Skript zur Vorlesung
Physikalische Chemie I
f¨ ur Studierende der Biochemie, Biologie und des Lehramts (3. Semester)
WS 2012/2013
von
PD Dr. Stephan A. B¨aurle
Skript erstellt von
M. Bernhardt, M. Hammer, M. Knorn
“Eine wirklich gute Idee erkennt man daran, dass ihre Verwirklichung von vorneherein ausgeschlossen erschien”,
Albert Einstein
Vorbemerkung
• Dieses Skript ist eine Abschrift des handschriftlichen Manuskripts “Aufschrieb PC1” der Vorlesung Physikalische Chemie I f¨ur Studierende der Biochemie, Biolo- gie und des Lehramts (3. Semester) von Priv.-Doz. Dr. Stephan A. B¨aurle und dient als Begleitmaterial zu dieser Vorlesung. Es erhebt keinen Anspruch auf Vollst¨andigkeit und kann Fehler enthalten, welche bei der ¨Ubertragung zustan- de kommen k¨onnen.
• Es soll nicht als Ersatz zum Besuch der Vorlesung sowie der ¨Ubungen dienen.
• Fehler und Anregungen k¨onnen gerne unter folgenden E-mail-Adressen1 gemeldet werden.
1stephan.baeurle@chemie.uni-regensburg.de, michaelhammer@hotmail.de
Inhaltsverzeichnis
1 Reaktionskinetik 5
1.1 Reaktionsgeschwindigkeit . . . 5
1.1.1 Konzentrationsabh¨angigkeit der Reaktionsgeschwindigkeit . . . . 6
1.1.2 Gesamt-Reaktionsordnung . . . 6
1.1.3 Umsatzvariable XA(t) . . . 6
1.2 Reaktionen 1. Ordnung in Bezug auf ein Edukt . . . 7
1.2.1 Allgemeine Formulierung . . . 7
1.2.2 Halbwertszeitt1/2 f¨ur Reaktionen 1. Ordnung . . . 8
1.2.3 Mittlere Lebensdauer τ eines Edukt-Teilchens . . . 9
1.3 Reaktionen 2. Ordnung in Bezug auf ein Edukt . . . 9
1.3.1 Allgemeine Formulierung . . . 9
1.3.2 Halbwertszeitt1/2 f¨ur Reaktionen 2. Ordnung in Bezug auf EduktA 10 1.4 Reaktionen der Gesamtordnung 2, jedoch jeweils 1. Ordnung in Bezug auf 2 Edukte . . . 11
1.4.1 Allgemeine Formulierung . . . 11
1.4.2 Halbwertszeitt1/2 (bez¨uglich Edukt A) f¨ur ungleiche Anfangskon- zentrationen . . . 13
1.5 Temperaturabh¨angigkeit der Reaktionsgeschwindigkeitskonstanten . . . . 13
1.5.1 Temperaturverhalten nach Arrhenius . . . 13
1.5.2 Moderne Gleichungen zur Beschreibung der Temperaturabh¨angigkeit der Geschwindigkeitskonstantenk . . . 16
1.6 Reaktionsmechanismus und Reaktionsordnung . . . 16
1.7 Hin- und R¨uckreaktionen 1. Ordnung (Gleichgewichtsreaktionen) . . . 17
1.8 Folgereaktionen . . . 19
1.9 Parallelreaktionen . . . 21
1.10 Folgereaktionen mit vorgelagertem Gleichgewicht und Quasistationarit¨at 23 1.11 Enzymreaktionen . . . 26
1.11.1 Michaelis-Menten-Kinetik . . . 27
1.11.2 Lineweaver-Burk-Gleichung . . . 29
1.12 Reaktionen an fester Katalysatoroberfl¨ache . . . 30
1.12.1 Fall 1: Adsorption/Desorption eines Gases A auf/von einer Kata- lysatoroberfl¨ache . . . 30
1.12.2 Fall 2: Unimolekulare Reaktion an der Katalysatoroberfl¨ache der adsorbierten Spezies . . . 33
1
Inhaltsverzeichnis 2
1.12.3 Fall 3: Bimolekulare Reaktion an der Katalysatoroberfl¨ache zwi-
schen adsorbierten Spezies . . . 34
2 Thermodynamik 39 2.1 Grundlagen und Wiederholung 1. Semester . . . 39
2.1.1 System und Umgebung . . . 39
2.1.2 Prozesse und Randbedingungen . . . 39
2.1.3 Innere und ¨außere Energie . . . 40
2.1.4 Nullter Hauptsatz . . . 41
2.1.5 Erster Hauptsatz . . . 41
2.1.6 Entropie . . . 42
2.1.7 Zweiter Hauptsatz . . . 45
2.1.8 Dritter Hauptsatz . . . 45
2.2 Gibbs’sche Fundamentalgleichungen und charakteristische Funktionen . . 47
2.2.1 Innere Energie U . . . 47
2.2.2 Enthalpie . . . 49
2.2.3 Helmholtz-Energie (freie Energie) F . . . 50
2.2.4 Gibbs’sche Energie (freie Enthalpie) G . . . 51
2.3 Thermodynamisches Gleichgewicht . . . 52
2.4 Gleichgewichte chemischer Reaktionen . . . 55
2.4.1 Gleichgewichtsbedingung und chemische Potentiale . . . 55
2.4.2 Gibbs’sche Reaktionsenergie . . . 57
2.4.3 Konzentrationsabh¨angigkeit des chemischen Potentials . . . 58
2.4.4 Thermodynamische Aktivit¨at verschiedener Stoffklassen . . . 59
2.4.5 Massenwirkungsgesetz . . . 60
2.5 Phasengleichgewichte . . . 63
2.5.1 Clausius-Clapeyron-Gleichung . . . 63
2.5.2 2-Phasen-Gleichgewichte . . . 66
2.5.3 Gibbs’sche Phasenregel . . . 70
2.5.4 Raoult’sches Gesetz . . . 71
2.5.5 Dampfdruckerniedrigung . . . 73
2.5.6 Siedepunktserh¨ohung und Schmelzpunktserniedrigung . . . 74
3 Elektrochemie 78 3.1 Elektrode (Halbzelle) . . . 78
3.2 Elektrochemische Zelle . . . 78
3.3 Galvanisches Element . . . 79
3.3.1 Physikalisch-chemische Prozesse an den Elektroden . . . 80
3.3.2 Zellspannung und elektromotorische Kraft . . . 81
3.3.3 Elektrodenpotential . . . 82
3.3.4 Elektrodenreaktion . . . 83
3.3.5 Zellreaktion . . . 85
3.4 Nernst-Gleichung . . . 86
3.5 Elektroden 2. Art . . . 88
Inhaltsverzeichnis 3
3.6 Elektrolyse-Zelle . . . 89
3.6.1 Faraday’sche Gesetze . . . 91
4 Leitf¨ahigkeit von Elektrolyten 92 4.1 Leitf¨ahigkeit . . . 92
4.1.1 Starke Elektrolyte . . . 93
4.1.2 Schwache Elektrolyte . . . 94
5 Transportvorg¨ange 96 5.1 W¨armeleitung . . . 96
5.2 Diffusion . . . 96
5.2.1 Fick’sche Gesetze . . . 96
5.2.2 Selbstdiffusion . . . 98 6 Physikalisch-chemische Konzepte in der Forschung 99
Empfohlene Literatur
1. Peter W. Atkins,
”Physikalische Chemie“, Wiley-VCH, ISBN-13:978-3527315468 2. Gerd Wedler,
”Lehrbuch der Physikalischen Chemie“, Wiley-VCH, ISBN-13:978- 3527310661
3. Wolfgang Bechmann, Joachim Schmidt,
”Einstieg in die physikalische Chemie f¨ur Nebenf¨achler“, Vieweg+Teubner, ISBN-13:978-3834806079
4. E. Zeidler,
”Teubner-Taschenbuch der Mathematik“, Vieweg+Teubner, ISBN-13:978- 3835101234
4
1 Reaktionskinetik
besch¨aftigt sich mit der Geschwindigkeit und dem Mechanismus einer Reaktion.
Aus kinetischen Messdaten werden ph¨anomenologische Geschwindigkeitsgesetze aufgestellt.
Chemische Reaktionsgleichung (allg. Form):
νAA+νBB+..→νPP +νQQ+..
mit A, B,.. = Ausgangsstoffe (Edukte)b
νA, νB..>0 = st¨b ochiometrische Koeffizienten der Edukte P,Q,.. = Endstoffe (Produkte)b
νP, νQ..>0 = st¨b ochiometrische Koeffizienten der Produkte
1.1 Reaktionsgeschwindigkeit
M¨ogliche Definition:
v? =− 1
νA ·dnA
dt =− 1
νB · dnB
dt =..= 1 νP · dnP
dt =..
mit dnA = ¨b Anderung der Stoffmenge nA des Edukts A im Zeitintervall dt (dni mit i=B,..,P,Q,.. entsprechend)
und v? >0.
Nachteil dieser Definition ist, dass die Reaktionsgeschwindigkeit v? von der Gr¨oße des betrachteten Reaktionssystems abh¨angt.
5
1.1. REAKTIONSGESCHWINDIGKEIT 6
Bessere Definition: f¨ur viele Anwendungen v = v?
V =− 1 νA ·
dnA
V
dt =− 1 νA · dcA
dt =..
mit V als Volumen des Reaktionssystems.
Vereinbarung: im Folgenden werden alle st¨ochiometrischen Reaktionsgleichungen so formuliert, dass νA = 1, d.h.
v =−dcA dt
1.1.1 Konzentrationsabh¨ angigkeit der Reaktionsgeschwindigkeit
Es gilt der allgemeine Ansatz (hier der Edukte):
v =k·cαA·cβB·...·cσS ·...
mit k = Geschwindigkeitskonstante der betrachteten Reaktionb
α = Reaktionsordnung in Bezug auf Reaktionspartnerb A (entsprechend f¨urβ, σ)
Anmerkung: α = 0 bedeutet, dass v nicht von der Konzentration des Stoffes A abh¨angt.
1.1.2 Gesamt-Reaktionsordnung
m=α+β+...+σ+..
bildet sich aus der Summe der Reaktionsordnungen aller Reaktionspartner (Edukte).
1.1.3 Umsatzvariable X
A(t)
Definition: Stoffmenge des Edukts Apro Volumen V, die zwischen t= 0 (Beginn der Reaktion) undt umgesetzt wird, d.h.:
XA(t) = nA(t= 0)−nA(t)
V =cA(t= 0)−cA(t) (1.1)
mit cA(t = 0) = Anfangskonzentration vonb A.
1.2. REAKTIONEN 1. ORDNUNG IN BEZUG AUF EIN EDUKT 7
1.2 Reaktionen 1. Ordnung in Bezug auf ein Edukt
Bei diesem Reaktionstyp ist die Reaktionsgeschwindigkeit zum Zeitpunkttproportional zur Konzentration eines Reaktionspartners zum Zeitpunktt.
Beispiele:
1. radioaktiver Zerfall (→ siehe ¨Ubungen)
2. Zerfall vieler Molek¨ule, die mehr als drei Atome besitzen, wie z.B.:
N2O5 →N2O4+1
2O2 (1.2)
Geschwindigkeitsgesetz:
v =−dcN2O5
dt =k·cN2O5
1.2.1 Allgemeine Formulierung
f¨ur den Fall, dass ein StoffAin einer Reaktion 1. Ordnung bez¨uglichcAverbraucht wird, d.h. entsprechend der Reaktionsgleichung
A−→k P
Geschwindigkeitsgesetz:
v =−dcA
dt =k·cA Integration des obigen Geschwindigkeitsgesetzes:
dcA
cA = d lncA=−k·dt Z lncA(t)
lncA(t=0)
d lncA=−k Z t
t=0
dt lncA(t)−lncA(t = 0) =−k·t Mit Abk¨urzung cA(t= 0) =a ergibt sich:
lncA(t) = lna−k·t
Graphische Darstellung: lncA(t) als Funktion von t ergibt eine Gerade mit der Steigung −k und dem Ordinatenabschnitt lna.
ln
cA(t) a
=−k·t
1.2. REAKTIONEN 1. ORDNUNG IN BEZUG AUF EIN EDUKT 8
Abb. 1.1: Konzentration des Edukts A als Funktion der Zeit im Falle einer Reaktion 0., 1.
und 2. Ordnung
Graphische Darstellung: ln (cA(t)/a) als Funktion vont ergibt eine Gerade mit der Steigung −k und dem Ordinatenabschnitt Null.
Aufl¨osen nach cA(t) liefert:
cA(t) =a·exp(−k·t) (1.3)
Das Abklingverhalten der Konzentration von A, ausgehend vom StartwertcA(t= 0) =a wird durch die Abklingfunktion f(t) = exp(−k·t) beschrieben, welche allein durch den Parameterk bestimmt wird (siehe Abb. (1.1)).
Beispiel Reaktionsgleichung (1.2):
cN2O5(t) = cN2O5(t= 0)·exp(−k·t)
1.2.2 Halbwertszeit t
1/2f¨ ur Reaktionen 1. Ordnung
Die Halbwertszeit entspricht dem Zeitintervall t1/2, in dem die Konzentration cA(t) auf den halben Wert abgefallen ist, wie z.B. von cA(t = 0) =a aufcA(t=t1/2) = a2
Allgemeiner Zusammenhang zwischen cA(t1), cA(t2) und Zeitdifferenz (t2 −t1) f¨ur Re- aktionen 1. Ordnung (Substraktion von integralem Zeitgesetz):
lncA(t1)
cA(t2) =k·(t2−t1)
1.3. REAKTIONEN 2. ORDNUNG IN BEZUG AUF EIN EDUKT 9
Einsetzen voncA(t2) = cA(t21) und t2−t1 =t1/2 liefert:
t1/2 = ln 2
k ≈ 0.7 k
Hieraus l¨asst sich ersehen, dass die Halbwertszeit t1/2 bei Reaktionen 1. Ordnung kon- zentrationsunabh¨angig ist.
1.2.3 Mittlere Lebensdauer τ eines Edukt-Teilchens
Die mittlere Lebensdauer τ eines Edukt-Teilchens ergibt sich aus den individuellen Le- bensdauern der Edukt-Teilchen τ1, τ2, τ3,..., gerechnet ab dem Zeitpunkt t = 0, wie folgt:
τ = τ1+τ2+τ3+..
N(t= 0)
wobei N(t=0) der Anzahl der Edukt-Teilchen beit = 0 entspricht.
F¨ur Reaktionen 1. Ordnung gilt:
τ = 1
k (1.4)
Einsetzen von Gleichung (1.4) in Gleichung (1.3) ergibt, dass zum Zeitpunkt t = τ die Anzahl bzw. Konzentration der Edukt-Teilchen auf den Bruchteil 1e ≈ 0.37 abgefallen ist.
1.3 Reaktionen 2. Ordnung in Bezug auf ein Edukt
Beispiel:
N O2 →N O+ 1 2O2 Geschwindigkeitsgesetz:
v =−dcN O2
dt =k·c2N O
2
1.3.1 Allgemeine Formulierung
f¨ur den Fall, dass ein StoffAin einer Reaktion 2. Ordnung bez¨uglichcAverbraucht wird:
v =−dcA
dt =k·c2A
1.3. REAKTIONEN 2. ORDNUNG IN BEZUG AUF EIN EDUKT 10
Integration des Geschwindigkeitgesetzes:
−dcA
c2A =−c−2A ·dcA=k·dt
− Z cA(t)
cA(t=0)=a
c−2A ·dcA=k· Z t
t=0
dt 1
cA(t)− 1
a =k·t 1
cA(t) = 1
a +k·t (1.5)
Graphische Darstellung: c1
A(t) als Funktion von t ergibt eine Gerade mit der Stei- gung k und dem Ordinatenabschnitt 1a.
Durch Aufl¨osen von Gleichung (1.5) nach cA(t) ergibt sich:
cA(t) =a· 1
1 +k·a·t (1.6)
Das Abklingverhalten der Konzentration von A, ausgehend vom Startwert cA(t= 0) = a, wird durch die Abklingfunktion f(t) = 1+k·a·t1 beschrieben, welche durch k und a bestimmt wird (siehe Abb. (1.1)) .
1.3.2 Halbwertszeit t
1/2f¨ ur Reaktionen 2. Ordnung in Bezug auf Edukt A
Allgemeiner Zusammenhang zwischencA(t1),cA(t2) und Zeitdifferenz (t2−t1) ergibt sich durch Subtraktion von Gleichung (1.5):
1
cA(t2) − 1
cA(t1) =k·(t2−t1)
Einsetzen voncA(t1 = 0) =a und cA(t2) = a2 sowiet2−t1 =t1/2 liefert:
t1/2 = 1 k·a
Hieraus ist ersichtlich, dass die Halbwertszeit t1/2 bei Reaktionen 2. Ordnung abh¨angig von der Anfangskonzentrationa ist.
1.4. REAKTIONEN DER GESAMTORDNUNG 2, JEDOCH JEWEILS 1.
ORDNUNG IN BEZUG AUF 2 EDUKTE 11
1.4 Reaktionen der Gesamtordnung 2, jedoch jeweils 1.
Ordnung in Bezug auf 2 Edukte
Beispiel:
H2+I2 →2HI Geschwindigkeitsgesetz:
v =−dcH2
dt =k·cH2 ·cI2
1.4.1 Allgemeine Formulierung
f¨ur den Fall, dass ein Stoff A in einer Reaktion 2. Gesamtordnung, jedoch 1. Ordnung in Bezug auf Edukt A und B verbraucht wird, gem¨aß folgender Reaktionsgleichung:
A+B →P (1.7)
Geschwindigkeitsgesetz:
v =−dcA
dt =k·cA·cB (1.8)
L¨osung f¨ur gleiche Anfangskonzentrationen:
cA(t= 0) =cB(t = 0) MitcA(t= 0) =a und cB(t= 0) =b folgt
a =b (1.9)
Aufgrund der St¨ochiometrie von Reaktion (1.7) gilt dann zu jedem Zeitpunkt t:
cA(t) = cB(t) (1.10)
Einsetzen in Gleichung (1.8) liefert:
v =−dcA
dt =k·c2A Durch Integration ergibt sich1:
cA(t) =a· 1 1 +a·k·t
1Vergleich mit Fall Reaktion 2. Ordnung in Bezug auf Edukt A in Gleichung (1.6)
1.4. REAKTIONEN DER GESAMTORDNUNG 2, JEDOCH JEWEILS 1.
ORDNUNG IN BEZUG AUF 2 EDUKTE 12
und wegen Gleichung (1.9) und Gleichung (1.10) cB(t) = b· 1
1 +b·k·t L¨osung f¨ur ungleiche Anfangskonzentrationen Es gilt:
cA(t = 0)6=cB(t= 0) d.h. a6=b
Aufgrund der St¨ochiometrie der Reaktion gilt hier zu jedem Zeitpunkt t:
cB(t) = cA(t) + (b−a) Einsetzen in Gleichung (1.8) liefert:
−dcA
dt =k·cA·[cA(t) + (b−a)]
| {z }
=cB(t)
Integriertes Geschwindigkeitsgesetz:
ln cA(t)
cA(t) + (b−a) = lncA(t)
cB(t) = lna
b −(b−a)·k·t (1.11) Durch Aufl¨osen von Gleichung (1.11) nach cA(t) ergibt sich:
cA(t) =a· (b−a)
b·exp((b−a)·k·t)−a =a·f(t)
Das Abklingverhalten der Konzentration vonA, ausgehend vom StartwertcA(t= 0) =a, wird durch die Abklingfunktionf(t) beschrieben, welche sowohl durch k als auch durch a und b bestimmt wird.
N¨aherung f¨ur Fall b a:
lncB(t)≈lnb und (b−a)≈b Damit ergibt sich aus Gleichung (1.11):
lncA(t) = lna−b·k·t Daraus folgt:
cA(t) =a·exp(−b·k·t)
1.5. TEMPERATURABH ¨ANGIGKEIT DER
REAKTIONSGESCHWINDIGKEITSKONSTANTEN 13
In diesem Fall ergibt sich n¨aherungsweise eine Reaktion 1. Ordnung, welche als eine sogenannte Reaktion pseudo 1. Ordnung bezeichnet wird (Vergleiche mit Reaktion 1. Ordnung (Gleichung (1.3))).
1.4.2 Halbwertszeit t
1/2(bez¨ uglich Edukt A) f¨ ur ungleiche Anfangskonzentrationen
Allgemeiner Zusammenhang zwischencA(t1), cA(t2), cB(t1), cB(t2) und (t2−t1):
1
b−a ·lncA(t1)·cB(t2)
cA(t2)·cB(t1) =k·(t2 −t1) Einsetzen voncA(t1) =a, cB(t1) =b, cA(t2) = a2, cB(t2) = b− a
2
| {z }
=cA(t2)
| {z }
=a 2
+(b−a)
und (t2−t1) =
tA1/2 liefert:
tA1/2 = 1 k · 1
b−a ·ln(2− a b)
Hieraus ist erkennbar, dasstA1/2 von beiden Ausgangskonzentrationen a und b abh¨angig ist.
1.5 Temperaturabh¨ angigkeit der
Reaktionsgeschwindigkeitskonstanten
1.5.1 Temperaturverhalten nach Arrhenius
Temperaturverhalten von k(t) wird in vielen F¨allen durch die sogenannte Arrhenius- Gleichung wiedergegeben:
d lnk
dT = Ea
R·T2 (1.12)
wobeiEa der Aktivierungsenergie der betrachteten Reaktion (n¨aherungsweise tempera- turunabh¨angig) entspricht.
Integration von Gleichung (1.12) liefert:
Z lnk(T)
lnk(T=∞)
d lnk = Ea
R Z T
T=∞
1 T2dT
1.5. TEMPERATURABH ¨ANGIGKEIT DER
REAKTIONSGESCHWINDIGKEITSKONSTANTEN 14
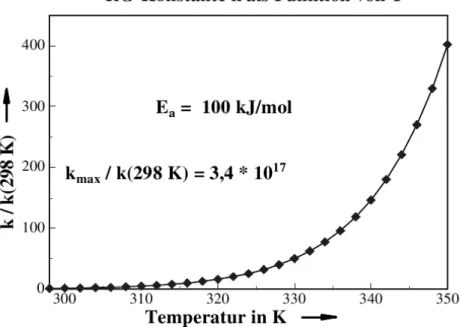
Abb. 1.2: Reaktionsgeschwindigkeitskonstante als Funktion der Temperatur unter der An- nahme, dass die AktivierungsenergieEa temperaturunabh¨angig ist.
lnk= lnk(T =∞)− Ea
R·T
wobei k(T = ∞) = kmax dem maximalen Wert der Geschwindigkeitskonstanten ent- spricht. Es folgt:
k(T) =kmax·exp
− Ea R·T
Anmerkung:H¨aufig wirdkmax auch als pr¨aexponentieller Faktor oder als Frequenzfak- tor A bezeichnet.
Division obiger Gleichung durch k(T = 298K) und Logarithmieren liefert die Gera- dengleichung:
ln
k(T) k(T = 298K)
=− Ea
R·T + ln
kmax
k(T = 298K)
Arrhenius-Auftragung:
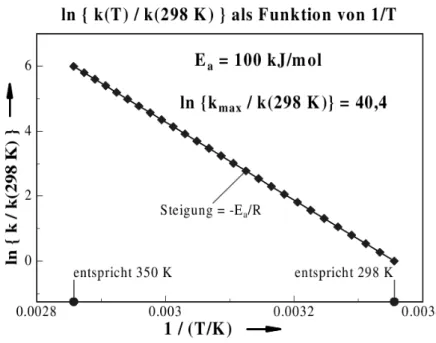
Auftragung von lnk gegen T1 zur graphischen Bestimmung der Aktivierungsenergie Ea (siehe Abb. 1.3 und 1.4 im Vergleich zu Abb. 1.2)
1.5. TEMPERATURABH ¨ANGIGKEIT DER
REAKTIONSGESCHWINDIGKEITSKONSTANTEN 15
Abb. 1.3: Bestimmung der Aktivierungsenergie Ea mit Hilfe der Arrhenius-Auftragung lnk gegen 1/T.
Abb. 1.4: Bestimmung des pr¨aexponentiellen Faktors durch Extrapolation von ln(k(T)/k(T = 298K)) auf 1/T=0.
Steigung der Geraden:
d lnk
d(T1) =−Ea R
1.6. REAKTIONSMECHANISMUS UND REAKTIONSORDNUNG 16
Dieses Verfahren wurde von dem schwedi- schen Physikochemiker Svante August Arr- henius (*1837-†1923) entwickelt. Er erhielt den Nobelpreis 1903.
1.5.2 Moderne Gleichungen zur Beschreibung der
Temperaturabh¨ angigkeit der Geschwindigkeitskonstanten k
lnk(T) =α+ β
T +γ·lnT mit α, β und γ als Konstanten, oder:
lnk(T) =a+b·Tc mit a, b und c als Konstanten.
1.6 Reaktionsmechanismus und Reaktionsordnung
Elementarreaktion: Teilschritt einer Gesamtreaktion, der nicht mehr in weitere Teil- schritte unterteilt werden kann.
Molekularit¨at: einer chemischen Reaktion beschreibt die Anzahl der Molek¨ule, die am geschwindigkeitsbestimmenden Schritt der Reaktion beteiligt sind.
An einer Elementarreaktion k¨onnen beteiligt sein:
• Ein Teilchen (Molek¨ul, Ion, Radikal, . . . ) → unimolekulare Reaktion.
Bsp.:
(CH2)3
| {z }
Cyclopropan
→CH3 −CH =CH2
| {z }
P ropen
1.7. HIN- UND R ¨UCKREAKTIONEN 1. ORDNUNG
(GLEICHGEWICHTSREAKTIONEN) 17
• Zwei Teilchen (Molek¨ule, Ionen, Radikale, . . . ) → bimolekulare Reaktion.
Bsp.: Esterverseifung
CH3COOCH2CH3
| {z }
Essigs¨aureethylester
+OH− →CH3COO−
| {z }
Acetation
+CH3CH2OH
| {z }
Ethanol
• Drei Teilchen (Molek¨ule, Ionen, Radikale, . . . ) → trimolekulare Reaktion.
Dieser Typ von Elementarreaktionen kommt nur sehr selten vor, da die Wahr- scheinlichkeit f¨ur den Zusammenstoß dreier Teilchen w¨ahrend der Reaktion sehr gering ist.
Anmerkung: Die Molekularit¨at einer Reaktion kann nur die Werte 1-3 annehmen.
H¨ohere Werte sind nicht bekannt. Besteht die Gesamtreaktion nur aus einem Elemen- tarschritt, so ist die Ordnung der Gesamtreaktion gleich der Molekularit¨at der Elemen- tarreaktion. Komplexe Reaktionen bestehen aus mehreren Elementarschritten. Sind alle Teilschritte einer komplexen Reaktion bekannt, kann die Gesamtkinetik aus der Kinetik der Elementarreaktionen abgeleitet werden.
1.7 Hin- und R¨ uckreaktionen 1. Ordnung (Gleichgewichtsreaktionen)
Schema:
A−←−−k−−hin→−
kr¨uck
P
F¨ur die Geschwindigkeit der Gesamtreaktion gilt:
−dcA
dt
| {z }
RGGesamt
=khin·cA(t)
| {z }
RGhin
−kr¨uck·cP(t)
| {z }
RGr¨uck
(1.13)
bzw.:
RGgesamt=RGhin−RGr¨uck (1.14)
wobei RG= Reaktionsgeschwindigkeit.b
F¨ur die Konzentrationen der Reaktionsteilnehmer gilt:
cP(t) =cP(t = 0) + [cA(t = 0)−cA(t)]
| {z }
Anteil von A, der zu P wegreagiert ist
=p+ [a−cA(t)] (1.15)
mit p=cP(t = 0). F¨ur die Anfangsbedingung p= 0 ergibt sich aus Gleichung (1.15):
cP(t) = a−cA(t) (1.16)
1.7. HIN- UND R ¨UCKREAKTIONEN 1. ORDNUNG
(GLEICHGEWICHTSREAKTIONEN) 18
Mit Gleichung (1.16) nimmt Gleichung (1.13) folgende Form an:
−dcA
dt =khin·cA(t)−kr¨uck·[a−cA(t)]
= [khin+kr¨uck]·cA(t)−kr¨uck·a
(1.17)
Grenzf¨alle von Gleichung (1.17):
1. Beginn der Reaktiont = 0: cA(t) = a
− dcA dt
t=0
=khin·a
F¨ur diesen Fall ergibt sich ein einfaches Geschwindigkeitsgesetz f¨ur eine Reaktion 1.
Ordnung ohne R¨uckreaktion.
2. Im chemischen Gleichgewicht gilt:
RGGesamt = 0 Aus Gleichung (1.14) folgt somit:
RGhin = RGr¨uck (1.18)
khin·[cA]Gleichgewicht = kr¨uck·[cP]Gleichgewicht
cP cA
Gleichgewicht
= khin
kr¨uck =K (1.19)
wobei K die kinetische Gleichgewichtskonstante der Reaktion darstellt.
Aus Gleichung (1.19) erhalten wir durch Logarithmieren und Differentiation:
d lnK = d ln khin
kr¨uck
Einsetzen der integralen Form der Arrhenius-Gleichung (1.12) f¨ur die Temperaturabh¨angigkeit von k:
khin(T) =kmax·exp
− Eahin R·T
kr¨uck(T) =kmax·exp
−Ear¨uck R·T
1.8. FOLGEREAKTIONEN 19
liefert:
d lnK
d(T1) =−Eahin−Ear¨uck R
Diese Gleichung beschreibt die Temperaturabh¨angigkeit der Gleichgewichtskonstanten K.
Auftragung von lnK gegen T1 liefert die Steigung d(1/Td lnK). Hieraus kann man die Dif- ferenz der AktivierungsenergienEahin−Ear¨uck der Gleichgewichtsreaktion bestimmen.
Durch Integration von Gleichung (1.17) ergibt sich:
cA(t) =a·
=0 f¨urt→∞
z }| {
khin
khin+kr¨uck ·exp [−(khin+kr¨uck)·t] + kr¨uck khin+kr¨uck
Nach langer Zeitt→ ∞wird das chemische Gleichgewicht der Reaktion erreicht und es folgt aus obiger Gleichung:
[cA]Gleichgewicht =a· kr¨uck kr¨uck+khin Mit Gleichung (1.16) ergibt sich:
[cP]Gleichgewicht=a−[cA]Gleichgewicht=a·
1− kr¨uck kr¨uck+khin
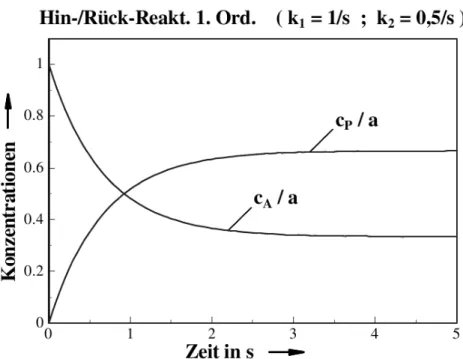
Das Zeitverhalten des Edukts A und Produkts P f¨ur eine Hin- und R¨uckreaktion 1.
Ordnung ist in Abb. (1.5) aufgetragen. Hieraus ist ersichtlich, dass sich nach langer Re- aktionsdauer ein Gleichgewicht der Konzentrationen der Reaktionsteilnehmer einstellt.
F¨ur den Quotienten der Konzentration von Produkt und Edukt im Gleichgewicht folgt somit:
cP cA
Gleichgewicht
= a·
1− k kr¨uck
r¨uck+khin
a· k kr¨uck
r¨uck+khin
= khin kr¨uck =K
Der Quotient der Konzentrationen im Gleichgewicht ist identisch mit der Gleichgewichts- konstanten bzw. dem Quotienten der Geschwindigkeitskonstanten der Reaktion.
1.8 Folgereaktionen
Schema:
A −k→1 P −k→2 Q
1.8. FOLGEREAKTIONEN 20
Abb. 1.5: Konzentration des Edukts A und Produkts P als Funktion der Zeit im Falle einer Hin- und R¨uckreaktion 1. Ordnung.
F¨ur den Fall, dass beide Teilschritte unimolekular ablaufen, gilt:
−dcA
dt =k1 ·cA mit der L¨osung:
cA(t) = a·exp (−k1·t) (1.20) F¨ur das Zwischenprodukt P gilt hingegen:
dcP
dt = k1·cA
| {z }
Bildung aus A
− k2 ·cP
| {z }
Reaktion zu Q
Einsetzen von Gleichung (1.20) ergibt:
dcP
dt +k2·cP =k1 ·a·exp (−k1·t) (1.21) (inhomogene Differentialgleichung 1. Ordnung)
L¨osen der Differentialgleichung (1.21) f¨urk1 6=k2 liefert:
cP(t) = a· k1 k2−k1 ·
exp (−k1·t)−exp (−k2·t)
1.9. PARALLELREAKTIONEN 21
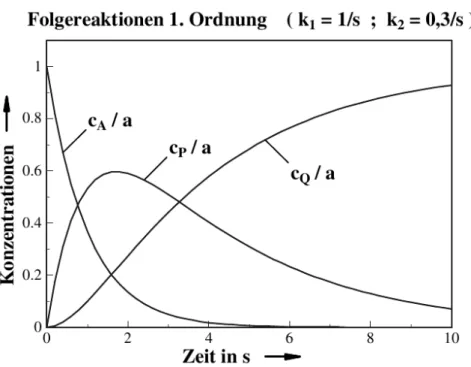
Abb. 1.6: Konzentration des Edukts A, Zwischenprodukts P und Folgeprodukts Q als Funk- tion der Zeit im Falle einer Folgereaktion 1. Ordnung.
F¨ur die Konzentration des Endprodukts Q gilt:
cQ(t) =a−[cA(t) +cP(t)]
Eine Auftragung des Zeitverhaltens der Konzentration f¨ur eine Folgereaktion 1. Ordnung in Bezug auf alle Reaktionsteilnehmer ist in Abb. (1.6) dargestellt.
1.9 Parallelreaktionen
Schema:
A−k→1 P A−k→2 Q
F¨ur den Fall, dass beide Elementarreaktionen unimolekular ablaufen, gilt:
dcP
dt =k1·cA (1.22)
dcQ
dt =k2·cA (1.23)
1.9. PARALLELREAKTIONEN 22
Abb. 1.7: Konzentration des Edukts A, Produkts P und des parallelentstehenden Produkts Q als Funktion der Zeit im Falle einer Parallelreaktion 1. Ordnung
sowie:
−dcA
dt = dcP
dt +dcQ
dt = (k1+k2)·cA Durch Integration ergibt sich die L¨osung:
cA(t) =a·exp
−(k1+k2)·t
(1.24) Verkn¨upfung von Gleichung (1.22) und Gleichung (1.23) ¨ubercA(t) liefert:
dcP = k1 k2 ·dcQ
Durch Integration erhalten wir:
cP(t) = k1
k2 ·cQ(t) (1.25)
Aufgrund der St¨ochiometrie gilt:
a=cA(t) +cP(t) +cQ(t) (1.26)
1.10. FOLGEREAKTIONEN MIT VORGELAGERTEM
GLEICHGEWICHT UND QUASISTATIONARIT ¨AT 23
Aus den Gleichungen (1.24), (1.25) und (1.26) folgt schließlich:
cP(t) = k1
k1+k2 ·a·
1−exp (−(k1+k2)·t)
cQ(t) = k2
k1+k2 ·a·
1−exp (−(k1 +k2)·t)
Eine Auftragung des Zeitverhaltens der Konzentrationen f¨ur eine Parallelreaktion 1.
Ordnung ist in Abb. (1.7) dargestellt.
1.10 Folgereaktionen mit vorgelagertem Gleichgewicht und Quasistationarit¨ at
Chemische Reaktionen in der Biologie und Biochemie bestehen in der Regel aus mehreren Teilreaktionen. Ein typisches Reaktionsschema lautet:
A −−→←−−k1
k−1
B −k→2 C
Unter der Annahme, dass alle Teilreaktionen Zeitgesetzen 1. Ordnung folgen, ergeben sich folgende differentielle Zeitgesetze:
dcA
dt =−k1·cA+k−1·cB (1.27) dcB
dt =k1·cA−k−1·cB−k2·cB dcC
dt =k2·cB Aufgrund der Stoffmengenerhaltung gilt außerdem:
cA+cB+cC =c0A
wobeic0A=cA(t= 0) =adie Anfangskonzentration von A darstellt (Schreibweise in der Biochemie gebr¨auchlich). Hieraus folgt:
dcA+ dcB+ dcC = 0
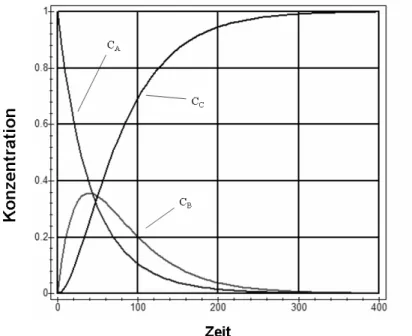
Obiges Gleichungssystem ist mit herk¨ommlichen analytischen Integrationsverfahren f¨ur den allgemeinen Fall nicht l¨osbar. Mit Hilfe numerischer Integrationsverfahren k¨onnen je- doch die Konzentrationsverl¨aufe der Reaktionsteilnehmer ermittelt werden (siehe hierzu Abb. (1.8)).
1.10. FOLGEREAKTIONEN MIT VORGELAGERTEM
GLEICHGEWICHT UND QUASISTATIONARIT ¨AT 24
Abb. 1.8: Konzentrationsverlauf des Edukts A, Zwischenprodukts B und des Endprodukts C als Funktion der Zeit bei einer Folgereaktion 1. Ordnung mit vorgelagertem Gleichgewicht (allgemeiner Fall).
Allgemeiner Fall: in Abb. (1.8) beobachtet man, dass cA exponentiell abnimmt und cB ein Maximum durchl¨auft. Dagegen cC zuerst langsamer als cB und nach dem Wen- depunkt exponentiell bis zum maximalen Wert ansteigt.
Wichtiger Spezialfall: wird in Abb. (1.9) gezeigt. Hier erfolgt Bildung von B aus A langsam. Dies stellt den geschwindigkeitsbestimmenden Schritt der Gesamtreaktion dar. Dann reagiert reaktives Zwischenprodukt B schnell entweder zu A zur¨uck oder zu C weiter, d.h.k−1, k2 k1. Dies hat zur Folge, dass die Konzentration von B auf sehr klei- nem Niveau gegen¨uber den anderen Konzentrationen bleibt. Hieraus resultiert eine sehr flache Kurve. In diesem Fall kann f¨ur Zwischenprodukt B ein quasistation¨arer Zustand angenommen werden:
dcB
dt =k1·cA−k−1·cB−k2·cB ≈0 (1.28) Durch Aufl¨osen obiger Gleichung nach cB ergibt sich:
cB =cA· k1 k−1+k2
1.10. FOLGEREAKTIONEN MIT VORGELAGERTEM
GLEICHGEWICHT UND QUASISTATIONARIT ¨AT 25
Abb. 1.9: Konzentrationsverlauf des Edukts A, Zwischenprodukts B und Endprodukts C bei einer Folgereaktion 1. Ordnung mit vorgelagertem Gleichgewicht. Spezialfall: Bil- dung von B aus A erfolgt langsam und B reagiert schnell entweder zu A zur¨uck oder zu C weiter. Konzentration von B bleibt sehr klein gegen¨uber den anderen Konzen- trationen. In diesem Fall kann ein quasistation¨arer Zustand f¨ur Zwischenprodukt B angenommen werden.
Einsetzen in Gleichung (1.27) liefert schließlich:
dcA
dt =−k1·cA+ k1·k−1
k−1+k2 ·cA=−
=k
z }| { k1·k2 k−1+k2·cA dcC
dt = k1·k2 k−1+k2
| {z }
=k
·cA
Durch Einf¨uhrung der Konstanten k = kk1·k2
−1+k2 ergibt sich n¨aherungsweise eine Differen- tialgleichung 1. Ordnung f¨ur Edukt A:
dcA
dt =−k·cA
mit integralem Zeitgesetz:
ln cA
c0A
=−k·t
1.11. ENZYMREAKTIONEN 26
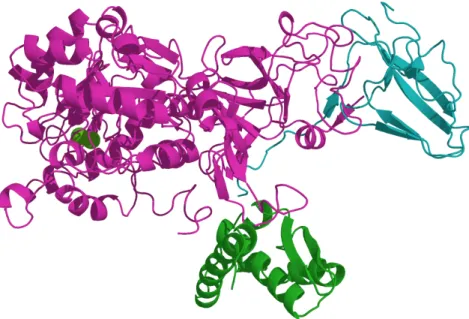
Abb. 1.10: Enzym Urease isoliert aus dem BakteriumKlebsellia Aerogenes. Es katalysiert die Hydrolysereaktion von Harnstoff zu Kohlenstoffdioxid und Ammoniak.
Dies entspricht n¨aherungsweise einer Reaktion 1. Ordnung folgenden Typs:
A−→k C
Obige Herleitung zeigt, dass eine Folge von Reaktionen, in der nur der langsamste Re- aktionsschritt f¨ur die Gesamtreaktion maßgeblich ist, n¨aherungsweise als eine Reaktion 1. Ordnung angesehen werden kann. Die quasi-station¨are N¨aherung kann in der Regel zur Behandlung von Reaktionen mit reaktivem Zwischenprodukt, wie z.B. von Enzym- und Radikalreaktionen, herangezogen werden.
1.11 Enzymreaktionen
Katalysatoren: sind Molek¨ule, die die Geschwindigkeit einer Reaktion erh¨ohen, jedoch aus der Reaktion unver¨andert hervorgehen.
Enzyme: sind biologische Katalysatoren auf Basis von Aminos¨auren (Proteine), welche bei der Steuerung des Stoffwechselgeschehens eine wichtige Rolle spielen.
Beispiel: Urease kommt besonders in Pflanzensamen und Bakterien vor (siehe Abb.
(1.10)). Sie katalysiert die Hydrolyse-Reaktion von Harnstoff:
H2N CON H2+ 3H2O −−−−→U rease 2N H4++OH−+HCO−3
1.11. ENZYMREAKTIONEN 27
1.11.1 Michaelis-Menten-Kinetik
nach Leonor Michaelis und Maud Menten 1913.
TypischesReaktionsschema einer Enzymreaktion:
S+E −−→←−−k1
k−1
ES −→kP P +E
wobei S = Substratb E = freies Enzymb
ES = Enzym-Substrat-Komplexb P = Produktb
Annahme: Hinreaktion sei 2. Gesamtordnung; die R¨uckreaktion sowie die Produkt- bildungsreaktion sei 1. Ordnung.
Differentielle Zeitgesetze f¨urES und P:
dcES
dt = k+1·cS·cE
| {z }
T1
−(k−1
|{z}
T2
+ kp
|{z}
T3
)·cES (1.29)
dcP
dt = kP ·cES (1.30)
wobei T1 = Bildung von ES aus E und Sb T2 = R¨b uckbildung von E und S T3 = Weiterreaktion zu P und Eb
F¨ur die Gesamtkonzentration des Enzyms gilt:
c0E =cE+cES (1.31)
1.11. ENZYMREAKTIONEN 28
Mittels Gleichung (1.31) k¨onnen wir cE aus Gleichung (1.29) eliminieren und wir erhal- ten:
dcES
dt =k+1·cS·c0E−(k+1·cS+k−1+kp)·cES (1.32) Bei Enzymreaktionen arbeitet man in der Regel mit sehr kleinen Enzymkonzentrationen, d.h. cS cES, und Substrat¨uberschußcS ≈c0s. F¨ur den Fall, daß der Enzym-Substrat- Komplex als reaktives Zwischenprodukt mit k−1, kP k+1 angenommen werden kann, kann die quasi-station¨are N¨aherung auf Gleichung (1.32) angewendet werden:
dcES dt ≈0 Durch Aufl¨osen nach cES ergibt sich:
cES = cS
cS+KM ·c0E (1.33)
wobei
KM = k−1+kP
k+1 (1.34)
die sog. Michaelis-Konstante darstellt. Einsetzen von Gl. (1.33) in Gleichung (1.30) liefert schließlich die Produktbildungsgeschwindigkeit:
vP = dcP
dt =kP ·c0E · cS
cS+KM Mit Hilfe der Definition f¨ur denMaximalwert von vP:
vPmax =kP ·c0E
welcher an der Substrat-S¨attigungsgrenze erreicht wird, ergibt sich schliesslich das soge- nannte Michaelis-Menten-Gesetz:
vP =vmaxP · cS
cS+KM (1.35)
wobeicS ≈c0S. Es stellt eine grundlegende Beziehung der Enzym-Kinetik dar ! Die sogenannte Wechselrate (turn-over-rate) des Enzyms:
kcat = vmaxP c0E
beschreibt wieviele Substratmolek¨ule von einem substratges¨attigten Enzymmolek¨ul pro Zeit umgesetzt werden.
1.11. ENZYMREAKTIONEN 29
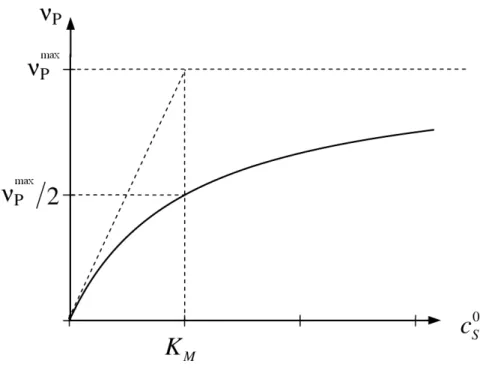
Abb. 1.11: Graphische Darstellung der Michaelis-Menten-Kinetik, in der die Produktbil- dungsgeschwindigkeit als Funktion der Gesamtkonzentration des Substrats auf- getragen wird.
Die ProduktbildungsgeschwindigkeitvP wird in enzymkinetischen Messungen als Funk- tion von c0S gemessen. Da das exakte cS w¨ahrend der Reaktion nur schwer bestimmbar ist, wird cS ≈ c0S angenommen und vP(c0S) aufgetragen (siehe Abb. (1.11)). Aus der Auftragung des Michaelis-Menten-Gesetzes ist erkennbar, dass bei c0S = KM die Pro- duktbildungsgeschwindigkeit vP = vmaxP /2 ist, daher wird die Michaelis-Konstante KM auch als Halbs¨attigungskonzentration bezeichnet !
1.11.2 Lineweaver-Burk-Gleichung
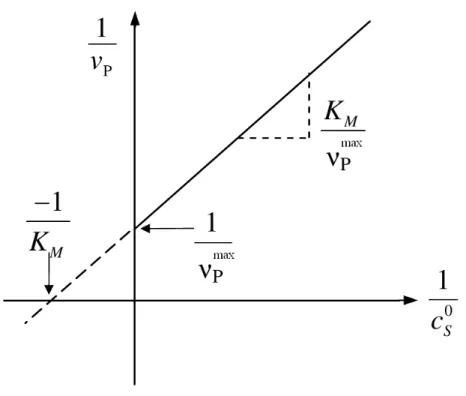
Um Meßwerte von vP auf Vertr¨aglichkeit mit der Michaelis-Menten-Kinetik zu pr¨ufen und gleichzeitig die Konstanten KM und vPmax zu bestimmen, formt man das Michaelis- Menten-Gesetz Gl. (1.35) so um, dass man eine Geradengleichung, die sog. Lineweaver- Burk-Gleichung, erh¨alt:
1
vP = KM vmaxP · 1
cS + 1 vPmax
mit cS ≈c0S. Auftragung von 1/vP gegen 1/c0S liefert eine doppelt-reziproke Auftragung (siehe Abb. (1.12)). Wenn experimentell ermittelte Wertepaare f¨ur v−1P und (c0S)−1 eine Gerade ergeben, dann gehorcht die Enzymreaktion einer Michaelis-Menten-Kinetik. Aus den Achsenabschnitten der Lineweaver-Burk-Auftragung lassen sich die Werte f¨ur die
1.12. REAKTIONEN AN FESTER KATALYSATOROBERFL ¨ACHE 30
Abb. 1.12: Doppelt-reziproke Auftragung nach Lineweaver-Burk zur Bestimmung der Michaelis-Konstanten KM und des Maximalwerts der Produktbildungsgeschwin- digkeit vmaxP .
Konstanten des Michaelis-Menten-GesetzesKM und vPmax ablesen.
1.12 Reaktionen an fester Katalysatoroberfl¨ ache
1.12.1 Fall 1: Adsorption/Desorption eines Gases A auf/von einer Katalysatoroberfl¨ ache
Reaktionsschema:
Adsorption: A(g)+ad-Platz −−→kAds A(ad) Desorption: A(ad) −−→kDes A(g)+ad-Platz
wobei ad-Platz ein Adsorptionsplatz an der Katalysatoroberfl¨ache darstellt.
Annahme: Die Teilreaktion der Adsorption sei bimolekular und die der Desorpti- on unimolekular. Zudem sind alle Adsorptionspl¨atze gleichwertig und besitzen daher die gleiche Besetzungswahrscheinlichkeit.
1.12. REAKTIONEN AN FESTER KATALYSATOROBERFL ¨ACHE 31
F¨ur die Geschwindigkeiten der Adsorptions- und Desorptionsteilreaktionen gilt dann:
RGAdsorption = 1
AKat · dnA(ad)
dt =kAds· pA·(1−θ) (1.36)
RGDesorption =− 1
AKat ·dnA(ad)
dt =kDes·θ (1.37)
wobei AKat = Oberfl¨b ache des Katalysators
dnA(ad) = ¨b Anderung der Stoffmenge von A am Adsorptionsplatz im Zeitintervall dt
pA = Partialdruck von A in der Gasphaseb
θ = Bedeckungsgrad (Maß f¨b ur die Oberfl¨achenkonz. der mit A-Teilchen) besetzten Adsorptionspl¨atze
1−θ = Maß f¨b ur die Oberfl¨achenkonz. der unbesetzten Adsorptionspl¨atze
Definition des Bedeckungsgrades:
θ = Zahl der besetzten Adsorptionspl¨. atze Gesamtzahl der Adsorptionspl¨. atze F¨ur das Adsorptions-Desorptionsgleichgewicht gilt:
RGAdsorption =RGDesorption (1.38)
Mit Gleichung (1.36) und Gleichung (1.37) folgt f¨ur obige Gleichung:
kAds· pA·(1−θ) =kDes·θ welche umgeschrieben werden kann als
θ
1−θ = kAds
kDes ·pA=α·pA (1.39)
mit
α= kAds kDes = 1
pA · θ 1−θ
Die Gr¨oße αentspricht dem Kehrwert des Partialdruckes p∗A, welcher bei Halbbesetzung der Adsorptionspl¨atze θ = 12 im System herrscht, d.h. α= p1∗
A.
Aufl¨osen von Gleichung (1.39) nach θ liefert die sog. Langmuir-Isotherme:
θ= α·pA 1 +α·pA
1.12. REAKTIONEN AN FESTER KATALYSATOROBERFL ¨ACHE 32
Abb. 1.13: Bedeckungsgrad der Katalysator-Oberfl¨ache als Funktion des Partialdrucks einer SubstanzAin der Gasphase wird durch die sog. Langmuir-Isotherme wiedergege- ben.
Graphische Auftragung von θ als Funktion von pA wird in Abb. (1.13) gezeigt.
Zwei Grenzf¨alle:
1. Kleine Partialdr¨uckepAp∗A:
θ ≈α·pA∝pA
Bei kleinen Partialdr¨ucken ergibt sich ein lineares Verhalten f¨ur den Bedeckungsgrad (siehe Abb. (1.13)) !
2. Große Partialdr¨uckepA p∗A:
1−θ ∝ 1 pA
⇒θ pA−→→∞ 1
Bei großen Partialdr¨ucken tendiert der Bedeckungsgrad gegen die S¨attigungsgrenze (sie- he Abb. (1.13)) !
1.12. REAKTIONEN AN FESTER KATALYSATOROBERFL ¨ACHE 33
Beweis:
1−θ = 1− α·pA
1 +α·pA = 1 1 + α
|{z}1 p∗
A
·pA
= 1
1 + ppA∗ A
= p∗A p∗A+pA
pAp∗A
≈ p∗A pA
⇒1−θ∝ 1
pA ⇒θp−→A→∞1
1.12.2 Fall 2: Unimolekulare Reaktion an der Katalysatoroberfl¨ ache der adsorbierten Spezies
Adsorptions-Desorptionsgleichgewicht:
A(g) +ad−P latz A(ad)
Folgereaktion:
A(ad)→k P(g) F¨ur die Reaktionsgeschwindigkeit gilt:
1 Akat
· dnP
dt =k·θ =k· α·pA 1 +α·pA
Die Geschwindigkeit der Folgereaktion an der Oberfl¨ache ist proportional zum Be- deckungsgradθ.
Zwei Grenzf¨alle:
1. Schwache Adsorption, d.h.α·pA1 bzw.pAp∗A: 1
Akat ·dnP
dt ≈k·α·pA
Bei schwacher Adsorption ergibt sich n¨aherungsweise ein Geschwindigkeitsgesetz 1. Ord- nung bzgl.pA !
1.12. REAKTIONEN AN FESTER KATALYSATOROBERFL ¨ACHE 34
2. Starke Adsorption, d.h.α·pA1 bzw. pAp∗A: 1
Akat
· dnP dt ≈k
Bei starker Adsorption ergibt sich n¨aherungsweise ein Geschwindigkeitsgesetz nullter Ordnung !
1.12.3 Fall 3: Bimolekulare Reaktion an der Katalysatoroberfl¨ ache zwischen adsorbierten Spezies
Adsorption-Desorptionsgleichgewicht von Spezies A und B an der Katalysatoro- berfl¨ache:
A(g) + ad-PlatzA(ad) B(g) + ad-PlatzB(ad) Folgereaktion:
A(ad) +B(ad)→k P(g)
Bei vielen Katalysereaktionen beruht der Mechanismus auf St¨oßen zwischen adsorbierten Spezies an der Katalysatoroberfl¨ache, wobei die Spezies um die gleichen Adsorptions- pl¨atze konkurrieren. Solche Reaktionen bezeichnet man als Langmuir-Hinshelwood- Reaktionen!
Aufgaben zur Kinetik:
1. Aufgabe: Reaktion erster Ordnung
14C ist ein radioaktives Kohlenstoffisotop, das nach einer Reaktion erster Ordnung mit einer Halbwertszeit von 5730a zerfällt (β- Zerfall). Es entsteht in der oberen Atmosphäre aus Stickstoff unter Einwirkung von Höhenstrahlung. Durch Entstehung und Zerfall stellt sich ein Gleichgewicht ein. Solange ein Organismus lebt, führt er immer weiter 14C durch Nahrung / Assimilation zu. Nach dem Absterben zerfällt 14C jedoch nur noch. Dies macht es möglich, über die verbleibende Radioaktivität, die zu der Menge an verbleibenden 14C proportional ist, das Alter von archäologischen Funden abzuschätzen.
Die Eismumie aus den Ötztaler Alpen weist nur noch eine restliche Radioaktivität von 52,7 % eines lebenden Menschen auf. Wann ist der Mann gestorben?
2. Aufgabe: Reaktionsumsatz einer Reaktion 2. Ordnung
Die Esterhydrolyse verläuft unter basischen Bedingungen irreversibel, da das für die Rückreaktion nicht weiter reaktive Säureanion entsteht. Bei der hier betrachteten Hydrolyse von Essigsäureethylester beträgt die Geschwindigkeitskonstante 0,11 L mol-1 s-1.
CH3COOC2H5(aq) + OH-(aq) CH3COO- (aq) + CH3CH2OH (aq)
Eingesetzt wurden die Edukte jeweils in einer Konzentration von 0,150 mol/l. Wie hoch sind die verbleibenden Esterkonzentrationen nach: 15 s, 15 min und 15 h ? Skizzieren Sie den Verlauf der Konzentrationen der Edukte und Produkte in der Reaktion in einem c über t Diagramm.
3. Aufgabe: Reaktionsordnung und Aktivierungsenergie
Das Molekül N2O5 zersetzt sich in einer Zerfallsreaktion zu N2O4 und O2. Bestimmen Sie die Ordnung der Reaktion durch eine geeignete Auftragung mit Hilfe der unten angegebenen Messdaten. Ermitteln Sie dann die Reaktionsgeschwindigkeitskonstante, die Halbwertszeit, sowie die Aktivierungsenergie der Reaktion.
45°C 65°C
t/s c(N2O5) / mol l-1 c(N2O5) / mol l-1
0 2,330 2,330
200 2,109 0,880
400 1,909 0,332
600 1,728 0,125
800 1,564 0,047
1000 1,416 0,018
4. Aufgabe: Hin- und Rückreaktion und Gleichgewicht
Bei der Bildung von HI in der Gasphase:
H2(g) + I2(g) 2HI(g)
wurde in einer Versuchsreihe bei einer Temperatur von 698,6K folgende Gleichgewichts- konzentrationen gemessen:
[H2] in Mol/l [I2]in Mol/l [HI] in Mol/l 4,5647 0,7378 13,5440
0,4789 0,4789 3,5341
1,1409 1,1409 8,4100
Dabei läuft die Reaktion in Form einer Elementarreaktion ab.
a) Bestimmen Sie die Gleichgewichtskonstante der Reaktion.
b) Wie verhalten sich bei Erreichen des Gleichgewichts die Reaktionsgeschwindigkeiten der Hin- und Rückreaktionen zueinander?
c) Wie verhalten sich die Geschwindigkeitskonstanten der Hin- und Rückreaktion zueinander?
5. Aufgabe: Folgereaktion erster Ordnung
Bei einem biotechnologischen Prozess entsteht aus der Substanz A das Produkt B, das selbst wieder zu dem Produkt C weiterreagiert. Die Reaktion soll nach folgendem Reaktionsschema ablaufen:
A B C
wobei kA und kB jeweils die Geschwindigkeitskonstanten der ersten und zweiten Teilreaktion darstellen. Jede Reaktionsstufe ist eine Reaktion erster Ordnung.
a) Geben Sie das differentielle Zeitgesetz für die Konzentration von B an, woraus sich folgendes integrales Zeitgesetz für cB ergibt:
cBt= kAc0A
kB−kA
e−kAt−e−kBt
wobei cA0 die Anfangskonzentration der Substanz A darstellt.
b) Leiten Sie einen Ausdruck für den Zeitpunkt her, bei dem die Konzentration von B ihr Maximum erreicht und berechnen Sie die maximale Konzentration von B.
c) Wie verhalten sich die beiden hergeleiteten Größen aus 5 a), wenn kB ansteigt während kA konstant gehalten wird ?
6. Aufgabe: Parallelreaktion erster Ordnung
131I ist ein radioaktives Iod-Isotop, das bei der Kernspaltung entstehen kann. 131I ist ein β- Strahler der mit einer Halbwertszeit von 8d zerfällt. Wird Iod vom Körper aufgenommen, lagert es sich hauptsächlich in der Schilddrüse ein. Die biologische Halbwertszeit beim Menschen in der Schilddrüse (durch Ausscheidung) beträgt 140d. Daraus folgt eine besonders starke Belastung der Schilddrüse bei Unfällen mit Radioaktivität, der man versucht mit Iod Tabletten entgegen zu wirken.
a) Berechnen Sie die effektive Halbwertszeit von 131I im menschlichen Körper.
b) Wie lange dauert es bis die mit 131I aufgenommene Radioaktivität in einem Menschen um 99 % abgenommen hat ?
7. Aufgabe: Konzentrationsverlauf und Quasistationärität
a) Geben Sie das Reaktionsschema an und skizzieren Sie die Konzentrationsverläufe der beteiligten Substanzen in Abhängigkeit der Reaktionsdauer für folgende Fälle:
i. Irreversible Parallelreaktion erster Ordnung ii. Hin-/Rückreaktion erster Ordnung
iii. Folgereaktion mit vorgelagertem Gleichgewicht
b) Welche Voraussetzungen müssen zur analytischen Lösung der differentiellen Zeitgesetze aus Aufgabe 7 a) iii. mit Hilfe der Quasistationaritätsbedingung erfüllt sein ?
8. Aufgabe: Folgereaktion und Quasistationärität
Aceton geht unter Säurekatalyse mit sich selbst eine Aldolkondensation ein. Dies geschieht nach folgendem vereinfachtem Reaktionschema:
O C H3 CH3
+ H+
k(+)
k(-)
OH C H2 CH3
+ H+
Aceton Enol
OH C H2 CH3
O C H3 CH3
+ k(p)
C H3
O
CH3 CH3
+ H2O
Enol Aceton Produkt (4-Methylpent-3-en-2on)
a) Leiten Sie unter Verwendung der Näherung des quasi-stationären Zustandes für das reaktive Zwischenprodukt (Enol) einen Ausdruck für die Konzentration des Enols her.
b) Leiten Sie daraus einen Ausdruck für die Produktbildungsgeschwindigkeit her.
9. Aufgabe: Michaelis-Menten-Kinetik
Das von Fleming entdeckte Antibiotikum Penicillin ist ein β-Lactam Antibiotikum, das bei gram-positiven Bakterien die Ausbildung der Zellwand verhindert. Manche Stämme von Staphylococcus aureus haben jedoch eine Gegenstrategie gegen das Penicillin entwickelt. Sie besitzen die β-Lactamase (auch als Penicillinase bekannt), die β-Lactam-Antibiotika, wie das Penicillin, hydrolysieren und somit unwirksam machen kann.
Im Rahmen einer Versuchsreihe sollte die Reaktionskinetik der Penicillinhydrolyse untersucht werden. Dazu wurde die von 1ng β-Lactamase (MW = 29,6 kDa) hydrolysierte Menge an Penicillin pro Minute in Abhängigkeit der Penicillinkonzentration in einem Reaktionsansatz von 1ml gemessen. Es wurden hierbei folgende Messdaten ermittelt:
Konzentration cPenicillin
[µM]
Produktbildungsgeschwindigkeit νP [nmol/min ml]
1 0,11
3 0,25
5 0,34
10 0,45
30 0,58
50 0,61
Nehmen Sie an, dass sich die Penicillinkonzentration während des Versuchs nicht merklich ändert.
a) Zeigen Sie anhand einer geeigneten Auftragung obiger Messwerte, ob die β-Lactamase der Michaelis-Menten-Kinetik gehorcht. Wenn ja, welchen Wert für die Michaelis- Menten-Konstante KM besitzt die β-Lactamase ?
b) Welchen Wert hat die maximale Produkbildungsgeschwindigkeit ?
c) Welche Wechselzahl hat das Enzym unter den angegebenen Bedingungen, wenn man davon ausgeht, dass das Enzym nur ein aktives Zentrum besitzt ?
2 Thermodynamik
2.1 Grundlagen und Wiederholung 1. Semester
2.1.1 System und Umgebung
System: Teil des Weltalls, der bei der Untersuchung betrachtet wird (siehe Abb. (2.1)).
Umgebung: Rest des Weltalls, der an das System angrenzt und gegebenfalls mit diesem in Wechselwirkung steht.
Ein Systembezeichnet man als:
• abgeschlossen bzw. isoliert, wenn weder Energie- noch Stoffaustausch mit der Umgebung stattfindet;
• geschlossen, wenn Energie- aber kein Stoffaustausch stattfindet;
• offen, wenn Energie- sowie Stoffaustausch stattfindet.
2.1.2 Prozesse und Randbedingungen
Prozesse, die an einem System ausgef¨uhrt werden, bezeichnet man je nach Randbedin- gungen als:
• isotherm: bei konstanter Temperatur;
• isobar: bei konstantem Druck;
• isochor: bei konstantem Volumen;
• adiabatisch: ohne W¨armeaustausch;
• reversibel: wenn sich, bei der Ausf¨uhrung, das System bei jedem Prozessschritt im Gleichgewicht befindet (kann n¨aherungsweise durch infinitesimale ¨Anderun- gen der Zustandsvariablen erreicht werden) und der Prozess auf demselben Wege vollst¨andig umkehrbarist;
• irreversibel: wenn die ¨Anderungen nichtumkehrbar sind.
39
2.1. GRUNDLAGEN UND WIEDERHOLUNG 1. SEMESTER 40
Abb. 2.1: Anschauliche Definition der Begriffe System, Umgebung und Weltall in der Ther- modynamik.
2.1.3 Innere und ¨ außere Energie
Innere Energie:
beschreibt die Gesamtenergie, die im System vorhanden ist, z.B. in Form kinetischer Energie (Bewegung) oder potentieller Energie (Anziehungs-/Abstoßungskr¨afte) der Mo- lek¨ule. Generell ist jede Energie, die nicht durch Bewegung des Gesamtsystems ver¨andert werden kann, zur inneren Energie zu z¨ahlen.
Außere Energie:¨
wird durch die Einwirkung ¨außerer Kr¨afte (z.B. Gravitationskraft) erzeugt und ver¨andert die Bewegung des Gesamtsystems, wie z.B. Lageenergie.