Aus der Klinik und Poliklinik für Innere Medizin III
(Direktor: Professor Dr. med. Wolfgang Herr) der Medizinischen Fakultät
der Universität Regensburg
Die Lokalisation von PD-1 während des „Kiss of Death“
Inaugural-Dissertation
zur Erlangung des Doktorgrades der Medizin der Medizinischen Fakultät
der Universität Regensburg
vorgelegt von
Lukas Kremmler
aus Würzburg
2013
Aus der Klinik und Poliklinik für Innere Medizin III
(Direktor: Professor Dr. med. Wolfgang Herr) der Medizinischen Fakultät
der Universität Regensburg
Die Lokalisation von PD-1 während des „Kiss of Death“
Inaugural-Dissertation
zur Erlangung des Doktorgrades der Medizin der Medizinischen Fakultät
der Universität Regensburg
vorgelegt von
Lukas Kremmler
aus Würzburg
2013
Dekan: Prof. Dr. Dr. Torsten E. Reichert
1. Berichterstatter: PD Dr. med. Christian Blank 2. Berichterstatter: Prof. Dr. med. Daniela Männel
Tag der mündlichen Prüfung: 13. März 2014
Inhaltsverzeichnis
1. Einleitung 1
1.1. Immunsystem . . . 1
1.1.1. Adaptive Immunität . . . 1
1.1.2. Lymphozyten . . . 2
1.2. T-Zell-Aktivierung . . . 3
1.2.1. Antigen-Erkennung . . . 3
1.2.2. TCR Signalvermittlung . . . 4
1.2.3. Immunsynapse . . . 4
1.2.4. Zytoskelett . . . 4
1.2.5. CD8+ Effektorfunktion . . . 5
1.2.6. Kostimulation . . . 6
1.2.7. Koinhibition . . . 6
1.3. Tumorgenese und Immunsystem . . . 8
1.3.1. Tumor-Antigene . . . 9
1.3.2. Immunantwort . . . 10
1.3.3. Tumor Immunescape . . . 11
1.3.3.1. Hemmung der Tumorerkennung . . . 11
1.3.3.2. Einfluss von Chemokinen und Liganden . . . 12
1.3.3.3. Fehlen von Kostimuli . . . 13
1.3.3.4. Zelluläre Inhibitoren . . . 13
1.4. Tumor-infiltrierende Lymphozyten . . . 14
1.4.1. Anergie . . . 14
1.4.2. Exhaustion . . . 15
1.4.3. Defekte in T-Zell-Signal und MTOC-Polarisierung . . . 15
1.5. Programmed Death Receptor 1 (PD-1) . . . 16
1.5.1. Struktur, Vorkommen und Signalwirkung . . . 16
1.5.2. PD-1-Liganden und zweiter Rezeptor . . . 17
1.5.3. PD-1 und Autoimmunität . . . 18
1.5.4. PD-1 in Infektion und Exhaustion . . . 19
1.5.5. PD-1 und Tumore . . . 20
1.5.6. Lokalisation von PD-1 . . . 21
1.6. Fragestellung dieser Promotion . . . 22
2. Material und Methoden 24 2.1. Material . . . 24
2.1.1. Tumorzelllinien . . . 24
2.1.2. Mausstämme . . . 24
2.1.3. Antikörper . . . 26
2.2. Methoden . . . 28
2.2.1. Zellkultur / Technik . . . 28
2.2.1.1. Zentrifugation . . . 28
2.2.1.2. Zellzählung mit Trypanblau . . . 28
2.2.2. Zellkultur . . . 28
2.2.2.1. Splitten von Tumorzelllinien . . . 28
2.2.2.2. Mycoplasmentest . . . 29
2.2.2.3. Auftauen und Einfrieren von Zelllinien . . . 29
2.2.3. Isolierung muriner Zellen . . . 29
2.2.3.1. Splenektomie aus Mäusen . . . 29
2.2.3.2. Gewinnung naiver T-Zellen aus Milzen . . . 29
2.2.3.3. Dynal-Kit . . . 30
2.2.3.4. SpinSep-Kit . . . 31
2.2.4. In vitroStimulation mit P815.B7-1 . . . 32
2.2.5. Analyse von Zellen mit Hilfe der Durchflusszytometrie . . . 32
2.2.5.1. Prinzip des FACS . . . 32
2.2.5.2. Antikörperfärbung auf murinen Zellen . . . 34
2.2.5.3. Labeling von 1B2 . . . 34
2.2.5.4. Analyse der T-Zell-Aufreinigung . . . 35
2.2.6. Beads . . . 35
2.2.6.1. Coating von Beads mit Antikörpern . . . 36
2.2.6.2. Stimulation von T-Zellen mit Beads für ELISA . . . 36
2.2.7. in vitro Assays . . . 36
2.2.7.1. Target Tumoren . . . 36
2.2.7.2. Zytokinproduktion – ELISA . . . 37
2.2.7.3. Proliferation – Tritium-Thymidine-Assay . . . 38
2.2.7.4. Zytotoxizität – LAMP-Test . . . 38
2.2.7.5. Zytotoxizität – Chrome-Release-Assay . . . 38
2.2.8. Immunfluoreszenz . . . 39
2.2.8.1. Labeling von Antikörpern mit Alexa Fluor . . . 39
2.2.8.2. Objektträger . . . 40
2.2.8.3. Immunfluoreszenz-Färbung . . . 40
2.2.8.4. Mikroskopie . . . 41
2.2.8.5. Analyse von Zellkonjugaten und Zellpolarisierungen . . . 41
2.2.9. Statistik . . . 42
3. Ergebnisse 43 3.1. T-Zell-Stimulation mit Beads . . . 43
3.2. T-Zell–Tumor–Interaktion . . . 47
3.2.1. T-Zell-Stimulation mit p815.B7-1 . . . 47
3.2.2. Funktionalitätsassays . . . 51
3.2.2.1. Verminderte Zytokinproduktion bei transgener Überex- pression von PD-1 . . . 52
3.2.2.2. Höchste Zellproliferationsrate bei PD-1-Abwesenheit . . 53
3.2.2.3. Höchste Zytotoxizität bei PD-1-Abwesenheit . . . 53
3.2.3. PD-L1 auf T-Zellen ohne Effekt auf T-Zell-Funktionalität . . . 54
3.2.4. Immunfluoreszenz-Mikroskopie . . . 57
3.2.4.1. PD-1-Expression auf aktivierten T-Zellen in Clustern . . 59
3.2.4.2. Lokalisation von PD-1 während des Zellkontakts . . . . 60
3.2.4.3. B16 Melanomzellen ohne PD-L1-Polarisierung . . . 63
3.2.5. Einfluss von PD-1 auf die Synapsenbildung . . . 64
3.2.5.1. PD-1 ohne Einfluss auf Konjugatbildung . . . 64
3.2.5.2. Effektivere Polarisierung von Granzym bei PD-1-Absenz 65 4. Diskussion 67 4.1. Sterische Inhibition des Beadcoatings durch PD-L1.Ig . . . 67
4.2. Exhaustion bei repetitiver Stimulation . . . 69
4.3. PD-1 vermittelte Inhibition der Effektorfunktionen . . . 71
4.4. PD-1 in Immunfluoreszenz-Mikroskopie . . . 74
4.5. Zellpolarisierung in Abhängigkeit der PD-1-Expression . . . 79
5. Zusammenfassung 81
Literaturverzeichnis 83
A. Danksagung 104
Abbildungsverzeichnis
1.1. Formation des Kiss of Death . . . 5
1.2. Kostimulatoren der B7-Familie . . . 7
1.3. PD-1 und Liganden . . . 18
1.4. PD-L1 Signalwirkung . . . 20
1.5. PD-1 Lokalisation . . . 22
2.1. Gewinnung und Aufbereitung von murinen T-Zellen . . . 30
2.2. T-Zell FACS Analyse . . . 33
2.3. CD8-Konzentration im FACS . . . 35
3.1. Zytokin-Produktion nach Bead-Stimulation . . . 43
3.2. Antikörper-Dichte auf Beads . . . 45
3.3. Zytokin-Produktion bei Isotypen Titration . . . 46
3.4. Phänotyp von p815.B7-1 . . . 47
3.5. Kinetik von PD-1 auf 2C TCRtg T-Zellen . . . 48
3.6. Kinetik von Aktivierungsmarkern auf 2C TCRtg T-Zellen . . . 49
3.7. Kinetik von CD80 und CD86 auf 2C TCRtg T-Zellen . . . 50
3.8. Expansionsfaktoren nach Stimulationsrunden . . . 51
3.9. Phänotyp von B16.SIY . . . 52
3.10. 2C Zytokin-Produktion und Proliferation . . . 53
3.11. 2C Zytotoxizität . . . 54
3.12. Phänotyp von PD-L1-defizienten T-Zellen . . . 55
3.13. Einfluss von lymphozytärem PD-L1 auf T-Zell-Proliferation . . . 56
3.14. Einfluss von lymphozytärem PD-L1 auf T-Zell-Funktionalität . . . 57
3.15. Immunfluoreszenz - PD-1 auf T-Zellen . . . 58
3.16. Immunfluoreszenz - PD-L1 auf T-Zellen . . . 59
3.17. Immunfluoreszenz - PD-1 und Tubulin . . . 60
3.18. Immunfluoreszenz - PD-1 und Tubulin . . . 61
3.19. Immunfluoreszenz - T-Zell-Rezeptor und Tubulin . . . 61
3.20. Immunfluoreszenz - PD-1 und CD3 . . . 62
3.21. Immunfluoreszenz - PD-L1 und Tubulin . . . 62
3.22. Immunfluoreszenz - PD-L1 auf Tumor . . . 63
3.23. Immunfluoreszenz - MTOC-Translokation . . . 64
3.24. Einfluss von PD-1 auf MTOC-Translokation . . . 65
3.25. Immunfluoreszenz - Granzym-Polarisierung . . . 66
3.26. Immunfluoreszenz - Granzym-Polarisierung . . . 66
4.1. Fluorochrom Spektren . . . 76
Tabellenverzeichnis
2.1. Tumorzelllinien . . . 24
2.2. Mausstämme . . . 25
2.3. FACS Antikörper . . . 26
2.4. Antikörper und Standards für ELISA . . . 27
2.5. Antikörper für Immunfluoreszenz . . . 27
2.6. Fluorochrome . . . 39
5.1. Abkürzungsverzeichnis . . . 103
1. Einleitung
1.1. Immunsystem
Das oberste Gebot eines jeden Organismus ist das Überleben in seiner Umwelt und die Wahrung seiner Integrität. Hinter den physischen Barrieren existiert in Mensch und Tier das Immunsystem, das sowohl gegen Gefahren von Außen, wie z.B. Bakterien, Viren, Pilzen oder Parasiten, als auch gegen potentiell bedrohliche Veränderungen von innen, wie z.B. die mögliche Entwicklung von Tumoren, schützen soll. Das Immunsystem, bestehend aus zellulären und molekularen Effektoren, kann dabei unterschieden werden in einen angeborenen, nativen und in einen erworbenen, adaptiven Anteil.
Die angeborene Immunität ermöglicht ab Existenzbeginn eine rasche, jedoch relativ un- spezifische Reaktion auf bis dato dem Organismus unbekannte Pathogene, bei der die Immuneffektoren verloren werden. Hierzu gehören unter anderem die Botenstoffe Zy- tokine, das Komplementsystem sowie natürliche Killerzellen (NK-Zellen), Granulozyten und Phagozyten.
Die adaptive, erworbene Immunität stellt die Antwort dar, die sich gezielt auf den Fremd- körper hin entwickelt und mit der Ausbildung von Gedächtniszellen eine rasche Antwort auf einen erneuten Befall ermöglicht. Diese Reaktion ist hoch-spezifisch und steigert sich bei wiederholter Exposition des Pathogens. [81]
1.1.1. Adaptive Immunität
Die adaptive Immunantwort erfolgt auf zwei Wegen: humoral und zellvermittelt. An- tikörper in Blutserum und Sekreten stellen die humorale Immunität dar. Sie werden von B-Lymphozyten produziert und neutralisieren Pathogene direkt oder markieren sie für die anschließende zellvermittelte Zerstörung. Die zelluläre Immunität besteht z.B.
aus T-Lymphozyten, die Fremdkörper oder infizierte körpereigene Zellen anhand ihres T-Zell-Rezeptors erkennen.
Antikörper und T-Zell-Rezeptoren sind spezifisch für bestimmte Epitope, also kleins- te Protein- oder Polysaccharidkomplexe. Bei der Generierung der antigen-bindenden
Region entstehen Kombinationen, die pro Individuum bis zu 109 verschiedene Epitope hoch-spezifisch erkennen.
Aufgrund der breiten Palette an detektierbaren Epitopen bedarf es im Gesunden eines Kontrollmechanismus, der eine Reaktivität gegen eigenes Gewebe verhindert. Diese To- leranzbildung findet zentral während der Lymphozytenschulung und in der Peripherie statt. Außerdem existieren zellulär- und zytokin-vermittelte Mechanismen, die eine kor- rekt gestartete Immunantwort auf das Pathogen beschränken und nach dessen Vernich- tung wieder beenden. Eine Störung der Kontrollen kann zu auto-reaktiven Erkrankungen führen. [1]
1.1.2. Lymphozyten
Lymphozyten werden unterteilt in B-Zellen und T-Zellen, die beide über spezifische Antigen-Rezeptoren verfügen. Zellen eines Klons besitzen dabei den gleichen Antigen- Rezeptor und unterscheiden sich damit von anderen Klonen mit unterschiedlichen Erken- nungssequenzen. B-Lymphozyten produzieren und sezernieren nach positiver Antigen- Erkennung Antikörper.
T-Lymphozyten, denen hier das Augenmerk gilt, erkennen Peptide auf der Zelloberfläche sowie solche, die im Serum an Haptene gebunden sind, und teilen sich wiederum in die Klasse der T-Helfer-Zellen und der T-Killer-Zellen auf. Oberflächenmarker, sogenannte
„Cluster of Differentiation“, helfen, die zwei T-Zell-Subpopulationen phänotypisch zu unterscheiden: während T-Helfer-Zellen CD4 exprimieren, findet sich auf den zytotoxi- schen T-Killer-Zellen vornehmlich CD8. CD4+Zellen reagieren mit MHC-II-Komplexen, CD8+ Zellen hingegen mit MHC-I.
Bei T-Helfer-Zellen führt die Antigen-Erkennung zur Produktion und Sekretion von Zy- tokinen, die das Immunsystem zu Spezialisierung und Vermehrung immun-kompetenter Zellen anstösst. T-Killer-Zellen sind in der Lage, Zielzellen bzw. Pathogene direkt zu neutralisieren. Des Weiteren existiert die Subgruppe der NKT- oder Natürliche Killer- T-Zellen, die im Gegensatz zu „normalen T-Zellen“ nicht an den MHC-Peptid-Komplex, sondern an den Komplex aus CD1d und darauf präsentierten Lipiden binden und nach erfolgreicher Erkennung zytolytisch wirken können. Als weitere besonders interessante T-Zell-Untergruppe gelten die regulatorischen T-Zellen (Treg), deren größte Populati- on sich durch den CD4+/CD25+/FoxP3+ Phänotyp auszeichnet. Diese Tregs sind über Effektoren wie TGF-β oder IL-10 in der Lage, die Reaktion des Immunsystems zu re- gulieren.
Der die T-Zellen charakterisierende T-Zell-Rezeptor (TCR) ist ein Heterodimer aus einer
alpha (α)- und einer beta (β)-Kette, die über Sulfid-Brücken kovalent verbunden sind.
Diese Ketten bestehen aus einem konstanten C- und einem variablen V-Teil; letzterer begründet die breite Variabilität und gleichzeitig hohe Spezifität unterschiedlicher T- Zell-Klone. Daneben existiert eine Population, die statt α- und β-Ketten sogenannte gamma (γ)- und delta (δ)-Ketten als Heterodimer im TCR einbauen; diese γ–δ–TCR T-Zellen kommen gehäuft in epithelialen Geweben vor, und sind nicht auf die Erkennung via MHC-Präsentation limitiert. [28]
Die im Knochenmark gebildeten T-Zellen migrieren zum Zweck der Reifung in den Thy- mus. Dort findet die Auslese optimaler Lymphozyten statt. Bei der positiven Selektion überleben nur die Lymphozyten, die schwache Bindungen mit Eigenpeptiden eingehen, während eine fehlende Peptid-Erkennung zur Apoptose führt: „Death by Neglect“. Die negative Selektion sortiert anschließend Lymphozyten aus, die eine zu hohe Reaktivität gegenüber eigenen Antigenen und damit ein auto-immunes Potential bieten. [152]
Die antigen-spezifische Aktivierung einer T-Zelle führt zur klonalen Expansion und der Bildung von Gedächtniszellen, die bei einem erneuten Auftreten des Pathogens eine schnelle, spezifische Immunantwort ermöglichen.
1.2. T-Zell-Aktivierung
1.2.1. Antigen-Erkennung
Kernhaltige Zellen präsentieren Bruchstücke ihrer Genprodukte auf ihrer Oberfläche.
Dazu werden zytosolische Proteine in Proteasomen in Peptide aufgespalten und im endo- plasmatischen Retikulum (ER) an Major Histocompatibility Complex Class I (MHC-I)- Proteine gekoppelt. Diese Komplexe werden via Golgi-Apparat und exozytotische Vesikel an die Zelloberfläche transportiert und von CD8+ T-Zellen ausgelesen.
Alternativ existieren professionelle antigen-präsentierende Zellen, z.B. dendritische Zel- len (DC), Makrophagen und B-Zellen, die extrazelluläre Proteine durch rezeptorvermit- telte Endozytose aufnehmen, prozessieren und schließlich an MHC-II-Moleküle gebunden auf der Zelloberfläche präsentieren. CD4+ T-Zellen erkennen diese MHC-II-Komplexe und verursachen bei Erkennung eines Fremdantigens eine Aktivierung des Immunsys- tems durch Ausschüttung entsprechender Zytokine, wie z.B. IL-2. Dies löst sowohl eine zelluläre als auch humorale Immunantwort aus. [31]
Für die Identifizierung der MHC-Peptid-Komplexe besitzen T-Lymphozyten den T-Zell- Rezeptor, mitα– undβ-Ketten sowie mit jeweils einer Konstanten C- und einer variablen
V-Region. Komplettiert wird der TCR-Komplex durch CD3 und ζ-Ketten. Jeder TCR ist hoch-spezifisch für ein Epitop. [161]
1.2.2. TCR Signalvermittlung
Eine positive Ligandenerkennung und -bindung des TCR-Komplexes bewirkt die Akti- vierung der T-Zelle. Zunächst werden ITAMs (Immunoreceptor tyrosine-based activation motifs) der CD3-Ketten von Lck (lymphocyte-specific protein tyrosine kinase) phospho- ryliert, die dann Zap70 (Zeta-chain-associated protein kinase 70) zum TCR rekrutieren.
Über mehrere Phosphorylierungs- und Komplexbildungsschritte wird das TCR-Signal in verschiedene Signaltransmitter und -verstärker wie DAG (Diacylglyerol), IP3 (Inositol- triphosphat), PKCθ (protein kinase-θ), Ras, PI3K (Phosphatidylinositol 3-kinase) und MAPK (Mitogen-activated protein kinase) übersetzt. [83, 109]
Eine adäquate Reaktion von Seiten der T-Zelle setzt eine Veränderung der Genexpres- sion mit vermehrter Interleukin-2-Transkription voraus. Dies wird vor allem durch die drei Transkriptionsfaktoren AP1 (Activator protein 1), NFAT (Nuclear factor of acti- vated T cells) und NF-κB (Nuclear factor-κB) bewirkt, die als Folge oben genannter Signaltransmitter aktiviert und in den Zellkern transloziert werden. [75]
1.2.3. Immunsynapse
Die Bindung des TCR an den Peptid-MHC-Komplex ist der erste Schritt bei der Ausbil- dung der immunologischen Synapse zwischen T-Zelle und antigen-präsentierender Zelle.
In enger Nähe akkumulieren aktivierte TCRs mit sogenannten Microclustern aus assozi- ierten Signalproteinen wie Zap70 und PI3K. [109, 138] Zusammen bildet sich der cSMAC (central supramolecular activation cluster), der eine Verstärkung der Signale bewirken soll. [30] Umgeben wird der cSMAC von Integrinen wie LFA1, die den Adhäsionsring pSMAC (peripheral supramolecular activation cluster) bilden. Zur Aufrechterhaltung der Stimulation werden weitere, im pSMAC gebildete, aktivierte TCR-Komplexe konti- nuierlich zum cSMAC verschoben. [19, 65, 165]
1.2.4. Zytoskelett
Die Aktivierung der T-Zelle löst eine Veränderung des Zytoskeletts aus. An der Immun- synapse findet eine rasante Polymerisation von Aktin und eine integrin-vermittelte Ad- häsion statt, die eine verstärkte Bindung der T-Zelle an die antigen-präsentierende Zelle
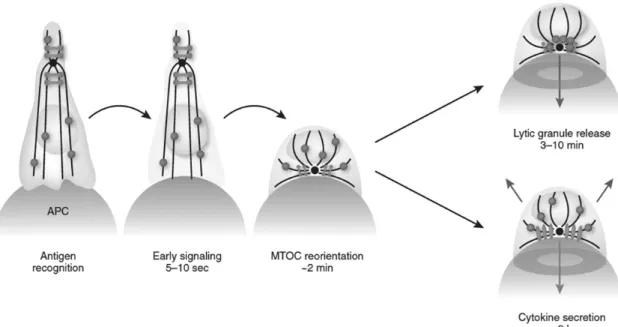
Abbildung 1.1.: Formation des „Kiss of Death“ zwischen Lymphozyt und antigen-präsentierender Zelle:
Nach Antigenerkennung kommt es zu einer Zellskelett-Transformation der T-Zelle durch Polarisierung des MTOC mit anschließender Sekretion von Zytotoxen und Zytokinen.
(nach: Huse et al., 2008 [76])
und eine Vergrößerung der Kontaktfläche ermöglicht. Gleichzeitig erhöht die Phospho- rylierung der MLCK (Myosin light chain kinase) die myosin-vermittelte Kontraktilität der Zelle. [13, 21]
Zur Ausbildung einer stabilen Immunsynapse gehört auch die Polarisierung des MTOC (Microtubule-organizing center), das nach TCR-Bindung direkt unter der Formation aus cSMAC- und pSMAC-Anteilen lokalisiert wird. [62] Das MTOC ist die zentrale Verbindung positiv geladener Enden von Microtubuli, deren negativ geladene Enden sich in die Peripherie der Zelle erstrecken. Dynein sorgt für die Mobilität des MTOC. Entlang dem mikrotubulären Zellskelett können lytische Granula, die Perforin oder Granzym B enthalten, zur Immunsynapse transportiert werden. Die Polarisation erlaubt so die gezielte Sekretion von zytotoxischen Substanzen und Zytokinen in den intrazellulären Zwischenspalt. Daher nennt man die Ausbildung dieser Kontaktstelle auch den „Kiss of Death“. [12, 76, 99]
1.2.5. CD8
+Effektorfunktion
Die CD8+zytotoxischen T-Zellen sind in der Lage, nach erfolgreicher Antigen-Erkennung, die Zielzelle mit zytolytischen Substanzen zu zerstören. Perforin und Granzym B sorgen
für eine Destabilisierung der Zellmembran. Über die Expression von Fas-Ligand CD95L und dessen Bindung an den Rezeptor Fas kann die Zielzelle durch Aktivierung von Cas- pasen zur Apoptose gebracht werden. [105] Die Sekretion von Zytokinen wie TNF-α, IL-2 oder INF-γ führt zur weiteren Aktivierung und Rekrutierung von Immunzellen.
Zusätzlich verursacht INF-γ eine gesteigerte Expression von MHC-I-Komplexen, was es den Lymphozyten erleichtert, die Zielzellen zu identifizieren. [26]
1.2.6. Kostimulation
Die Zwei-Signal-Hypothese besagt, dass für eine optimale T-Zell-Stimulation neben dem TCR-Signal ein zweiter, kostimulatorischer Oberflächenrezeptor notwendig ist, um eine Proliferation und die Ausführung der Effektorfunktion auszulösen. [100] Bei Ausbleiben eines solchen zweiten Signals begibt sich die T-Zelle in einen Zustand der Anergie, auf den im Weiteren noch eingegangen wird, oder kann sogar apoptotisch werden. Lange war in der Forschung die Zwei-Signal-Theorie besetzt durch die Moleküle CD28 und CTLA-4.
Tatsächlich ist wohl der wichtigste Mitspieler der lymphozytäre Rezeptor CD28, der die vor allem von professionellen APCs präsentierten Liganden B7-1 (CD80) und B7-2 (CD86) bindet. Das CD28-Signal bewirkt eine Phosphorylierung von Src-Kinasen und dadurch eine Rekrutierung weiterer Moleküle der Aktivierungskaskade wie PI3K. Der Effekt liegt wahrscheinlich eher in einer quantitativen Steigerung des TCR-Signals, als in einer qualitativ anderen Signalvermittlung. [4, 75]
Daneben exprimieren T-Zellen auch nach Aktivierung den CD40-Liganden (CD40L), der wiederum den Rezeptor CD40 auf APCs bindet und dort wiederum zu erhöhter B7-1 und B7-2 Expression und damit verstärkter T-Zell-Stimulation führt. Ferner er- folgt eine T-Zell-Aktivierung über den Kostimulus ICOS (inducible costimulus), der an den von APCs exprimierten ICOS-Liganden ICOS-L bindet und zu Proliferation und Zytokin-Produktion auf Seiten der Lymphozyten führt. Darüber hinaus wurden in den letzten Jahren noch weitere Kostimuli beschrieben, unter anderem die Familie der TNF- Rezeptoren. [34]
1.2.7. Koinhibition
Während die T-Zell-Aktivierung die oben genannte Kostimulation voraussetzt, gibt es im Sinne von Antagonisten ebenso inhibitorische Rezeptoren. Physiologischer Hinter- grund der Koinhibition im Gesunden, also die Hemmung einer Immunaktivierung, ist
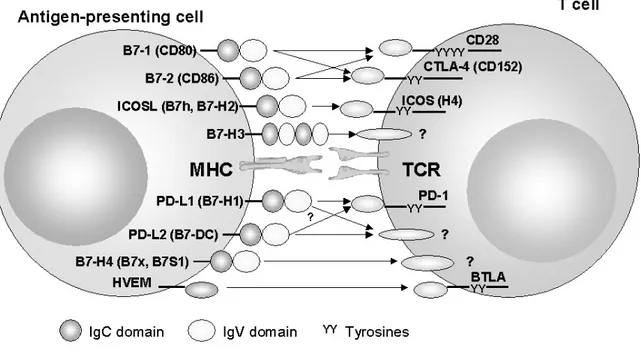
Abbildung 1.2.: Kostimulatoren der B7-Familie: Signale über CD28 und ICOS wirken kostimulatorisch, während Signale über CTLA-4 oder PD-1 koinhibitorisch in die Signalkaskade eingrei- fen. (nach: Blank et al., 2005 [16])
das Ziel, überschießende Immunantworten zu unterbinden bzw. im physiologischen Ma- ße verlaufen zu lassen, da es sonst zu autoimmun vermittelten Schädigungen kommen kann.
Die Koinhibition erfolgt beispielsweise über Rezeptoren auf Lymphozyten mit sogenann- ten ITIMs (= Immunoreceptor tyrosine-based inhibitory motif) und über die Rekrutie- rung von inhibitorischen Phosphatasen wie SHP-1, die die Fortleitung des TCR-Signal unterdrücken. Beispiele für solche koinhibitorischen Rezeptoren sind CTLA-4 (Cytotoxic T-Lymphocyte Antigen 4, CD152), ein Rezeptor aus der B7-Familie, sowie BTLA (B and T lymphocyte attenuator, CD272) und PD-1 – auf letzteren wird weiter unten im Detail eingegangen.
Die Funktionsweise am Beispiel von CTLA-4: Nach T-Zell-Aktivierung wird CTLA-4 auf der Zelloberfläche hochreguliert und bindet hoch-affin Liganden, die sonst über CD28 kostimulatorisch wirken, wie z.B. B7-1 (CD80) und B7-2 (CD86). [162] Es folgt wie
bei CD28 die Phosphorylierung durch Src-Kinasen und eine Kopplung mit SHP2 und PI3K. Die T-Zell-Aktivierung wird wohl durch die Dephosphorylierung membran-naher TCR-Signaltransmitter gehemmt, während zusätzlich die Bindung des Kostimulus CD28 in der Immunsynapse durch die höhere Affinität von CD80/CD86 gegenüber CTLA- 4 verhindert wird. [172] Es wurde ferner das Reverse-Stop-Signal-Modell beschrieben:
CTLA-4 verhindert das Stop-Signal des TCR, das die T-Zell-Motilität hemmt und damit die Zelladhäsion mit der APC ermöglicht. Die Kontaktzeit zwischen T-Zelle und APC reicht dadurch für eine effektive TCR-Aktivierung nicht aus. [137, 141] Daneben sind auch sogenannte ITSM (= Immunoreceptor tyrosine-based switch motifs) bekannt, die eine vielfältigere Rezeptor-Signal-Verarbeitung ermöglichen, da sie in Abhängigkeit der Aktivierung sowohl Kinasen als auch Phosphatasen rekrutieren können. [155]
Zusammengefasst dienen diese inhibitorischen Rezeptoren dem Schutz des Organismus vor einer autogenen Schädigung. Jedoch bietet die Hemmung des Immunsystems in einem solchen Maße auch attraktive Ansatzpunkte für pathologische Vorgänge. Es ist z.B. auffällig, dass der Ligand für PD-1, PD-L1, vermehrt auf Tumorzellen sowohl in Tiermodellen als auch im Menschen nachgewiesen werden konnte. Es ist also naheliegend, dass es zu einer Interaktion zwischen Tumorgewebe und Immunsystem kommen muss.
1.3. Tumorgenese und Immunsystem
Hanahan und Weinberg haben in zwei Übersichtsarbeiten die Markenzeichen der Tumor- entstehung („Hallmarks of Cancer“) zusammengefasst: Aufrechterhaltung von prolifera- tiven Signalen, Vermeidung von Wachstumshemmnissen, Widerstand gegenüber dem Zelltod, Erlangung von replikativer Unsterblichkeit, Induktion von Gefäßbildung und Aktivierung von Invasion und Metastasierung („sustaining proliferative signaling, eva- ding growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis“). Darüber hinaus und für diese Arbeit von existentieller Bedeutung ist der Einfluss des Immunsystems und damit das Ziel des Tumors, eben diesem zu entgehen. [70, 71]
Die Theorie einer fortwährenden Immunosurveillance, also der Überwachung des Or- ganismus mit Suche und Begrenzung von Zellveränderungen durch das Immunsystem, wurde erstmals Anfang des 20. Jahrhunderts von Paul Ehrlich formuliert. [48] Diese wur- de später von Thomas und Burnet um die Hypothese von immunogenen Tumorantigenen und der dadurch ermöglichten, frühzeitigen Tumorabstoßung erweitert. [23, 22, 123] Es konnte weiterhin in Mausmodellen gezeigt werden, dass sowohl die Absenz von T-Zellen
als auch ein Defekt im IFN-γ-Rezeptor mit erhöhter Inzidenz an Neoplasien einhergeht.
[84]
Für die Immunosurveillance werden drei Stadien postuliert: die Vernichtung des Tumors (Elimination), das Gleichgewicht (Equilibrium) und die Immunflucht (Escape). Die Eli- mination des Tumors setzt die Erkennung voraus, sowohl durch die angeborene als auch durch die adaptive Säule des Immunsystems. Es schließt sich die Aktivierung des Immun- systems mittels Zytokinen, Antigen-Präsentation in den drainierenden Lymphknoten, Bildung von tumor-antigen-spezifischen T-Zellen und schließlich deren Einwanderung in den Tumor an. [46, 149] Daneben kommen auch nicht immunogene Kontrollsysteme wie intrinsische DNA-Reparatur oder Apoptose nach DNA-Schädigung zur Geltung.
Wenn aber die Elimination nicht vollständig erfolgreich ist, kommt es häufig zur Selektio- nierung von Tumorzellen. Dies bedeutet, dass Tumorzellen überleben, die aufgrund einer geringeren Immunogenität der Abstoßung durch das Immunsystem entgehen, oder die aufgrund des Selektionsdrucks Kontrollmechanismen der Zellproliferation überwinden konnten. Solange sich also Schutzmechanismen und tumor-abstoßende Immunreaktion sowie die Weiterentwicklung des Tumorgewebes die Waage halten, spricht man von der Phase des Equilibriums.
Kann die Immunreaktion das Wachstum des Tumors und dessen Metastasierung schließ- lich nicht mehr im Rahmen halten, spricht man von Immunescape. [91]
1.3.1. Tumor-Antigene
Gesunde Zellen werden bezüglich Lebensdauer, Wachstum, Funktionsweise und Zelltei- lung dergestalt reguliert, dass sie ihren spezifischen Zweck im Organismus erfüllen und sich im Gleichgewicht mit anderen Zellarten befinden. Jede Zelle präsentiert auf ihrer Oberfläche Strukturen, sogenannte Antigene, die zur Erkennung und Kommunikation mit anderen Zellen dienen. Diese Antigene können aus Proteinen, Lipiden oder Kohlen- hydraten bestehen.
Bei Malignomen ist die Zell-Steuerung defekt, was zu unbändigem Wachstum, der In- filtrierung gesunden umliegenden Gewebes und der Bildung von Metastasen an anderer Stelle führen kann. Tumore sind also Fremdkörper aus körpereigenen Zellen. Dies er- schwert die Erkennung als Pathogen, da das Antigen-Profil der Tumorzellen dem von gesunden Zellen stark ähnelt.
Charakteristische Antigene auf Tumoren stellen hingegen einen Ansatzpunkt für das Im- munsystem und damit auch Immuntherapien dar. Man kann diese Antigene hinsichtlich Ursprung und Art unterscheiden: [35]
Tumor-spezifische Antigene sind Folge von DNA-Mutationen, Spleiss-Varianten oder chromosomaler Translokationen. Deren Genprodukte werden auf der Zelloberfläche prä- sentiert und können dort von Immunzellen als fremd erkannt werden, da diese Anti- gene in gesunden Körperzellen nicht vorkommen. Ein Beispiel hierfür ist das Prostata- spezifische Antigen PSA. Auch können Strukturen, die eigentlich nur in der Embryoge- nese oder bei der Differenzierung eine Rolle spielen und in reifen Geweben supprimiert werden, durch gestörte Zellprozesse von Tumoren wieder produziert und exprimiert wer- den, beispielsweise HER-2/neu. [10, 18]
Zu den tumor-assoziierten Antigenen gehören Proteine, die im Organismus bereits be- kannt sind: Gewebe-spezifische Differenzierungsantigene kommen nur in einer bestimm- ten Gewebeart vor, jedoch auf gesunden wie auch auf Tumorzellen. Ein Beispiel ist MART1/Melan-A, das von Melanozyten und Melanomen exprimiert wird. Auf ähnliche Weise kann eine Störung der Gen-Steuerung zu einer verstärkten oder aberranten Ex- pression von Proteinen führen, die von gesunden Zellen zwar auch produziert, aber in deutlich geringerer Konzentration exprimiert werden. [124]
Besonders tumor-spezifische, aber auch tumor-assoziierte Antigene stellen also ein po- tentielles Ziel für die Immunabwehr dar.
1.3.2. Immunantwort
Die Schritte einer effektiven Tumor-Immunantwort beginnen mit der Erkennung eines Tumorantigens. Im Folgenden besteht die resultierende Antwort – neben dem angebo- renen Immunsystem mit z.B. Natürlichen Killerzellen – u.a. aus der klonalen Expansion von CD8+ T-Zellen mit Spezifität für ein tumor-assoziiertes Epitop, oder auch aus der Produktion tumor-spezifischer Antikörper. [80, 163] Man kann die Reaktion des Immun- systems auf einen Tumor in zwei Phasen aufteilen. In den sekundären Lymphorganen, z.B. den Lymphknoten, findet mittels Antigen-präsentierender Zellen die „Priming“- Phase statt, in der T- und B-Zellen oder auch Makrophagen geschult werden. Im Tumor selbst kommt es zur „Effector“-Phase, in der die tatsächliche Antwort auf die Tumorzel- len abläuft. Für Letztere müssen aktivierte Effektor-T-Zellen an und in die Tumorstelle wandern und Kontakt zu Antigen-exprimierenden Tumorzellen herstellen, um dann – die volle Funktionalität vorausgesetzt – die Abstoßung und Vernichtung der Tumorzel- len durchzuführen. [59] Insgesamt besteht der Ablauf der lymphozytären Reaktion initial aus der Aktivierung, dann der Expansion eben dieser aktivierten spezifischen Lympho- zyten, der Effektorantwort im Sinne der Tumorlyse (was jedoch die Überwindung einer möglichen Immunsuppression erfordert), sowie der Ausbildung von Memory T-Zellen.
Die Migration oder das sogenannte „Homing“ von aktivierten T-Zellen in Tumorgewe- be kann in einigen Krebsarten wie Melanomen, Kolon- oder Ovarkarzinomen spontan beobachtet werden. Die Beobachtung dieser lymphozytären Infiltration war der Grund- stein für die Forschung mit adoptiver T-Zell-Therapie, bei der tumor-spezifische T-Zellen ex vivokultiviert und anschließend in den Patienten re-infundiert werden. [45, 59] Auch wenn einige Tumorarten zu einer Rekrutierung von CD8+T-Lymphozyten führen, konn- ten bislang in den meisten Tumormetastasen diese Infiltrate nicht beobachtet werden.
Eine wichtige Rolle bei der effektiven Anlockung von Immunzellen spielen Chemokine, wie z.B. MCP-1, MIP-1α, IP-10 und Gamma-Interferon (IFN-γ).
1.3.3. Tumor Immunescape
Die exakten Vorgänge, mit denen der Tumor sich dem Immunsystem entzieht, sind noch nicht vollständig verstanden. Es wird postuliert, dass neben den Tumorzellen selbst auch das zelluläre Umfeld, das Tumor-Microenvironment, zur Immunescape beiträgt.
[59, 60, 179] Generell stehen dem Tumor zwei Wege zur Verfügung: sich nicht zu erkennen zu geben oder sich aktiv selbstzuverteidigen, durch Hemmung der Immunaktivierung oder durch Abwehr bereits aktivierter tumorspezifischer Zellen. Dem gegenüber steht das Phänomen von tumor-assoziierten Entzündungsreaktionen, die bei manchen Tumorarten der Tumorgenese vorausgehen, in anderen Fällen z.B. durch Zytokin-Produktion Teil der Tumorproliferation sind. [27] Daneben sind die bereits oben genannten Mechanismen im Sinne der „Hallmarks of Cancer“ natürlich essentiell für die Tumorpersistenz.
1.3.3.1. Hemmung der Tumorerkennung
Sich dem Zugriff des Immunsystems zu entziehen, beginnt damit, dass manche Tumoren eine mechanische Hürde, nämlich eine Kapsel, ausprägen, die den Zugang von Immun- zellen und dadurch die Erkennung des Tumors durch das Immunsystem verhindern.
Die verminderte Expression von MHC-I-Komplexen auf Tumoren wurde in mehreren Modellen beschrieben. Die MHC-Veränderung reduziert die Erkennung und Vernich- tung durch CD8+zytotoxische T-Zellen. [122] Die eingeschränkte Expression kann durch Genmutationen, die bei der Karzinogenese vorkommen, oder auch durch defekte Regula- tion und Prozessierung der MHC-Antigene verursacht sein. In kolorektalen Karzinomen konnte beispielsweise eine β2-Mikroglobulin-Mutation und eine Herabregulierung von LMP7/TAP2 als Auslöser des MHC-I-Verlusts identifiziert werden. [25] Daneben wur- de der Verlust von MHC-I nach Immuntherapie in einem Mausmodell beschrieben, der
durch ex vivo Kultivierung wieder rückgängig gemacht werden konnte und somit even- tuell vom Tumor-Microenvironment beeinflusst wird. [6]
Veränderte Expression von tumor-assoziierten Antigenen ist ein weiterer Mechanismus, mit dem sich Tumorzellen dem Zugriff von antigen-spezifischen Immunzellen entziehen.
Derartige Herunterregulierungen oder komplette Antigen-Verluste können unabhängig von der MHC-Veränderung auftreten und sind in einem Tumor oftmals heterogen ver- teilt. [88] Beispielsweise konnte der Verlust vom tumor-assoziierten Antigen MART-1 mit einer Tumorprogression in Korrelation gesetzt werden. [40] Ebenso wie der MHC- Verlust tritt der Antigenverlust v.a. nach initialer Immunreaktion oder Immuntherapie auf, im Sinne einer Selektionierung.
In humanen Pankreas-Karzinomen wurde der Verlust der CD3 zeta-Kette (CD247) in tumor-infiltrierenden Lymphozyten beobachtet. [167] Da diese Kette Teil des TCR- CD3-Komplexes und damit des TCR-Signalweges ist, führt das Fehlen von CD3 zeta zu verringerter T-Zell-Funktionalität. Das Auftreten dieser Veränderung konnte u.a. in Korrelation mit erhöhten Konzentrationen von IL-10 gesetzt werden. [6]
Die Steuerung der Zellbiologie in gesunden Organismen erfolgt unter anderem über Apoptose, also den gesteuerten Zelltod. Dieser wird z.B. über Fas, den Rezeptor für den Fas-Liganden FasL, oder über TRAIL-Rezeptoren (TNF-related apoptosis-inducing ligand receptors) vermittelt. In Tumoren konnte, um diesen Zelltod-Signalen zu ent- gehen, veränderte Expressionsmuster der Rezeptoren aber auch beteiligter Proteine wie cFLIP beobachtet werden.[37, 106] Gleichzeitig nutzen manche Tumoren wie z.B. Zervix- Adenokarzinome die Expression des Fas-Liganden, um Immunzellen in die Apoptose zu bringen und sich selbst vor der Vernichtung zu schützen. [85]
1.3.3.2. Inhibition und Einfluss durch Faktoren, Chemokine und Liganden Des Weiteren produzieren Tumore und ihr Microenvironment Zytokine und Chemokine, die das Immunsystem modulieren. Hervorzuheben sind Interleukin-10 (IL-10), Trans- forming Growth Factor β (TGF-β) und Vascular Endothelial Growth Factor (VEGF).
[98] In nicht-kleinzelligen Lungenkarzinomen konnte beispielsweise ein Übergewicht von immun-inhibitorischen Botenstoffen im Vergleich zu Immun-Stimulanzien festgestellt werden. [104]
VEGF fördert nicht nur die Gefäßeinsprossung und Blutversorgung des wachsenden Tumors, sondern verhindert auch die Differenzierung von dendritischen Zellen, blockiert den Transkriptionsfaktors NF-κB und hemmt damit die Immunantwort. [103, 121]
IL-10 wirkt als immun-inhibitorisches Zytokin, indem es die Antigen-Präsentation in
Dendritischen Zellen und dadurch die Aktivierung von CD8+ T-Zellen verhindert, sowie die Produktion von IL-12 hemmt. [39, 153] Es konnte auch gezeigt werden, dass bereits die Rekrutierung von DCs unter IL-10 inadäquat ist.
TGF-β spielt eine wichtige Rolle bei der Kontrolle des Zellzyklus samt Zellteilung und -differenzierung. Beispielsweise löst die Bindung von TGF-β an den Rezeptor mittels Mitogen-aktivierter Proteinkinase (MAP-Kinase) die Apoptose der Zelle aus. Außerdem inhibiert TGF-β die Aktivierung von Lymphozyten. [64] Bei der Tumorgenese kommt es oftmals zu einer Mutation des TGF-β-Signalweges, so dass eine Kontrolle des Zell- zyklus und der Proliferation misslingt. [90] Gleichzeitig führt eine verstärkte Sekretion von TGF-β zur Angiogenese, zur Produktion von IL-10 und zur Aktivierung immun- inhibitorischer regulatorischer T-Zellen. [17, 126]
Zuletzt können Tumoren sich direkt gegen antigen-spezifische T-Zellen verteidigen, in- dem sie inhibitorische Moleküle wie CTLA-4 (siehe oben, Koinhibition) oder PD-L1 exprimieren. [41]
1.3.3.3. Fehlen von Kostimuli
Trotz Anwesenheit von tumor-spezifischen Immunzellen und Expression von MHC und Antigenen können viele Tumore proliferieren. Dies kann am Fehlen von kostimulatori- schen Signalen liegen, die notwendig für die T-Zell-Aktivierung sind, wie z.B. CD28 als Rezeptor für CD80 und CD86. Entsprechend der Zwei-Signal-Hypothese treten T-Zellen bei Ausbleiben der Kostimulation in den Zustand der Anergie ein und der Tumor erreicht so eine antigen-spezifische Toleranz. [3, 143]
1.3.3.4. Zelluläre Inhibitoren
Als Kontrollinstanz des Immunsystems existieren sogenannte regulatorische T-Zellen, kurz Tregs, eine T-Zell-Subpopulation mit charakteristischer Expression von CD4, CD25 (IL-2 Rezeptor) und FoxP3. Sie sollen überschießende Immunantworten und eine Aktivie- rung von weiteren T-Zellen bremsen. Während dieser Mechanismus Autoimmun-Schäden verhindert, stellt es einen Fluchtmechanismus für Tumoren dar und ermöglicht deren Progression. [139, 159] Beispielsweise konnten erhöhte Konzentrationen von TGF-β se- zernierenden Tregs in Tumorinfiltraten von epithelialen Malignomen und deren immun- inhibitorische Wirkung nachgewiesen werden. [160, 171]
1.4. Tumor-infiltrierende Lymphozyten
Untersuchungen von Tumorproben konnten die Existenz von tumor-infiltrierenden Lym- phozyten (TIL) und deren Antigen-Spezifität nachweisen. Dennoch resultiert diese Ak- tivierung des Immunsystems nicht immer in einer erfolgreichen Abstoßung des Tumors.
Auch in Experimenten ex vivo waren die TILs oftmals unfähig, zytotoxisch zu wirken.
[67, 136] Die Sekretion von zytolytischen Substanzen wie Perforin oder Granzym B, sowie die Fähigkeit, Zielzellen zu erkennen und an diese zu binden, war reduziert. Dane- ben wurden in TILs auch verminderte Zytokin-Produktion und Proliferation beobachtet.
[132]
Die mangelhafte Perforin-Produktion könnte dabei im natürlichen Zellzyklus begründet sein: nach der kompletten Veräußerung vorhandener zytolytischer Speichergranula ist vor erneuter toxischer Funktionalität die Regeneration im Sinne des Zellzyklus notwendig.
[59] Dafür sprechen die Ergebnisse von Experimenten, in denen ex vivo eine starke T- Zell-Aktivierung erreicht wurde über Bindung von CD3/CD28, und die eine zunehmende Granula-Expression in Memory-Zellen und TILs zur Folge hatte. [60, 107]
1.4.1. Anergie
Man spricht vom Übergang der T-Zelle in den Zustand der Anergie, wenn der T-Zell- Rezeptor sein Antigen erkennt und darüber aktiviert wird, eine Kostimulation z.B. über CD28 aber ausbleibt. Da auch eine erneute Erkennung des Antigens im Sinne einer Restimulation oder ein späterer Kostimulus die Anergie nicht wieder aufheben kann und kein IL-2 produziert wird, sind derartig anerge Lymphozyten nicht in der Lage – trotz ihrer Antigenspezifität – eine effektive Abstoßung herbeizuführen. [50, 150, 164]
Gajewski setzte ein Mausmodell mit 2C T-Zell-Rezeptor transgenen T-Zellen ein, bei der die Stimulierung der T-Zellen ausschließlich über das allo-antigene MHC Ld ohne eine B7-vermittelte Kostimulation ablief. Phänotypisch waren die T-Zellen nach Tumorkon- takt mit einer hohen CD44-Expression aktiviert, jedoch waren die T-Zellenex vivo nach TCR/CD28-Stimulation nicht in der Lage, Zytokine zu produzieren. Die Kultivierung mit Phorbolmyristat-Acetat und Ionomycin (PMA/I), das die proximale Signalübermitt- lung des T-Zell-Rezeptors umgeht, führte zur normalen Zytokin-Ausschüttung, was dem Verhalten klassisch anerger Lymphozyten entspricht.[51, 59]
1.4.2. Exhaustion
Rafi Ahmed beschrieb die funktionelle Erschöpfung von T-Zellen in chronischen Infek- tionen, die sogenannte „Exhaustion“. [176] Diese führt zum zunehmenden Verlust der Zytokin-Produktion von IL-2, TNF-α und IFN-γ, sowie der Zytotoxizität ex vivo. [89]
Ebenfalls war die Reaktivität auf IL-7 und IL-15 vermindert. [169] „Exhausted“ CD4+ T-Zellen produzieren weniger IL-2 und TNF-α, dafür größere Mengen des immunsup- pressiven IL-10. [20] Gen-Analysen legten Veränderungen in „exhausted“ T-Zellen offen, die v.a. Transkriptionsfaktoren und für Chemotaxis und Migration notwendige Genex- pressionen betreffen. Außerdem zeigten sich Defizite im Zellmetabolismus und Energie- haushalt. Ein besonderes Merkmal ist die hohe Expression von PD-1, auf das später noch eingegangen wird. [170]
Tumor-infiltrierende T-Zellen demonstrieren in Tumoren mit hoher Antigendichte ein vergleichbares Verhalten wie virus-spezifische T-Zellen bei chronischen Infektionen, mit vergleichbarem Verlust der Effektorfunktion und ähnlichem Phänotyp. [89]
1.4.3. Defekte in T-Zell-Signal und MTOC-Polarisierung
Frey et al. untersuchten im Mausmodell den Mechanismus der TCR-Signal-Blockade als Ursache für ineffektive Tumorabstoßung. Direkt nach Gewinnung von Lymphozy- ten aus Tumormaterial war die lytische Funktion in vitro aufgehoben. Mikroskopische Analysen zeigten, dass die TILs nicht in der Lage waren, lytische Granula und den MTOC an die Immunsynapse mit Zielzellen zu polarisieren.[133] Der Defekt wurde auf die Blockade des antigen-abhängigen TCR-Signals zurückgeführt. [93] Damit einher ging auch eine verringerte Adhäsion der TILs an die Tumorzellen. [92] Initiale TCR-Signale führten zur raschen Aktivierung und Verschiebung von inhibitorisch wirksamen SHP- 1-Phosphatasen. Nach Kultivierung ex vivo, ohne Tumorkontakt, konnten die TILs die Blockade des TCR-Signalwegs und damit ihre Funktionalität wiedergewinnen. Ebenso führte aber auch die erneute Kokultivierung mit Tumorzellen zu einem erneuten Funk- tionsverlust. Daraus postulierten Frey et al. einen schnell wirksamen Inhibitor, der nach Antigen-Erkennung aktiv wird. [166]
1.5. Programmed Death Receptor 1 (PD-1)
1.5.1. Struktur, Vorkommen und Signalwirkung
Programmed Death Receptor 1 (PD-1) wurde erstmalig 1992 in einem T-Zell-Hybridom beschrieben, dessen Zellen sich in der Apoptose, dem programmierten Zelltod, befan- den. Hierbei wurde das PD-1 kodierende Gen Pdcd1 hochreguliert [77]. Pdcd1 liegt bei Mäusen auf Chromosom 1 und bei Menschen auf Chromosom 2 und enthält 5 Exons.
Das daraus kodierte Protein aus 288 Aminosäuren gehört zu den Typ I Transmembran- Proteinen. Die Struktur setzt sich zusammen aus einer extrazellulären IgV-ähnlichen Immunglobulin-Domäne, einer Verbindungsdomäne, einer Transmembran-Domäne so- wie einer Intrazellulär-Domäne, die zwei Tyrosin-gebundene Signalmotive trägt: Immu- noreceptor tyrosine-based inhibitory motif (ITIM) und Immunoreceptor tyrosine-based switch motif (ITSM). [66, 101]. Aufgrund diesen Designs, das in ähnlicher Form bei CD28, ICOS und CTLA-4 vorherrscht, wurde PD-1 zu der CD28-Familie gezählt. Aller- dings bestehen klare Unterschiede: CD28-Mitglieder existieren meist als Dimere, wäh- rend PD-1 als Monomer exprimiert wird. Auch die Anordnung von ITIM und ITSM sind so nicht in CD28-Proteinen zu finden. [118]
Exprimiert wird PD-1 auf der Zelloberfläche von CD4+ und CD8+ T-Zellen, B-Zellen, Natürlichen Killer (NK) T-Zellen, aktivierten Monozyten sowie dendritischen Zellen (DC). Ruhende, naive T-Zellen exprimieren kein PD-1, die Expression ist aber durch Aktivierung induzierbar [5, 77, 113] und wird durch Anwesenheit von Tumornekrose- Faktor TNF verstärkt. [118]
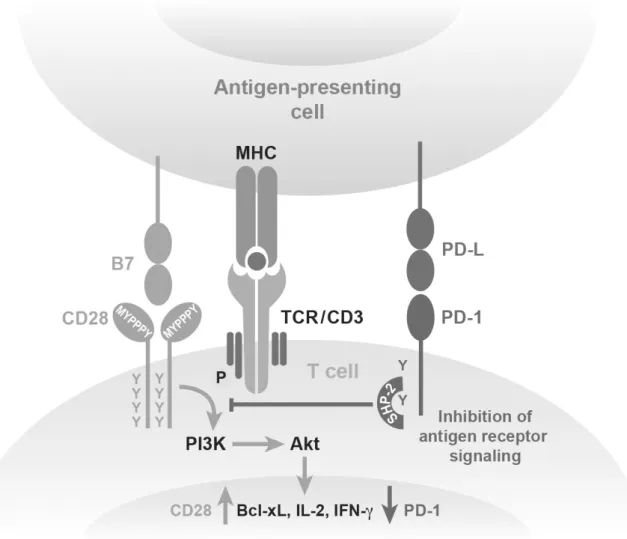
Die Ligandenbindung am extrazellulären Teil von PD-1 führt zur Phosphorylierung der zwei Tyrosin-Reste auf der intrazellulären Seite. Daraufhin wird die Phosphatase SHP-2 (Src homology 2-containing tyrosine phophatase 2), sowie in geringerem Maße SHP-1 an die Zytoplasma-Domäne gebunden, wo sie bei gleichzeitiger Aktivierung des T-Zell- Rezeptors Zwischenprodukte der TCR-Signalkette wie Syk und Phosphatidylinositol- 3-OH-Kinase (PI3K) dephosphorylieren und damit inaktivieren können. [119, 146] Da- durch wird die Phosphorylierung des Moleküls Akt, das PI3K bei der Signalübertragung folgt, verhindert. Ebenfalls inhibiert wird die Phosphorylierung von CD3-ζ, ZAP70 und PKC-θ sowie die Aktivierung von Erk [86, 125]. SHP-1 ist bekannt als negativer Regu- lator der Zellaktivierung, während SHP-2 einerseits im Zusammenhang mit Zellaktivie- rung durch Hemmung von negativen Regulatoren mittels Dephosphorylierung, als auch negativer Beeinflussung von IL-6 und dem Wachstumshormon GH beschrieben wurde.
PI3K-Aktivierung und nachfolgend Phosphorylierung von Akt (Proteinkinase B) resul-
tieren in einer erhöhten Expression von Glucosetransportern in der Zellmembran und in erhöhter glykolytischer Enzymaktivität. Diese Bereitstellung von Energie ist die Grund- lage für die aktivierte Zelle, Effektorfunktionen zu übernehmen und proliferieren zu können. [53] Die Blockade dieser Signalkette führt demnach zur Reduktion des Glucose- Stoffwechsels, aber auch der Genkodierung von Bcl-xL, einem Apoptose-hemmenden Protein. [134] Ferner resultiert die PD-1-vermittelte Zellhemmung in reduzierter Zytokin- Produktion (v.a. IFN-γ, TNF-αund IL-2) und in geringerer zellulärer Proliferation.
Der inhibitorische Effekt ist dabei indirekt proportional zur Stärke der TCR-Stimulation, kann aber auch durchbrochen werden, v.a. wenn es zu einer STAT5-Aktivierung kommt, wie es bei IL-2, IL-7 und IL-15 Signalen der Fall ist. [11, 29, 55] Auch Stimulierung mittels CD28, das zur Zellexpansion und zur Aktivierung von anti-apoptotischen und zytokin-enkodierenden (IL-2, IFN-γ) Genen führt, kann der Hemmung durch PD-1 ent- gegenwirken. [55]
1.5.2. PD-1-Liganden und zweiter Rezeptor
Es sind zwei Liganden für PD-1 beschrieben. Programmed Death Receptor 1 Ligand 1, kurz PD-L1 (B7-H1; CD274) ist wie PD-1 ein Typ I Transmembran-Protein mit einer Länge von 290 Aminosäuren. Das auf Chromosom 19 (Maus) bzw. 9 (Mensch) befindli- che Gen Cd274 besteht aus 7 Exons und kodiert für eine IgV- und eine IgC-Domäne. Das Molekül hat eine weite Verbreitung auf hämatopoetischen sowie nicht-hämatopoetischen Zellen. [42, 55] Es kommt vor auf T-Zellen, B-Zellen, dendritischen Zellen, Makropha- gen, mesenchymalen Stammzellen, Knochenmark-entstammenden Mastzellen, vaskulä- rem Endothel, Epithel, Keratinozyten, Muskelzellen, Hepatozyten, Pankreasinselzellen, Astrozyten, in der Plazenta und auf der Cornea des Auges. [49, 142, 173] Die Anwe- senheit von IFN-α, -β und -γ aber auch IL-10 triggert eine verstärkte Expression von PD-L1. [145]
PD-L2 (B7-DC; CD273) wird durch das Gen Pdcd1lg2 (7 Exons) kodiert und ist ebenfalls ein Typ I Transmembran-Protein, zeigt aber eine deutlich geringere Verbreitung im Vergleich zu PD-L1. Es ist hauptsächlich auf dendritischen Zellen, Makrophagen und kultivierten Knochenmark-abstammenden Mastzellen zu finden. [111] PD-L2 trägt wie PD-L1 extrazellulär eine IgC- und IgV-Domäne. PD-L2 wird nach Stimulierung durch IL-4, NF-κB, IFN-γund GM-CSF (Granulocyte/Monocyte Colony-Stimulating Factor) aufreguliert. [127]
Ferner konnte ein zweiter Rezeptor für PD-L1 identifiziert werden. Es handelt sich mit B7-1 (CD80) um ein bereits bekanntes Mitglied der B7-Superfamilie. B7-1 bindet PD-
L1 mit einer höheren Affinität als PD-1/PD-L1, jedoch mit einer geringeren Affinität als B7-1/CD28. PD-L1 kann somit sowohl über PD-1 als auch über B7-1 inhibitorisch wirken. [24, 86]
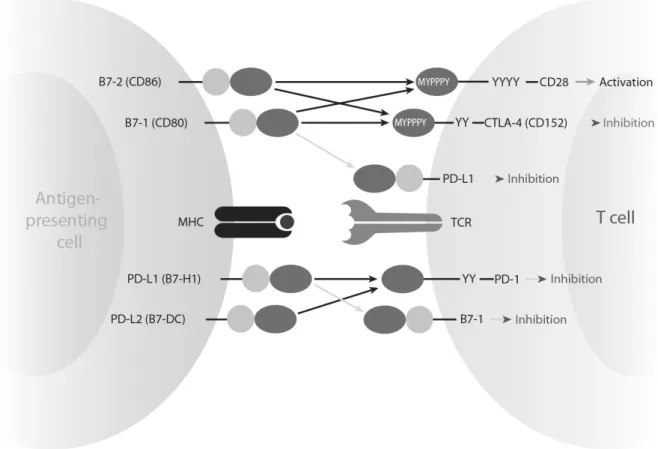
Abbildung 1.3.: Ligandenpaare der B7-Familie zwischen T-Zelle und APC: PD-1-Aktivierung bzw. PD- L1-Bindung führen zur T-Zell-Inhibition. (nach: Keir et al., 2008 [86])
1.5.3. PD-1 und Autoimmunität
Die Bildung zentraler Toleranz des Immunsystems im Thymus wird von PD-1 Signalen beeinflusst, wobei sowohl die positive als auch die negative Selektionierung reifender Thymozyten betroffen ist und das Fehlen von PD-1 oder seinen Liganden zu veränderten Phänotypen der Lymphozyten führt. [14, 87, 114]
Auch bei der peripheren Toleranz ist PD-1 beteiligt: entgehen T-Zellen, die eine zu hohe Eigen-Reaktivität haben, der Selektionierung im Thymus, so können diese über PD- 1/PD-L1-Interaktion gehemmt werden. Dies bedeutet umgekehrt, dass das Fehlen von PD-1 zu einer defekten Regulierung der Autoimmunität führt.
In Modellen konnte der Einfluss von PD-1 auf Autoimmun-Erkrankungen gezeigt wer- den. PD-1 defiziente C57BL/6 Mäuse entwickelten beispielsweise Glomerulonephritiden,
während Balb/c Mäuse ohne das Pdcd1-Gen an autoimmun-dilativen Kardiomyopathien erkrankten. [116, 117] Die Antikörper-Blockade von PD-1 verursachte eine Exazerbation des autoimmun-vermittelten Diabetes mellitus. [8] Ebenso nahmen entzündliche Infil- trate im zentralen Nervensystem im EAE-Mausmodell – für Multiple Sklerose – zu, wenn anti-PD-1 oder anti-PD-L2 Antikörper verabreicht wurden. Dies zeigt, dass sich eine auto-immun vermittelte Erkrankung wie die Multiple Sklerose bzw. EAE im Tier- modell bei defekter PD-1-abhängiger Immunregulierung verschlechtert aufgrund einer überschießenden Immunreaktion. Hingegen verbesserten dendritische Zellen, die PD-L1 überexprimieren, die auto-immune Enzephalitis und konnten die Inflammation eindäm- men [74].
Eine punktuelle Veränderung der Pdcd1-Genstruktur, ein sogenannter Single nucleoti- de polymorphism (SNP) wird assoziiert mit dem Auftreten von systemischem Lupus erythematodes, Diabetes mellitus Typ I und Encephalitis disseminata [97, 130].
1.5.4. PD-1 in Infektion und Exhaustion
Auch virale und bakterielle Infektionen führen im idealen Fall zu einer Erkennung und Vernichtung des Pathogens durch das Immunsystem. PD-1 vermittelte T-Zell-Inhibition kann jedoch bei der Etablierung chronischer Infektionen eine Rolle spielen. [169] Eine hohe PD-1-Expression auf HIV-spezifischen CD8+ Lymphozyten korreliert beispielswei- se mit einer erhöhten Viruslast und reduzierter CD4-Zahl. [44] Versuche, die PD-1- Inhibition mit Antikörper-Therapie oder PD-1 defizienten T-Zellen zu durchbrechen, führten allerdings nicht nur zu verbesserter Immunantwort bei HIV, Hepatitis B und C, HTLV, Helicobacter pylori und weiteren Erkrankungen, sondern auch zu Schädigung von gesundem Gewebe. [79]
Ahmed et al. konnte im LCMV-Mausmodell zeigen, dass in akuten Infektionen virus- spezifische CD8+T-Zellen anfangs PD-1 exprimierten, nach Ausheilung PD-1 herabregu- lierten, und im Folgenden virus-spezifische Gedächtniszellen bildeten. Die chronische In- fektion hingegen führte zu konstanter, im Verlauf nicht rückläufiger Expression von PD-1 auf T-Zellen, die zwar virus-spezifisch, nach einiger Zeit aber nur noch mono-funktionell (entweder Produktion von IFN-γoder TNF-α) waren. 80 Tage nach Infektion waren die T-Zellen größtenteils unfunktionell, „exhausted“. Wurde in diesen Tieren der PD-1/PD- L1-Signalweg durch Zugabe von Antikörpern blockiert, konnte eine Wiederherstellung der Effektorfunktion und eine verbesserte Viruskontrolle beobachtet werden. [9, 170]
Abbildung 1.4.: Signalwirkung von PD-L1: Ligandeninteraktion von PD-L1/PD-1 führt zu einer Unter- brechung der Antigenrezeptor-Signalkaskade und damit zu einer Hemmung der T-Zell- Funktionalität. (nach: Keir et al, 2008 [86])
1.5.5. PD-1 und Tumore
Eine besonders ausgeprägte Rolle spielt die Interaktion zwischen PD-L1 und PD-1, wie oben bereits erwähnt, als Mechanismus des Immunescape von Tumoren.
PD-L1 wird in einer relativ großen Anzahl verschiedener Tumorgewebe exprimiert, wie z.B. in Malignomen in Brust, Lunge, Colon, Ovar, Haut (Melanome), Blase, Leber, Ma- gen, Gehirn (Gliome), Niere, Pankreas und Thyreoidea. In Ovarkarzinomen hemmt PD- L1 die Fähigkeit von CD8+ T-Zellen, in das Tumorgewebe einzuwandern. In Myelomen korreliert die PD-L1-Expression mit einer schlechteren Prognose. Vergleichbare Aussagen wurden zu Nicht-kleinzelligen Bronchialkarzinomen, Magen- und Nierenzellkarzinomen veröffentlicht. [95, 158] In anderen Veröffentlichungen divergieren die Ergebnisse hin- sichtlich PD-L1-Expression auf Tumorzellen und prognostischer Relevanz, wie es die
Veröffentlichungen von Hino et al. (2010), Gadiot et al. (2011) und Taube et al. (2012) demonstrieren. [57, 72, 156]
Im Tiermodell verhindert PD-L1 auf Tumoren die Aktivierung von T-Zellen und damit die Tumorzell-Lyse; in manchen Fällen kommt es zur vermehrten Apoptose von tumor- spezifischen T-Zellen. [41, 69]
Die Blockade des PD-1/PD-L1-Signalwegs führt in vitro zu einer verbesserten T-Zell- Aktivierung [36, 41, 86, 95, 157].In vivoführt der Einsatz von blockierenden anti-PD-L1 Antikörpern bei Mäusen sowie ein PD-1 defizienter Genotyp zu verbesserter Antitumor- Immunantwort, teilweise mit kompletter Tumorabstoßung. [15, 73, 78]
Auch tumor-assoziierte dendritische Zellen können PD-L1 auf ihrer Oberfläche hochre- gulieren. Die Blockade mittels eines murinen anti-PD-L1 Antikörper kann hierbei die DC-vermittelte T-Zell-Aktivierung verbessern. [36]
Die Analyse von tumor-infiltrierenden Lymphozyten (TIL) zeigte in Untersuchungen, dass TIL im Vergleich zu Lymphozyten in gesundem Gewebe oder im peripheren Blut mehr PD-1 exprimieren. Da auch tumor-spezifische T-Zellen im peripheren Blut we- niger PD-1 exprimierten als tumor-spezifische TIL, legen diese Daten nahe, dass die Expressionszunahme von PD-1 durch das Microenvironment induziert wird. Die Unter- suchung der Funktionalität zeigte, dass auch ein TCR-unabhängiger, starker Stimulus (PMA/Ionomycin) in den tumor-spezifischen, PD-1 positiven TIL keine vergleichbare Zytokin-Produktion wie in PD-1 negativen TIL mehr auslösen konnte: die Effektorfunk- tion war zu stark via PD-1 gehemmt. [86]
1.5.6. Lokalisation von PD-1
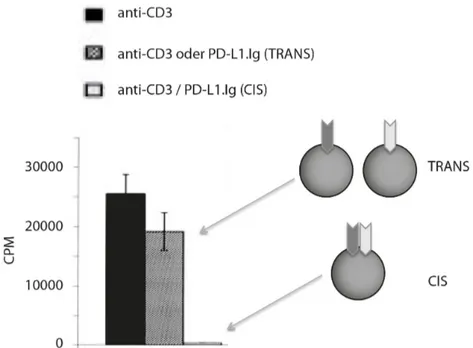
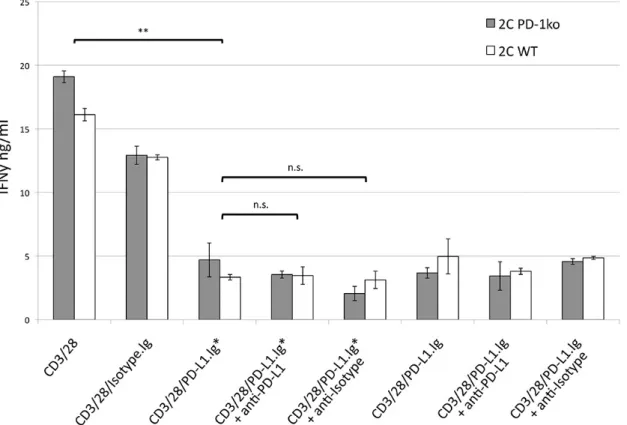
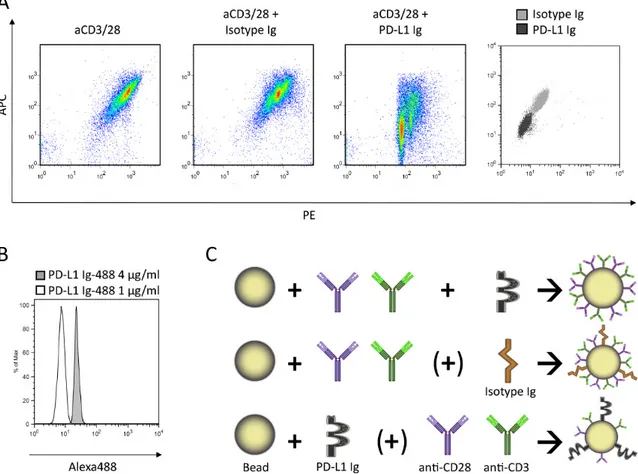
Bennett et al. setzten in Experimenten sogenannte Beads ein, d.h. protein-beladbare Mi- krosphären aus Eisenpartikeln, um die Lokalisation von PD-1 und dem T-Zell-Rezeptor und eine eventuelle Abhängigkeit von einander zu untersuchen. Die Ergebnisse legten na- he, dass PD-1 sich in unmittelbarer Nachbarschaft zum Antigenrezeptor befinden muss, um seine inhibitorische Wirkung zu entfalten: Nur Beads, die sowohl einen TCR-Stimulus (anti-CD3 Antikörper) als auch den Liganden für PD-1 gleichzeitig auf der Oberfläche trugen (CIS-Konfiguration), führten zu reduzierter T-Zell-Aktivierung. Waren PD-L1 und anti-CD3 hingegen auf getrennten Beads (TRANS-Konfiguration) gebunden, fand keine Inhibition statt. [11] (Abbildung 1.5)
Auch Untersuchungen von Chemnitz et al., die die Rekrutierung von SHP-1 und SHP- 2 zu PD-1 untersuchten, legten nahe, dass das PD-1-Signal in direkter Nähe zu CD3 oder CD28 initiiert werden muss, da andernfalls keine Hemmung der T-Zell-Antwort
Abbildung 1.5.: PD-1/TCR-Kolokalisation als Voraussetzung für PD-L1-vermittelte Inhibition: Da aus- schließlich simultane Präsentation von PD-1- und TCR-Stimuli auf gemeinsamem Bead zur T-Zell-Hemmung führten, während separate Präsentation auf getrennten Beads die- sen Effekt nicht zeigte, postulierte Bennett et al. die Kolokalisation von TCR und PD-1 als Voraussetzung für eine PD-L1-vermittelte T-Zell-Hemmung. (nach: Bennett et al, 2003 [11])
stattfindet. [32]
Untersuchungen der intra- und extrazellulären Lokalisierung von PD-1 in der Arbeits- gruppe von James Allison zeigten, dass PD-1 in CD4+ T-Zellen sowohl an der Plasma- Membran, in der Nähe des Golgi-Apparates und am Trans-Golgi-Netzwerk zu finden ist. Gehen die CD4+ T-Zellen dann physischen Kontakt mit einer dendritischen Zelle ein, zeigt sich initial eine Akkumulation von PD-1 an der Synapse, ähnlich wie CD28, allerdings verteilt sich im Zeitverlauf PD-1 dann wieder diffuser auf der Zelloberfläche.
Die Konzentrierung von PD-1 an der Zellkontaktstelle war dabei abhängig von der Prä- senz von PD-L1 und PD-L2, wobei PD-L2 die größere Affinität besitzt und zu einer verstärkten Akkumulation führt. [127]
1.6. Fragestellung dieser Promotion
In Zusammenschau der bisherigen Forschungsergebnisse und Datenlage ist also festzu- halten, dass eine Interaktion von PD-L1 und PD-1 eine Rolle im Immunescape Mecha- nismus von Tumoren spielt. Dies wird unterstrichen durch die Expressionsmuster von
PD-1 auf tumor-infiltrierenden Lymphozyten und PD-L1 auf einer Breite an Tumorar- ten. Die Lokalisationsanalysen mit Bead-Zell-Konjugaten von Bennett et al. legen dabei eine Ortsabhängigkeit bzw. eine Kolokalisation von PD-1 und dem T-Zell-Rezeptor nahe.
Unter der Annahme, dass eine Modifikation des PD-L1/PD-1-Signals einen Ansatzpunkt für eine mögliche Tumortherapie darstellt, soll im Mausmodell der Einfluss von PD-1 auf die T-Zell-Funktionalität durch genetische Variationen (Knockoutsetting, Wildtyp, Überexpression) verglichen werden. Unklar ist auch bislang, welche Rolle die Lokalisation von PD-1 während der Interaktion zwischen T-Lymphozyt und Tumorzelle spielt und ob das Phänomen, das bei Bead-Zell-Analysen beobachtet wurde, auch hier nachvollzogen werden kann. Diesbezüglich soll auch untersucht werden, ob die beschriebene Koloka- lisation von PD-1 mit dem TCR auch eine Veränderung der Oberflächen-Expression bedingt, also ob und wenn ja, wann im Laufe der T-Zell-Aktivierung PD-1 divers oder eher akkumuliert exprimiert wird. Da die T-Zell-Effektorfunktionalität abhängt von der adäquaten Modifikation des Zellskeletts und der Ausbildung der Synapse, also des „Kiss of Death“, stellt sich hier die Frage, ob die Inhibition durch PD-1 möglicherweise einer insuffizienten Ausbildung eben dieses „Kiss of Death“ zu Grunde liegt. Und schließlich ist natürlich von Interesse, ob – wenn PD-1 aller Wahrscheinlichkeit nach kolokalisiert mit dem TCR und somit eine aktive Bewegung stattfindet – auch der Tumor eine Verände- rung seiner PD-L1 Expression bzw. des Expressionsmusters im Sinne eines „Counterat- tack“ herbeiführt, um den Immunescape zu optimieren. Zusammengefasst ist somit das Ziel dieser Promotionsarbeit die Analyse der Lokalisation von PD-1 auf T-Lymphozyten während der Interaktion mit Tumorzellen und der dadurch bedingte Einfluss auf die T- Zell-Funktionalität.
2. Material und Methoden
2.1. Material
2.1.1. Tumorzelllinien
Tabelle 2.1.: Tumorzelllinien
Tumor Tumorart Maus Background Literatur/Anmerkung
p815 Mastozytom H-2d DBA/2 ATCC
p815.B7-1 Mastozytom H-2d DBA/2 Gajewski et al. [58]
mit B71 transfiziert HTR.c Mastozytom H-2d DAB/2 Gajewski et al. [58]
Subklon von p815, bildet solide Tumo- renin vivo
MC57-SIY Fibrosarkom H-2b C57BL/6J Spiotto et al. [151]
transfiziert mit SIYRYGGL EL4 T-Zell-Lymphom H-2b C57BL/6N ATCC
chemisch induziert B16.SIY Melanom H-2b C57BL/6J Blank, Brown et al.[15]
transduziert mit dem Peptid SIYRYGGL-eGFP
B16.SIY E12 Melanom H-2b C57BL/6J transduziert mit dem Peptid SIY- RYGGL ohne GFP
zur Verfügung gestellt von Lisa Borkner
2.1.2. Mausstämme
Experimente mit murinen Zellen wurden an der Universität Regensburg und am NKI (Nederlands Kanker Instituut) in Amsterdam durchgeführt. Die Tiere wurden in IVC (individually ventilated cages)-Käfigen, gemäß den Richtlinien des Deutschen bzw. Nie- derländischen Tierschutzgesetzes gehalten. In Regensburg wurde dem Trinkwasser stan-
dardmäßig konzentrierte Salzsäure HCl (1ml/l Wasser) zugesetzt. Zur Vermeidung no- sokomialer Infektionen wurde zusätzlich Cotrim K (144mg Cotrimoxazol (Kombination aus Trimethoprim und Sulfamethoxazol)/250ml Wasser) zugegeben.
Tabelle 2.2.: Mausstämme
Maus Haltung Erstbeschreibung Herkunft
C57BL/6 Kauf - Janvier, Le Genest-St-
Isle, Frankreich C57BL/6 PD1tg Zucht, IVC Keir et al. [87] zur Verfügung gestellt
durch A. Sharpe, Ei- genzucht
2C TCRtg RAG2-/- Zucht, IVC Kranz et al. [96];
Chen et al. [33]
Zur Verfügung gestellt durch T. Gajewski;
Eigenzucht
2C TCRtg RAG2-/- PD1-/- Zucht, IVC Nishimura et al. [115] Zur Verfügung gestellt durch T. Honjo; Ei- genzucht
2C TCRtg RAG2-/- PD1tg Zucht, IVC - Eigenzucht
C57BL/6 PD-L1-/- IVC - Zur Verfügung gestellt
durch Universität Würzburg
2C TCRtg RAG2-/- Die beiden Gene RAG-1 und RAG-2 (Recombinase Activati- on Gene) werden zur Rekombination der T- und B-Zell-Rezeptoren benötigt. Bei einem Defekt in einem der beiden Gene werden keine funktionellen Rezeptoren gebildet und es entstehen keine reifen T- oder B-Zellen. RAG1-/- und RAG2-/- Mäuse sind daher von Geburt an lymphopen [108, 148].
2C TCRtg RAG2-/- Mäuse tragen zusätzlich zur RAG-Defizienz die Informati- on für einen transgenen TCR, was zur Ausreifung einer monoklonalen T-Zell- Population führt. Dieser transgene CD8+ T-Zellklon ist spezifisch sowohl für das artifizielle SIY-Peptid (SIYRYGGL), welches auf dem syngenen MHC-I-Komplex H-2Kb präsentiert wird, als auch für das ubiquitär vorkommende p2Ca-Peptid (LSPFPFDL) der α-Ketoglutarat-Dehydrogenase im allogenen MHC-I-Komplex H-2Ld [96, 144]. Auch in diesem Mausstamm werden aufgrund des genetischen Defektes im RAG2-Gen alle T- und B-Zellen während der Thymusreifung deple- tiert, mit Ausnahme der T-Zellen, die den transgenen Rezeptor exprimieren. Da
die eingefügte genetische Information für den T-Zell-Rezeptor CD8-restringiert ist, liegen in der Peripherie daher ausschließlich 2C TCRtg CD8+ T-Zellen vor.
2C TCRtg RAG2-/- PD1-/- 2C TCRtg RAG2-/- wurden mit PD1-/- Mäusen ge- kreuzt. Die PD-1-Defizienz ist bedingt durch eine Veränderung im PD-1 kodie- renden Gen PDCD1, das zu einem Defekt in der Transmembrandomäne von PD-1 führt. Dadurch wird zwar PD-1 transduziert, aber nicht auf der Zelloberfläche prä- sentiert. Intrazellulär ist aber immer noch die extrazelluläre Domäne vorhanden und nachweisbar.
2C TCRtg RAG2-/- PD1tg Ebenso wurden 2C TCRtg RAG2-/- Mäuse mit BL/6 PD1tg Mäusen gekreuzt. Die so entstandenen Mäuse haben ausschließlich T-Zellen mit dem 2C TCR und zeigen bereits im naiven T-Zell-Stadium eine konstitutive Überexpression von PD-1.
2.1.3. Antikörper
Tabelle 2.3.: FACS Antikörper
Isotyp Klon Herkunft Fluorochrom Hersteller Verd.
anti-CD3 rIgG1,κ 145-2C11 Ratte F, APC BD Pharmingen 1:80 anti-CD4 rIgG2a,κ RM4-5 Ratte F, PE, APC BD Pharmingen 1:80 anti-CD8 rIgG2a,κ 53-6.7 Ratte F, PE, APC BD Pharmingen 1:80
anti-CD28 hIgG 37.51 Hamster PE ebioscience
anti-CD44 rIgG2a,κ IM7 Ratte PE BD Pharmingen 1:80
anti-CD62L rIgG2a,κ Mel-14 Ratte PE BD Pharmingen 1:80
anti-CD80 Hamster PE 1:80
anti-CD86 Ratte PE 1:80
anti-CD107a rIgG2a,κ 1D4B Ratte F BD Pharmingen 1:200
anti-PD1 hIgG J43 Hamster PE ebioscience 1:50
anti-PD-L1 rIgG2a,κ MIH5 Ratte PE ebioscience 1:80
anti-H-2Kb Biotin 1:50
anti-H-2Ld Maus purified 1:1
anti-mouse Ig Ziege PE Southern Biotech 1:20
anti-mouse Ig Ziege F BD Pharmingen 1:50
Isotyp rIgG2a,κ R35-95 Ratte F, PE, APC BD Pharmingen
anti-1B2 - - Maus F, AF647 Kranz et al.[96] 1:50
2.4G2 - - - - ATCC -
Streptavidin - - - PE BD Pharmingen 1:50
Tabelle 2.4.: Antikörper und Standards für ELISA
BD Pharmingen Herkunft Klon Konz.
Capture anti-Maus IL-2 Ratte JES6-1A12 1µg/ml
Detektion anti-Maus IL2-bio Ratte JES6-5H4 0,1µg/ml
Standard rMu IL2 0,02µg/ml
Capture anti-Maus IFN Ratte R4-6A2 1µg/ml
Detektion anti-Maus IFN-bio Ratte XMG1.2 0,1µg/ml
Standard rMu IFN 0,1µg/ml
Tabelle 2.5.: Antikörper für Immunfluoreszenz
Isotyp Klon Herkunft FluorochromHersteller Verd.
anti-CD3 rIgG1,κ 145-2C11 Ratte FITC BD Pharmingen 1:30 anti-CD8 rIgG2a,κ 53-6.7 Ratte FITC BD Pharmingen 1:50
anti-PD1 hIgG J43 Hamster purified,
Biotin
ebioscience 1:30 anti-PD-L1 rIgG2a,κ MIH5 Ratte purified ebioscience 1:30
anti-1B2 - - Maus F, purified Kranz et al.[96] 1:50
anti-Granzyme B Hase purified abcam 1:100
anti-Perforin Ratte purified abcam 1:100
anti-Tubulin DM1A Maus purified NeoMarkers 1:100
anti-hamster IgG Ziege Biotin ebioscience 1:30
anti-mouse Ig Ziege FITC Southern Biotech 1:100
anti-rabbit IgG Ziege Alexa 488 1:100
anti-rat IgG G28-5 Ziege FITC BD Pharmingen 1:100
Isotyp rIgG2a,κ R35-95 Ratte purified BD Pharmingen
Isotyp hIgG Hamster purified ebioscience
Isotyp mIgG1,κ Maus purified
Isotyp mIgG2a,κ Maus Biotin BD Pharmingen
Isotyp rIgG2a,κ Ratte Biotin ebioscience
Streptavidin - - - Cy5 Caltag 1:50
Streptavidin - - - FITC BD Pharmingen 1:50
2.2. Methoden
2.2.1. Zellkultur / Technik
Alle Arbeiten mit murinen Tumorzelllinien und Primärkulturen sowie alle funktionellen Analysen wurden stets unter sterilen Bedingungen durchgeführt (LaminaAir HB2448, Heraeus).
Die Inkubation von Tumorzelllinien, Primärkulturen und funktionellen Analysen erfolgte in einem Brutschrank (Heraeus 6000) bei einer Luftfeuchtigkeit von 95%, einem CO2- Gehalt von 7,5% sowie bei einer konstanten Temperatur von 37°C.
2.2.1.1. Zentrifugation
Soweit nicht anders angegeben, wurden alle Zentrifugationsschritte sowohl für Tumorzel- len als auch für murine Primärzellen bei 4°C für fünf Minuten bei 330×g durchgeführt.
2.2.1.2. Ermittlung der Lebendzellzahl mittels Trypanblau-Färbung
Zellsuspensionen wurden 1:1 mit einer Trypanblaulösung (Trypan Blue Stain 0,4%, Gib- co, Invitrogen) verdünnt und in einer Neubauer-Zählkammer mit Hilfe eines Mikroskops (Leitz DMRB, Leica) ausgezählt. Der Farbstoff Trypanblau wird ausschließlich von Zel- len mit fehlender Membranintegrität aufgenommen. Daher erscheinen tote Zellen blau, lebende Zellen hingegen ungefärbt. Berechnet wird die Zellzahl nach folgender Formel:
Anzahl lebender Zellen ×Verdünnungsfaktor × 0,01
Anzahl der Großquadrate = Zellzahl×106/ml (2.1)
2.2.2. Zellkultur
2.2.2.1. Splitten adhärenter und nicht-adhärent wachsender Tumorzelllinien Alle Tumorzelllinien wurden ca. zweimal pro Woche bei einer Konfluenz von etwa 90%
abgeerntet. Dazu wurde zunächst das Medium der adhärent wachsenden Kulturen ab- genommen und der Zellrasen wurde einmal mit PBS gespült. Das Ablösen der Tumor- zellen erfolgte durch eine ca. dreiminütige Inkubation bei RT mit 3ml 1× = 10% Tryp- sin/EDTA (10fach, PAN Biotech) in PBS. Die Suspension aus abgelösten Zellen wur- de mit 10ml Medium verdünnt und abzentrifugiert. Eine neue Zellkulturflasche wurde schließlich – je nach Bedarf und Zelllinie – mit 1/5 bis 1/20 der abgeernteten Zellen angeimpft.
Nicht-adhärent wachsende Kulturen wurden zweimal pro Woche abzentrifugiert und mit Medium gewaschen. Die neue Kultur wurde ebenfalls – je nach Bedarf und Zelllinie – mit 1/5 bis 1/20 der abgeernteten Zellen angeimpft.
2.2.2.2. Mycoplasmentest
In Regensburg erfolgte in regelmäßigen Abständen die Überprüfung der verwendeten Tumorzelllinien auf Mycoplasmen mit Hilfe des VenorGEM-Tests (Minerva Biolabs). In Amsterdam wurden die Zellkulturen mittels des MTC-NI-Schnellnachweissystems (My- coplasma Tissue Culture NI, Gen-Probe) getestet.
2.2.2.3. Auftauen und Einfrieren von Zelllinien
Zellaliquots (1–5×106), die in flüssigem Stickstoff eingefroren waren, wurden bei Raum- temperatur aufgetaut und unverzüglich in 10ml Medium überführt. Nach Zentrifugation und einem Waschschritt mit Medium wurden die Zellen in eine Zellkulturflasche mit Medium überführt. Aliquots neu aufgetauter Tumorzellen wurden nach zwei bis drei Passagen erneut eingefroren. Dazu wurden nach dem Abernten Zellaliquots mit 1–5×106 Zellen in 1ml Einfriermedium zunächst bei –80°C eingefroren und nach zwei Tagen zur längeren Lagerung in flüssigem Stickstoff eingelagert.
2.2.3. Isolierung muriner Zellen
2.2.3.1. Splenektomie aus Mäusen
Um T-Zellen aus Mäusen zu isolieren, wurden die Tiere zuerst mittels CO2-Narkose getötet und in eine sterile Werkbank transferiert. Dort wurden die Tiere mit 70% Ethanol gewaschen. Mit einem ersten Set, bestehend aus Pinzette und Schere, wurde die Haut samt Fell in Höhe des linken Rippenbogens angehoben und aufgeschnitten. Das darunter liegende Bauchfell wurde mit einem zweiten, mit Ethanol gereinigten Set eröffnet und die Milz entnommen. Diese wurde in ein mit gekühltem Medium gefülltes 15ml Röhrchen überführt.
2.2.3.2. Gewinnung naiver T-Zellen aus Milzen
Für die Isolierung von T-Zellen aus Milz und Lymphknoten wurden die entnomme- nen Organe zunächst steril über ein Zellsieb (Cell Strainer 100µm, BD Falcon) in ei- ner Petrischale (Easy Grip Petri Dish, Falcon) zerrieben, um eine Einzelzellsuspension
herzustellen. Alternativ wurde das Zellsieb direkt auf ein 50ml-Röhrchen (BD Falcon) aufgesetzt, so dass die Zellsuspension direkt in den Behälter tropfte. Die Suspension wurde erneut über ein Zellsieb pipettiert, um Gewebeteile zu entfernen. Nach einem Waschschritt wurden die Splenozyten in dem jeweils benötigten Puffer zur Zellaufreini- gung oder Stimulation aufgenommen. Sowohl naive polyklonale als auch monoklonale T-Zellen wurden mit Hilfe der negativen Zellseparation isoliert. Im Rahmen dieser Arbeit kamen zwei verschiedene Separationssysteme zur negativen Zellisolierung zum Einsatz, das Dynal-System und das SpinSep-System.
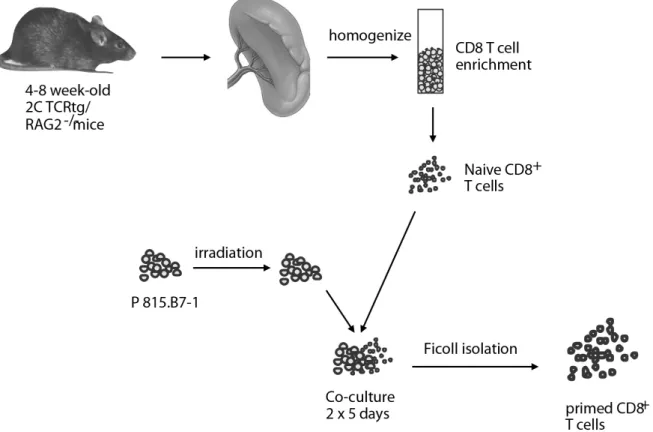
Abbildung 2.1.: Gewinnung und Aufbereitung von murinen T-Zellen: Nach Tötung durch CO2-Narkose und Entnahme der Milz wurden T-Zellen mit Hilfe von Isolierungskits gewonnen, auf- gereinigt, die CD8+ Konzentration durchflusszytometrisch bestimmt und anschließend durch Kokultivierung mit bestrahlten Zieltumorzellen zur Proliferation gebracht.
2.2.3.3. Dynal-Kit: Magnetische Zellseparation
Die Separationsmethode von Dynal (Dynal Mouse CD8+ Negative Isolation Kit, Invi- trogen Dynal) beruht auf der Kopplung magnetischer Polystyrol-Beads mit Zellen. Ein Cocktail aus verschiedenen Ratte-Antikörpern markiert zunächst alle Zellen, die entfernt werden sollen. Die Beads, die mit polyklonalen sheep-anti-rat IgG Antikörpern beschich-