A. von hoher Genauigkeit, da Zweifahanregun-
gen T
2
unddie nihtverbundene Vierfahanregung T 2
2
den wihtigsten Beitrag
zurKorrelation liefern.AuÿerdemführendieEinfahanregungen dazu,dassdie
HF-MOsrelaxierenundsomitleihteFehlerinderReferenzwellenfunktionaus-
gleihen können.
Die CCSD-Energie ergibt sih aus (2.25) und die Amplitudengleihungen
erhält man wieder durh Projektion von Hexp(T)j
0
i mit angeregten Kon-
gurationenvon links.Beispielsweise erhält manaus(2.24)
h u
m
jHexp(T
1 +T
2 )j
0
i = E
CCSD h
u
m
j exp(T
1 +T
2 )j
0 i
= E
CCSD t
u
m
; (2.27)
wasnahTaylorentwiklungderExponentialfunktionundAusnutzenderTatsa-
he, dassHnurEin- und Zweielektronenoperatoren enthält,für dielinke Seite
h u
m jHj
0 i +
X
a X
r t
r
a h
u
m jHj
r
a i+
X
a;b X
r;s U
rs
ab h
r
a jHj
rs
ab i +
X
a;b;
X
r;s;t U
rst
ab h
r
a jHj
rst
ab
(2.28)
ergibt,wobeidieKoezienten U dieClusteramplituden enthalten.
Alternative Formulierungmittels Hausdor-Entwiklung. Umdendi-
rektenVergleihmitderspäterbeshriebenenrelativistishenRELCCSD-Methode
zuermöglihen,sollhiernoheinealternativeFormulierungderCC-Gleihungen
beshrieben werden. Durh Anwendung des Hausdor-Lemmas auf den Aus-
drukexp( T)Hexp( T)
exp( T)Hexp(T) = H+[H;T℄+ 1
2
[[ H;T℄;T℄
+ 1
3!
[[[H;T℄;T℄;T℄ (2.29)
+ 1
4!
[[[H;T℄;T℄;T;T℄+:::;
undMultiplikationmith
0
joderbeliebigenangeregtenKongurationen
rst:::
ab:::
vonlinkserhältmanimCCSD-Fall(T=T
1 +T
2
)undfürkommutierendeAm-
plitudenoperatoren([ T
;T
℄=0)dieEnergie-und dieAmplitudengleihungen
h
0
jH+[ H;T℄+ 1
2
[ [H;T℄;T℄+ 1
3!
[[[H;T℄;T℄;T℄
+ 1
4!
[[[H;T℄;T℄;T;T℄j
0
i = E
CCSD
;(2.30)
rst:::
ab:::
H+[H;T℄+ 1
2
[[H;T℄;T℄+ 1
3!
[[[ H;T℄;T℄;T℄
+ 1
[[[ H;T℄;T℄;T;T℄j
0
i = 0: (2.31)
Der normalgeordnete Hamiltonoperator lautet inzweiterQuantisierung
H = H H
SCF
= X
I F
I
I E
I
I +
1
4 X
I;J X
KL
hIKkJLi E IK
JL
| {z }
W
(2.32)
mit den Orbitalanregungsoperatoren
E I
J
=e I
J +e
I
J
(2.33)
und
E IK
JL
=E I
J E
K
L Æ
K
J E
I
L
: (2.34)
Mit dieser Denition lässt sih die erste CCSD-Amplitudengleihung bequem
als
A
I
WT
1
+WT
2
+WT
1 T
2 +
1
2 WT
2
1 +
1
3!
WT 3
1 j
0 i
(
I
A )t
A
I
= 0 (2.35)
shreiben.
Perturbative Behandlung der Dreifahanregung: CCSD(T). In der
Praxis zeigtsih,dassEinbeziehung von Dreifahanregungen dieCCSD-Ergeb-
nisse signikant verbessert und in vielen Fällen nahezu FCI-Qualität erhalten
werdenkann.PerturbativeBerüksihtigung derverbundenenDreifahanregun-
gen führtzurCCSD(T)-Methode,dieallgemeinalsbeste praktishanwendbare
Single-Referenz-Korrelationsmethode gilt [31℄.
Die(T)-KorrekturbestehtdabeiauszweiTeilen.ZunähstwirdderEinuss
derTriplesausdemMP4-Energieausdrukfür Triples
T( 4)=
X
I<J<K X
A<B<C
W ABC
IJK
2
D ABC
IJK
(2.36)
berehnet. Dabeisind
W ABC
IJK
= 1
4
^
P ABC
IJK (
X
D
hBCk DKit AD
IJ
X
E
hECk JKit AB
IE )
; (2.37)
D ABC
IJK
=
I +
J +
K
A
B
C
: (2.38)
^
P ist einAntisymmetrisierungsoperator
^
P ABC
IJK
=
^
P
IJK
^
P ABC
; (2.39)
^
P
IJK
IJK = IJK IKJ+JKI JIK+KIJ KJI; (2.40)
und
^
P ABC
ergibt sihanalog. Fernerist
h BCkDKi
BC
r 1
jDKi
BC
r 1
jKDi : (2.41)
Dannwirdnoh einStörungstermfünfter Ordnung addiert,
T( 5)= X
I<J<K X
A<B<C V
ABC
IJK W
ABC
IJK
D ABC
IJK
(2.42)
mit
V ABC
IJK
= 1
4
^
P ABC
IJK
hBCk JKit A
I
: (2.43)
Insgesamt ergibt sih dieCCSD(T)-Energie nah
E
CCSD(T)
=E
CCSD
+T(4)+T(5): (2.44)
Verlässlihkeit derCoupled-Cluster-Methode. Bei derAnwendung von
Single-Referenz-CC-Methoden ist zu beahten, dass CC durh die Einfahan-
regungen zwar Dezite der Referenzwellenfunktion deutlih mildern, aber bei
ausgeprägtem Multireferenzharakter niht mehr ausgleihen kann. Ein Maÿ
fürdie Gültigkeitder Single-Referenz-Behandlungist dieT
1
-Diagnose
T
1
= kt
1 k
p
n
; (2.45)
wobei t
1
der Vektor der Singles-Amplituden und n die Anzahl an korrelierten
Elektronenist.FürdieGültigkeitvonSingle-Referenz-CCSDsollteT
1
kleinerals
0,02sein undfür CCSD(T) können selbstbei Werten um0,04noh Ergebnisse
vonMRCI-Qualität erwartetwerden.
2.2.4 Dihtefunktionaltheorie (DFT)
GrundlagefürdieDFT-MethodeistdasHohenberg-Kohn-Theorem,dasbesagt,
dassdie elektronishe Grundzustandsenergie vollständigdurh dieElektronen-
dihtebestimmt wird.
StattderWellenfunktion verwendetmanbeiDFTdarumdieElektronen-
dihte
( x
1 )
Z
dx
2 Z
dx
3
Z
dx
n j (x
1
;x
2
;:::;x
n )j
2
(2.46)
undminimiert dasEnergiefunktional
E
DFT
[℄=T
s
[ ℄+E
ne
[℄+J[℄+E
x
[℄; (2.47)
wobei T
s
diekinetishe Energie, E
ne
dieKern-Elektron-Wehselwirkung, J die
Coulomb-Abstoÿung der Elektronen und E
x
das Austaush-Korrelationsfunk-
tionalist.
Ein enormerVorteildieserMethode liegtdarin, dass statt der3nKoordi-
natenin nurdreiRaumkoordinatenenthältdamitwirddieDimensionalität
derzuevaluierendenIntegraledrastishreduziertundmankannaufnumerishe
Integration zurükgreifen.
DasProblembeiDFTist jedoh,dassdieexaktefunktionale Formvon E
x
nihtbekanntist.Wäresiebekannt,könntemitDFTdieexakteGrundzustands-
DFT-Methodenentwiklung istdaherdieHerleitunggeeigneter Formelnfürdas
Austaush-Korrelationsfunktional.
InderKohn-Sham-TheoriewirddieElektronendihteinEinelektronenfunk-
tionen,denKohn-Sham-Orbitalen expandiert.Auf diesemWegeerhältmandie
den SCF-Gleihungen ähnelnden Kohn-Sham-Gleihungen
^
h
KS
i
=
i
i
: (2.48)
Wie beiderSCF-Methode werden dieKohn-Sham-Orbitale inBasisfunktionen
expandiert und diesoerhalteneMatrixgleihung iterativgelöst.
Zwei gängige Austaushkorrelationsfunktionale sind das BH-LYP- und das
B3-LYP-Funktional [33℄. Dabei handelt essih umsog. Hybridfunktionale, die
zumTeilden exaktenHF-Austaushterm,zumanderenModelltermeenthalten.
ImB3-FunktionalsindbeispielsweiseandereFunktionalewie E
x
ausLSDAund
B88, E
ausLSDA und GGAsowie derexakte HF-Austaush mit untershied-
lihen Wihtungen enthalten. Die genaueGestaltdereinzelnen Funktionale er-
gibt sih durhAnpassung an exakteLösungen bzw. hohgenaue Lösungendes
elektronishenProblemsfüreinfaheModellsystemewiedasfreieElektronengas
(LSDA).
DieQualitätvonDFT-ErgebnisseniststarkvonderArtdesSystemsundder
WahldesFunktionals abhängig. Mitden gängigen Funktionalen BH-LYP oder
B3-LYP erhält man für kovalent gebundene Systeme in der Regel Ergebnisse
von MP2-Qualität [28℄.
Relativistishe Methoden
Die moderne Physik ist für diePhysiker viel zu shwer.
David Hilbert
3.1 Die Dira-Gleihung
FürSystememitshwerenAtomenoderzurhohgenauenBeshreibungkleinerer
Systeme ist es notwendig, Eekte aus derspeziellen Relativitätstheorie (SRT)
zuberüksihtigen.Die beiden zentralen Postulate der SRTsind
1. Konstanzder Lihtgeshwindigkeit inallen Inertialsystemenund
2. Gleihheit allerphysikalishen Gesetze inallen Inertialsystemen.
AusdemzweitenPostulatfolgtinsbesondere,dassphysikalisheGesetzeLorentz-
kovariant sein müssen.Mankannzeigen, dassdieseBedingung erfülltist, wenn
das Gesetz Welttensoren der selben Stufe verbindet [34℄. Die Shrödingerglei-
hung erfülltdiese Bedingung niht, da sie zweite Ableitungen nah denOrts-,
abernur eineerste Ableitung nahderZeitkoordinate enthält,und kanndaher
niht als relativistishe Wellengleihung verwendet werden.
ZurBeshreibungvonSpin- 1
2
-Teilhen dientimRahmenderrelativistishen
Quantenmehanik die Dira-Gleihung, diefür einfreies Teilhen inHamilton-
Form[35 37℄
p
0
= ~ ~p+m 2
; p
0
=ih
t
(3.1)
lautet. Dabei ist ~p der dreikomponentige Impulsvektor, ~ = (
1
;
2
;
3 )
T
und
und sindMatrizenmit
=
1
2 0
0 1
2
(3.2)
i
=
0
i
i 0
;i=1;2;3: (3.3)
Die
i
sinddiebekannten Pauli-Matrizen. ZustandsvektorderDira-Gleihung
ist dervierkomponentigeVektor
= 0
B
B
1
2
3
4 1
C
C
A
; (3.4)
ein sog. vierkomponentiger Spinor (4-Spinor).
1
Wie in Postulat 2 derSRT ge-
fordert, istdieDira-Gleihung Lorentz-kovariant.
Shreibt mandie Dira-Gleihung inMatrixformauf,
0
B
B
p
0
m 0 p
z
( p
x ip
y )
0 p
0
m (p
x +ip
y
) p
z
p
z
(p
x ip
y
) p
0
+m 0
(p
x +ip
y
) p
z
0 p
0 +m
1
C
C
A 0
B
B
1
2
3
4 1
C
C
A
=0;
(3.5)
erkennt man,dassdieKomponentenvon durhdieAuÿerdiagonalenelemente
gekoppeltsind.Häug werden
1 und
2
zueinem2-Spinor L
unddieübrigen
beiden Komponenten zu S
zusammengefasst. Man spriht dann in quanten-
hemishen Berehnungen von der sog. groÿen und der kleinen Komponente L
bzw. S,diegröÿtenteils den elektronishen bzw. positronishen AnteilderWel-
lenfunktion beshreiben. Beispielsweise ist der4-Spinor
+
= 0
B
B
1
0
~p
jEj+m0
0 1
C
C
A e
i(pr) i E
h t
(3.6)
eine spezielle Lösung der Dira-Gleihung für das freie Teilhen, die in der
nihtrelativistishen Theorie einer elektronishen Lösung mit -Spin entspre-
hen würde. In der relativistishen Theorie ist noh ein positronisher Anteil
eingekoppelt.
Die elektronishen Lösungen haben positive Energieeigenwerte, positroni-
she Lösungennegative.DaeinelektronisherZustanddemnah sofortineinen
positronishen Zustand kollabieren würde, postulierte Dira, dass im Vakuum
sämtlihe negativen Energieniveaus mit Elektronen besetzt sind (Elektronen-
see).
Imäuÿeren Feld wirddieDira-Gleihung unter Substitution
p
0
!p
0 e
(3.7)
~ p!~p
~
A
~ (3.8)
1
UnterSpinorenversteht man Gröÿen,dieunterRotation inderSU(2)bzw. SU(4)das
zu
p
0 e
~
~ m
=0: (3.9)
Da die Potentialterme in Atomen und Molekülen niht explizit zeitabhängig
sind, kann die Dira-Gleihung durh Festlegung auf ein Bezugssystem (Kern-
ruhesystem) inzeitunabhängige Formumgewandelt werden:
m 2
+~~+e
=E : (3.10)
FürMoleküle inBO-Näherung (A=0,e=V)ergibt sih
E+V m 2
(~p)~
(~~p) E+V +m 2
L
S
=0: (3.11)
Da Raum- und Spinanteile gekoppelt sind (~~p), kommutieren s^ und
^
l niht
mitH, sondern nur derGesamtdrehimpuls
^
j =
^
l+s.^
3.1.1 Symmetrie im relativistishenFall
Transformationsverhalten von Spinoren unter Zeitumkehr. Für zwei-
komponentige Spinorenlässt sih derZeitumkehroperator als
^
K = i
2
^
K
0
(3.12)
shreiben. Dabei ist
2
eine Paulimatrix (häug auh
y
genannt) und
^
K
0 ist
der Operator der komplexen Konjugation. Den entsprehenden Operator für
vierkomponentige Spinoren erhält maneinfahüber dasTensorprodukt
1
2
^
K: (3.13)
Man kann nun Paare von Spinoren so konstruieren, dass sie durh Anwen-
dungdesZeitumkehroperators auseinanderhervorgehen:
^
K
I
=
I
(3.14)
^
K
I
=
I
(3.15)
Dergequerte Spinor
I
und sein ungequertes Pendant
I
werdendann zu-
sammen als Kramerspaar bezeihnet. Das einfahste Kramerspaar stellen die
Spinorbitale mit -und -Spindar.
AusnutzungderbeshriebenenKramerssymmetrieführtdazu,dassbestimm-
teMatrixelemente inderKramersbasisaufeinanderzurükgeführtwerdenkön-
nen.Beispielsweise können beim später beshriebenen relativistishen Coupled
Cluster die Amplituden für gequerte und ungequerte Spinoren in Beziehung
TransformationsverhaltenvonSpinorenunterRotation. Spin- 1
2
-Spino-
renji(stehtdabeifüreinenbeliebigenSpinzustand)transformierensihunter
Rotation umden Winkel umeineallgemeine Ahsen^ gemäÿ
D
^ n
( )ji = exp
i
~n^
2
j i
=
1os
2
i~n^sin
2
ji (3.16)
mit ~ = (
1
;
2
;
3 )
T
:
Für Rotationenumdiez-Ahse^ ergibt sih
ji!
e i=2
hji
e i=2
h ji
; (3.17)
wobei/ dies
z
-Quantenzahlen 1
2
darstellen. Unter Rotation um2 umdie
^
z-Ahsegehen2-SpinoreninihrNegatives überunderst unterRotation um4
erhält man die Identität. Dieshat zur Folge, dass jedem Element der Gruppe
SO(3) (Rotationen in drei Dimensionen) zwei Elemente der SU(2) (Gruppe
derspeziellen unitären Transformationen inzweiDimensionen - zuihr gehören
Rotationenim Spin- 1
2
-Raum) entsprehen.
Doppelgruppen. Zur gruppentheoretishen Beshreibung von Spinfunktio-
nen werden daherDoppelgruppen eingeführt, in denen dieRotation um4 als
Identität festgesetzt ist, die Rotation um 2 ist in den Doppelgruppen eine
gewöhnlihe Rotation. Die Gruppenordnung verdoppelt sih im Vergleih zur
ursprünglihen Punktgruppe,dajedesElement derPunktgruppe mitder Rota-
tionum2 verknüpft werdenkann.ImFolgenden werden Doppelgruppen stets
mit demSymboldereinfahenMolekülprunktgruppe miteinerhohgestellten2
dargestellt, z.B. D
2h
Punktgruppe, D 2
2h
Doppelgruppe.
Als irreduzible Darstellungen (Irreps) ergeben sih für Doppelgruppen ne-
ben den bosonishen Irreps der Punktgruppe zusätzlihe fermionishe Irreps,
nah denen sih Spinfunktionen mit halbzahligem Gesamtdrehimpuls transfor-
mieren. Bosonishe Irreps beshreiben die Transformation der Funktionen mit
ganzzahligemGesamtdrehimpuls.
Während zur Klassikation der Symmetrie von Wellenfunktionen für Ato-
me und zweiatomige Moleküle meist die Termsymbole 2S+1
X
J
bzw. der Wert
der z-Komponente des Gesamtdrehimpulsvektors = + verwendet wird,
beshreibt mandieZustandssymmetrien inmehratomigen Molekülenüber ihre
Doppelgruppensymmetrie. Dabei wird zunähst die Darstellung D S
des Spin-
multipletts S in der jeweiligen Doppelgruppe bestimmt. Das direkte Produkt
von D S
mit derirreduziblenDarstellungderRaumsymmetrie desnihtrelativi-
stishenZustandsliefertdannnahAusreduktiondieirreduziblenDarstellungen
derrelativistishen Zustände.
DieDarstellung D S
kannfolgendermaÿen erhaltenwerden: Die Komponen-
ten von S transformieren sih unter Rotation wie die eines sphärishen Ten-
sors; nutzt man aus, dass sih die Kugelähenfunktion Y M
J
( ;) wie die
M
J
-te Komponente eines sphärishen Tensors J-ter Stufe transformiert, kann
dasTransformationsverhaltenderSpinfunktionüberden-abhängigenTeilvon
Y M
J
J
ausgedrüktwerden[38℄:
M
S
=e iM
S
; : Raumanteil: (3.18)
Der Charakter der Darstellung unter Rotation umeinen Winkel ' ergibt sih
demnah zu
'
= S
X
MS= S e
iM
S '
: (3.19)
Ausreduktionliefert die irreduziblen Darstellungen,nah denen sih die D S
in
derjeweiligen Doppelgruppetransformiert.
FürvieleFällesinddieD S
inSpinkorrelationstafelnzusammengetragen(sie-
hez.B.[37,39℄),wieinTabelle3.1fürdieDoppelgruppenC 2
2v
undD 2
2h
.Daraus
könnendieausreduziertenDarstellungenfürdieS entnommenwerden.DieDar-
stellungbeliebigerDrehimpulseJ lässtsihgemäÿD J
=D S=J
ebenfallsdaraus
entnehmen.
D S
C 2
2v
D 2
2h
D 0
A
1
A
g
D 1=2
E
1=2
E
g=2
D 1
A
2 B
1 B
2
B
1g B
2g B
3g
D 3=2
2E
1=2
2E
g=2
D 2
2A
1 A
2 B
1 B
2 2A
g B
1g B
2g B
3g
.
.
.
.
.
.
.
.
.
Tabelle 3.1: Korrelation derDarstellung D S
desSpinmultipletts 2S+1 inden
Doppelgruppen C 2
2v
und D 2
2h .
3.2 Methoden der relativistishen Quantenhemie
3.2.1 Die Vielteilhen-Diragleihung
EineLorentz-kovariante Formulierung derVielteilhen-Dira-Gleihung ist bis-
lang niht gelungen und stellt immer noh ein aktives Forshungsgebiet der
Quantenfeldtheorie dar. Für die in quantenhemishen Berehnung verlangte
Genaugkeit genügt es aber, eine näherungsweise Lorentz-kovariante Gleihung
zuverwenden. DabeiwirdderVielteilhen-Hamiltonoperator angesetztals [40℄
H= X
i h
D (i)+
X
i<j
V(i;j) (3.20)
mitdem Einelektronen-Dira-Hamiltonoperator
h (i)=~ ~p +m 2
+V ( r): (3.21)
V ( r
i
) istdasKernpotential fürTeilhen iundV (i;j) beshreibt dieElektron-
Elektron-Wehselwirkung.GängigistdieVerwendungdesDira-Coulomb-Opera-
tors
h
DC
=h
D (i)+
1
r
ij
(3.22)
oderdesDira-Coulomb-Gaunt-Operators
h
DCG
=h
D (i)+
1
r
ij
~
i ~
j
r
ij
: (3.23)
Auf dieVerwendung desexakterenDira-Coulomb-Breit-Operators
h
DCB
=h
D (i)+
1
r
ij
~
i ~
j
2r
ij (~
i ~r
ij )(~
j ~r
ij )
2r 3
ij
(3.24)
wirdmeistverzihtet,dadiezuberehnendenIntegralewegendesdrittenTerms
(Eihterm) sehr aufwändig zu berehnen sind und sih für die Quantenhemie
dershwerenElemente kaumwihtige Beiträge mehrergeben.
3.2.2 Die Dira-Hartree-Fok SCF-Näherung
DieDira-Hartree-Fok-Näherung(DHF)[41℄ähneltformalsehrihremnihtre-
lativistishenAnalogon,derSCF-Methode.WiederwirddieGesamtwellenfunk-
tionalsSlaterdeterminanteangesetztundausderVariationdesEnergiefunktio-
nals unter Variation derOrbitaledie DHF-Gleihungen hergeleitet.
Ein wesentliher Untershied zwishen DHF und HF liegt in der Form der
Orbitale,dieimvierkomponentigenFormalismusnatürlih4-Spinorensind.Zum
Erhalt der relativistishen Dira-Fok-Roothaan-Gleihungen werdendie groÿe
unddiekleineKomponenteinderBasisvon2-Spinoren 4L
bzw.
4S
expandiert.
Unter Verwendung des Dira-Coulomb-Hamiltonoperators erhält man für den
i-ten Spinor [13℄
V LL
N +J
LL
K LL
LS
K LS
SL
K SL
V SS
N +J
SS
K SS
2 2
S SS
C L
i
C S
i
=
S LL
0
0 S
SS
C L
i
C S
i
i
(3.25)
wobei die VektorenC die Koezienten derBasissatzexpansion enthalten, und
S dieÜberlappmatrizen sind. Als DHF-Energieergibt sih[42℄
E DC
= D
LL
V LL
+D SS
V SS
+D SL
LS
+D LS
SL
2m 2
D SS
S SS
+ 1
2
D LL
I LL;LL
D LL
+D LL
I LL;SS
D SS
+D SS
I SS;LL
D LL
+D LS
I SL;LS
D SL
+D SL
I LS;SL
D LS
SS SS;SS SS
wobei D;V ; und S Dihte-, Potentialenergie-, kinematisher Impuls und
Überlappmatrizen sind. I ist dieMatrixderZweielektronenintegrale mit
I VW;XY
ij;kl
= Z
dr
1 Z
dr
2
Vy
i (1)
W
j ( 1)
Xy
k (2)
Y
l (2)
r
12
Æ
VW Æ
XY
Z
dr
1 Z
dr
2
Vy
i ( 1)
Y
l ( 1)
Xy
k (2)
W
j ( 2)
r
12
Æ
VY Æ
XW
:(3.27)
DieZweielektronenmatrix istblokdiagonal inden Komponenten.
3.2.3 Expansion im Basissatz
Im Untershied zu ihrem nihtrelativistishen Pendant werden relativistishe
Basissätze natürlih aus relativitishen Optimierungen erhalten. Die Beshrei-
bung relativistisher Eekte kann es notwendig mahen, bei der Konstruktion
der Basissätze auf besondere Eigenshaften zu ahten. Beispielsweise ist zur
korrekten Beshreibung der Spin-Bahn-Aufspaltung des p-Orbitals in den Ele-
mentenderdrittenHauptgruppeeineVergröÿerungderAnzahlanp-Funktionen
imVergleih zurnihtrelativistishen Basisnötig [43℄.
Zwishen groÿer undkleiner Komponente gilt dieBeziehung
S
=
1
E+V +m 2
(~~p) L
; (3.28)
die auh als kinetishe Balane bezeihnet wird. Ein Basissatz für die kleine
KomponentelässtsihdemnahdurhAnwendung desDierentialoperatorsauf
die Basisfunktionen der groÿen Komponente erhalten. Hält ein Basissatz die
Bedingung derkinetishen Balaneniht ein, kommt eszur Kontamination der
elektonishenLösungmitpositronishenZuständenundzueinem beshränkten
variationellen Kollaps.
Eine gängige Methode zur Erzeugung kinetish balanierter Basissätze ist
die Erzeugung der Basis für die kleine Komponente aus den Funktionen der
groÿen Basis durh Anwendung des Dierentialoperators. Dabei entsteht aus
jedem Drehimpuls `in der groÿen Basis je eine kleine Funktion mit ` 1 und
`+1, wie aus der Rodrigues-Formel für assoziierte Legendre-Funktionen er-
sihtlih ist [44℄. Des Weiteren werden zur exakten Einhaltung der kinetishen
Balane häug unkontrahierte Basissätze verwendet. Die sih ergebenden Ba-
sissätze für die kleine Komponente können wegen der höheren Entartung der
(`+1) -Funktionendoppelt sogroÿwie diedergroÿenKomponente werden.
Hauptproblem bei der praktishen Durhführung von DHF-Berehnungen
ist wegen der Gröÿe des Basissatzes für die kleine Komponente die Bereh-
nung der Zweielektronenintegrale der kleinen Komponente. Bei Verbindungen
mitnureinemshwerenElementkönnendieI SS;SS
-Integralevernahlässigtwer-
den, ohne dass daraus merklihe Fehler resultieren, denn diese Integralklasse
trägt formell nur zur Ordnung 4
zur Energie bei.
2
Werden jedoh Molekü-
le mit mehreren shweren Zentren betrahtet, führt die starke Population der
2
kleinenKomponentedazu,dassdieI SS;SS
-Integralenihtmehr vollständigver-
nahlässigt werden können. Die weitgehende Lokalisation der Dihte D SS
in
Kernnähe kann ausgenutzt werden, um den Beitrag der SS;SS-Integrale zur
DHF-EnergieineinenEin-undeinenMehrzentrenbeitragzuzerlegen.Vernah-
lässigtmandieAustaushwehselwirkung zwishenDihten, dieanuntershied-
lihenZentrenlokalisiertsind,kannderMehrzentrenbeitragdurhreineelektro-
statishe Wehselwirkung beshrieben werden, im allgemeinen Fall also durh
Multipolentwiklung. Abbruh derMultipolentwiklung nah dem dominanten
Ladungs-Ladungs-WehselwirkungstermführtzurSimpleCoulombiCorretion
(SCC)[45℄.Im VergleihzurDHF-Energieohne I SS;SS
-Berüksihtigung liefert
sie den Energiebeitrag
E=
X
A E
SS
A +
1
2 X
A6=B q
S
A q
S
B
R
AB
: (3.29)
Hierbei ist E SS
A
der lokale Energiebeitrag der Integrale I SS;SS
am Zentrum
A und q S
A
ist die Ladung inder kleinen Komponente amAtom A. Numerishe
Tests belegen,dass Verwendung der SCC für zweiatomige Moleküle der 6. Pe-
riodeFehler von biszu 510 5
a:u:verursahen können,wasaber imVergleih
zuanderenFehlernundvorallemdurhdieerzielte BeshleunigungderBereh-
nungen um einen Faktor 3 und Verringerung des benötigten Speiherplatzes
vertretbar ist.
3.2.4 Vierkomponentiges relativistishes Coupled-Cluster
IndiesemAbshnitt wirddierelativistishe CCSD(T)-Variante,wie sieimPro-
gramm RELCCSD [46,47℄ implementiert ist,skizziert. Um denVergleih mit der
nihtrelativistishen Versionmöglihsteinfahzu mahen,halteih mih inder
Notationmöglihstan 2.2.3.3. Zur Untersheidung von besetzten Spinorenvon
Kramers-Paaren werdendie ersten mit I;J;K ;L, dieletzteren mit i;j;k;l und
unbesetzte Spinoren mit A;B;C ;D (a;b;;d) bezeihnet. Beliebige Spinoren
werden alsP;Q;R ;S (p;q;r;s) notiert.
WiediemeistennihtrelativistishenKorrelationsmethodenkannauhCou-
pled Cluster ohne gröÿere Probleme für vierkomponentige Berehnungen mo-
diziert werden, wenn von Elektron-Positron-Paarerzeugun g abgesehen wird
(No-Pair-Näherung). Dazu muss lediglih statt mit Molekülorbitalen mit Mo-
lekülspinoren gearbeitetwerden. Beahtet werdenmuss, dassin derrelativisti-
shen Theorie keine Spinsummation durhgeführt werden kann, da sih Spin-
und Raumsymmetrie niht getrennt behandeln lassen. Stattdessen wird mit
Kramers- undDoppelgruppensymmetrie gearbeitet.
Grundidee beim vierkomponentigen relativistishen Coupled Cluster (REL-
CCSD) ist wieder die Erzeugung von Anregungen bis zur maximalen Ordnung
durh Anwendung einerExponentialfunktion desClusteroperators. Die sih er-
gebenden Gleihungen sind formell ganz ähnlih den nihtrelativistishen CC-
Gleihungen.
terQuantisierung inderBasis derMolekülspinoren:
H NP
= X
I;A Z
A
I
^
E I
A +
1
4 X
I;J X
A;B V
IJ
AB
^
E AB
IJ
: (3.30)
HierbeistelltZdieEinelektronenintegrale,V dieantisymmetrisiertenZweielek-
tronenintegrale und
^
E Anregungsoperatoren dar. In dieser Formulierung un-
tersheiden sih nihtrelativistishe und relativistishe NP-Theorie nur in der
Denitionvon Z undV.
Amplitudengleihungen lassensihanalogzurnihtrelativistishen Methode
z.B.durh Hausdor-Expansionvonexp( T)H NP
exp( T)undMultiplikation
mit Bra-Vektoren angeregter Kongurationen von links erhalten. In der REL-
CCSD-Implementierung[46,47℄ wirdalsCCSD-Energiegleihung
F I
A 2
X
K ;C F
K
C T
A
K T
C
I +
X
C H
A
C T
C
I
X
K H
K
I T
A
K +
X
K ;C H
K
C T
AC
IK +T
A
K T
C
I
(3.31)
+ X
K ;C V
AK
IC T
C
K +
X
K ;C<D V
AK
CD
CD
IK
X
K<L;C V
KL
IC
AC
KL
= 0
mitden Intermediaten
F Q
P
=Z Q
P +
X
K V
QK
PK
(3.32)
AB
IJ
=T AB
IJ +T
A
I T
B
J T
A
J T
B
I
(3.33)
H A
C
=F A
C
X
K<L;D V
KL
CD
AD
KL
(3.34)
H K
I
=F K
I +
X
L;C<D V
KL
CD
CD
IL
(3.35)
H K
C
=F K
C +
X
L;D V
KL
CD T
D
L
(3.36)
unddenClusteramplitudenT A
K
usw.verwendet. DieseFormulierungist äquiva-
lent zu den nihtrelativistishen CCSD-Gleihungen, was wegen der formellen
Übereinstimmung der verwendeten Hamiltonoperatoren auh zu erwarten ist.
Die etwas kompliziertere Struktur der Gleihung (3.31) im Vergleih zu (2.31)
ergibtsihausVerwendungdesOperatorsW imFallderFormulierungderniht-
relativistishen Gleihung. Die hier gewählte Formulierung lässt sih natürlih
besserimplementieren.
AusnutzungderKramers-Symmetrie liefert fürgeshlossenshalige Systeme
letztlih folgendeRelationen für dieClusteramplituden:
t a
i
=t a
i
; t a
i
= t a
(3.37)
t ab
ij
=t a
b
i
j t
a
b
ij
=t ab
i
j
t a
b
i
j
=t ab
ij t
a
b
ij
= t ab
i
j :
(3.38)
Alle anderen t
2
-Amplituden können aus Antisymmetrieeigenshaften erhalten
werden.
Dieerste Amplitudengleihung wirddann zu
f a
i 2
X
k;
f k
t
a
k t
i +
X
h
a
t
i X
h h
k
i
+ X
k;
h k
_
t a
ik +t
a
k t
i
+ X
k;;
h k
t ab
ik
+ X
k;
_
ak
i t
k +
X
k;;
ak
i t
k +
X
k;<d _
ak
d _
d
ik
(3.39)
+ X
k;<d ...
ak
d ...
a
kl +
X
k;;d
ak
d
d
ik
X
k<l ; _
kl
i _
a
kl
X
k<l ; ...
kl
i ...
t a
kl
X
k;l ;
kl
i
a
kl
= 0;
wobeidiePunktein _
t,
tund ...
t
AbkürzungenfürdieAnzahlanuntershiedlihen
Kramerspaaren int sind und ,_ und ...
Abkürzungen für antisymmetrisierte
Zweielektronenintegralesind(diePunktegebendiePermutationssymmetriean).
Weiterhingilt
_
ab
ij
= _
t ab
ij +t
a
i t
b
j t
a
j t
b
i
(3.40)
ab
ij
=
t ab
ij +t
a
i t
b
j
(3.41)
...
ab
ij
= ...
ab
ij
(3.42)
unddieh sindKramers-undpermutationssymmetrishe Analogaderoben ein-
geführten Terme H. Durh Verwendung von Kramerssymmetrie kann der Re-
henaufwand umeinen Faktor von a.2 verringert werden.
DerAusdruk für dieTriples-Korrektur ist wieder ganz analog zur nihtre-
lativistishen Variante
E (T)
=
X
I<J<K X
A<B<C W
ABC
IJK W
ABC
IJK Y
ABC
IJK
D ABC
IJK
; (3.43)
wobei W und D wie im nihtrelativistishen Fall deniert sind. Für RELCCSD
wurde zusätzlih dieneue CCSD-T-Methode mit
Y ABC
IJK
=P
IJK P
ABC
T AB
IJ T
C
K +
1
3 P
IJ T
A
I T
B
J T
C
K
(
I +
J +
K
A
B
C )
(3.44)
implementiert,dieimVergleihzuCCSD(T)noheinenzusätzlihenTermfünf-
Für relativistishes CCSD(T) gelten dieselben Regeln wie für sein nihtre-
lativistishesPendant: Beihinreihend guterQualitätderHF-Determinanteals
Referenz sind Ergebnisse mit nahezu FCI-Qualität erzielbar. Dabei sollte die
T
1
-Diagnoseunter 0,04liegen.
Leider ergibt sih bei den shweren Elementen, für die eine relativistishe
Beshreibung nötig wird, auh die Notwendigkeit zur Korrelation von outer-
Core-Elektronen, z.B. mindestens der d-Elektronen im Tl. Zudem hat man es
in der relativistishen Theorie i.A. mit niedrigerer Symmetrie als in nihtre-
lativistisher Betrahtung zu tun, so dass niht mehr so viele Integrale aus
Symmetriegründen null sind. Vor dem selben Hintergrund nimmt der Multi-
referenzharakter mit der Stärke der relativistishen Eekte zu. Dabei kommt
esvor allem wegender Spin-Bahn-Aufspaltungzur Kopplungder HF-Referenz
mitZuständen anderer Spinmultiplizität.
Stabilität des isolierten Dianions
in der Gasphase
Untersuhung des isolierten Al 2
4
Darin besteht das Wesen der Wissenshaft. Zuerst denkt man an
etwas, das wahr sein könnte. Dann sieht man nah, ob es der Fall
ist und im allgemeinen ist es niht der Fall. BertrandRussell
4.1 Problemstellung
Bei derUntersuhung derTrielidluster auf Aromatizität stützen sih mehrere
Autoren auf quantenhemishe Berehnungen am isolierten Dianion Al 2
4 und
seinenshweren Homologen[2 5,7,9℄. Allerdingsist bekannt,dassessihbeim
Al 2
4
um ein thermodynamish instabiles Molekül handelt, daswahrsheinlih
metastabilund eheralsResonanzzustand [48℄ zu beshreiben ist.
Wie Vergleihe zwishen quantenhemishen Berehnungen für gebundene
Zustände (Bound-State-Beshreibung) und quantenstreutheoretishe Untersu-
hungen für solhe Resonanzzustände zeigen, kann die Bound-State-Beshrei-
bung quantitativ und sogar qualitativfalshe Ergebnisse liefern, da es zurMi-
shungderWellenfunktiondesquasi-gebundenenZustandsmitPseudo-Kontinu-
umszuständen 1
unterhalb desResonanzzustands kommt [49℄, Abb.4.1.
Bei der Rehtfertigung der Bound-State-Beshreibung des Dianions beruft
mansihdarauf,dassdieseswiediemeistenmehrfahgeladenenAnionen(Mul-
tiply Charged Anions, MCAs) in derGasphase thermodynamish instabil, be-
züglih Elektronenverlust jedoh metastabil sei [50℄. Dies begründet man wie
folgt:WährenddasbindendePotentialbzw.dieattraktiveCoulomb-Wehselwir-
kungdiePotentialhyperähe fürElektronenverlustbeiNeutralmolekülen bzw.
Monoanionen dominiert, überwiegt im Fall der MCAs die repulsive Coulomb-
WehselwirkungimlangreihweitigenTeilderHyperähe.DurhÜberlagerung
von kurzreihweitigem bindendem mit langreihweitigem repulsivem Potential
kommt es zur Ausbildung einer Potentialbarriere, der repulsiven Coulombbar-
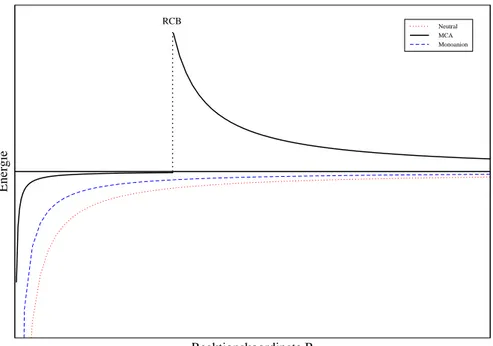
riere(RCB), Abb.4.2.
1
DieBezeihnungalsPseudo-Kontinuumzuständerührtdabeidaher,dassdieWellenfunk-
tionender freienElektroneninderBasissatz-Bound-State-BeshreibungdurhdiuseBasis-
funktionenapproximiertwerden, dieein diskretes Energiespektrum aufweisen. Würden zur
BeshreibungderfreienElektronenebeneWellenoderwieinderzeitabhängigenTheorieWel-
lenpaketeverwendet,soergäbesiheinehtesEnergiekontinuum.
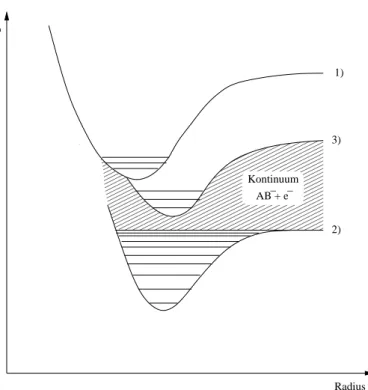
00000000000000000 00000000000000000 00000000000000000 00000000000000000 00000000000000000 00000000000000000 00000000000000000 00000000000000000 00000000000000000 00000000000000000 00000000000000000 00000000000000000 00000000000000000
11111111111111111 11111111111111111 11111111111111111 11111111111111111 11111111111111111 11111111111111111 11111111111111111 11111111111111111 11111111111111111 11111111111111111 11111111111111111 11111111111111111 11111111111111111
Energie
Radius 2)
1)
3)
Kontinuum AB + e − −
Abbildung4.1:PotentialkurvenfüreinhypothetisheszweiatomigesMolekülAB
(1)undsein Mono-(2)bzw. Dianion(3).DasDianionistthermodynamish in-
stabilbezüglihElektronenverlust.Shwingungsniveaussinddurhwaagerehte
Linien angedeutet. Wie man sieht, sind die Zustandsniveaus des Dianions von
einemKontinuumvonZuständendesMonoanionsmitfreiemElektronumgeben.
DurhÜberlappungderWellenfunktionen kanndaherdasDianionohneHinde-
rung ineine Kontinuumslösung übergehen: Das Dianiongeht Autodetahment
zumMonoanion mit freiemElektron ein.
Reaktionskoordinate R
Energie
Neutral MCA Monoanion
RCB
Abbildung 4.2: Shematishe Potentialkurven für Elektronenverlust
bei (a) Neutralmolekülen (AB ! AB +
+e ), (b) einfah geladenen
Anionen (AB ! AB+e ), und () mehrfah geladenen Anionen
AB n
! AB (n 1)
+e
.Eingezeihnet sind nur die im jeweiligen Kurven-
bereih dominierenden Potentiale. Zur Ausbildung der repulsiven Coulomb-
Barriere(RCB)kommt esbeiMCAsdurh Überlagerungderkurzreihweitigen
attraktiven und langreihweitigen repulsiven Wehselwirkung.