Regulation von PD-L1 und dessen zellintrinsische Funktion für die Vermittlung von Radioresistenz im Kopf-Hals-Tumor
212
0
0
Volltext
(2)(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
Abbildung
+7
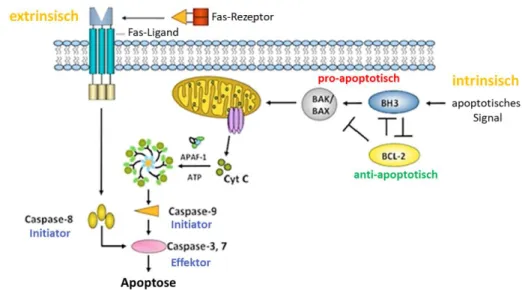

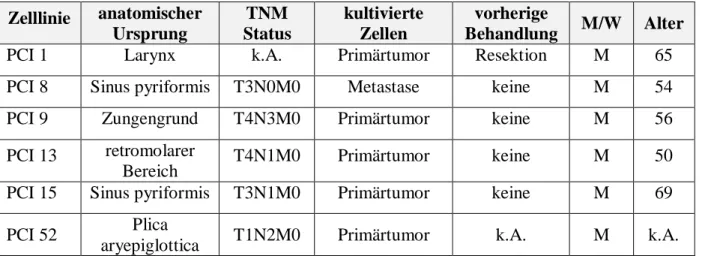
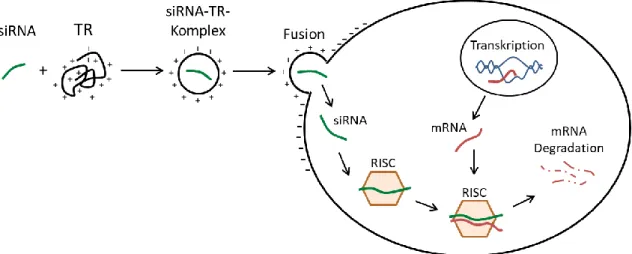
ÄHNLICHE DOKUMENTE
Das Kopf-Hals-Tu- morzentrum am Klinikum Bremen-Mitte bietet als Zusammenschluss der Kliniken für Hals-Nasen-Ohren-Heilkunde sowie Mund-, Kiefer- und Gesichtschirurgie ei-
„Fachlichen Anforderungen an Kopf-Hals-Tumor-Zentren“ definierten Qualitätskriterien erfüllt. Das Kopf-Hals-Tumor-Zentrum Bremen-Mitte erhält daher die
Die Angabe des Medians für Zähler und Nenner bezieht sich nicht auf ein bestehendes Zentrum, sondern gibt den Median aller Zähler der Kohorte und den Median aller Nenner der
Während bei fortgeschrittenen Tumoren häufig eine kombinierte Therapie aus operativer Resektion mit anschließender adjuvanter Radio- oder Radiochemotherapie (RCT) erfolgt und
Auf einen förmlichen Kaufvertrag wurde in beiderseiti- gem Einverständnis verzichtet, AF wies Svenja aber darauf hin, dass der Kaufvertrag nur unter der Bedingung gelten solle,
Für die Chirurgie des Mittelohres und der seitlichen Schädelbasis stehen alle verfügbaren Techniken der Mittelohr- und Schädelbasischirurgie zur Verfügung. Die überwiegende
Patienten mit einer Läsion un- ter 4 cm im größten Durchmesser, die mit einem einzigen Bestrahlungsfeld be- handelt werden konnten, wiesen eine hö- here Remissionsrate auf
Was man nicht sieht Wie Kopf- schmerzen, Gleichgewichtsprobleme und die anderen vielfältigen Phäno- mene genau zustande kommen, ist nicht geklärt und wird häufig als di-
