Quantenchemische Untersuchungen zur
Z/E-Photoisomerisierung von
Stilben und Azobenzol
Diplomarbeit
vorgelegt von Kathleen Gollnisch
aus Malchin
Quantenchemische Untersuchungen zur
Z/E-Photoisomerisierung von
Stilben und Azobenzol
von
Kathleen Gollnisch
Diplomarbeit in Chemie angefertigt am
Institut für Theoretische Chemie und Computerchemie vorgelegt der
Mathematisch-Naturwissenschaftlichen Fakultät der Heinrich-Heine-Universität Düsseldorf
Hiermit versichere ich, die vorliegende Arbeit eigenständig und ausschließlich mit Hilfe der im Literaturverzeichnis aufgeführten Hilfsmittel erstellt sowie Zitate kenntlich gemacht zu haben.
Düsseldorf, den 30.03.2010
Referentin: Frau Prof. Dr. Christel M. Marian
Für meine Eltern
Inhaltsverzeichnis
1 Einleitung 13
2 Quantenchemie 21
2.1 Born-Oppenheimer-Näherung . . . 22
2.2 Variationsprinzip . . . 24
2.3 Hartree-Fock . . . 25
2.4 Elektronenkorrelation . . . 26
2.5 DFT . . . 27
2.6 MRCI . . . 28
2.7 DFT/MRCI . . . 30
3 Intramolekulare Relaxationsprozesse 31 4 Theoretische Methoden 33 5 Stilben 35 5.1 Grundzustand trans-Stilben . . . 36
5.1.1 Geometrieoptimierungen . . . 36
5.1.2 Charakterisierung der elektronischen Zustände . . . 37
5.2 S1-Zustand trans-Stilben . . . 40
5.2.1 Geometrieoptimierungen . . . 40
5.2.2 Charakterisierung der elektronischen Zustände . . . 42
5.2.2.1 Einfluss von Rydbergfunktionen . . . 44
5.2.2.2 Einfluss des Lösungsmittels . . . 44
5.2.3 Spektren von trans-Stilben . . . 45
5.2.3.0.1 Absorptions- und Emissionsbanden . . . 45
5.3 Grundzustand cis-Stilben . . . 47
5.3.1 Geometrieoptimierungen . . . 47
5.3.2 Charakterisierung der elektronischen Zustände . . . 48
5.4 S1-Zustand cis-Stilben . . . 50
5.4.1 Geometrieoptimierungen . . . 50
5.4.2 Charakterisierung der elektronischen Zustände . . . 51
5.4.2.1 Einfluss von Rydbergfunktionen . . . 54
5.4.2.2 Einfluss des Lösungsmittels . . . 54
5.4.3 Spektren von cis-Stilben . . . 55
5.4.3.0.1 Absorptions- und Emissionsbanden . . . 55
5.5 Triplett - T1-Minimum . . . 57
5.5.1 Geometrieoptimierungen . . . 57
5.5.2 Charakterisierung der elektronischen Zustände . . . 59
5.5.2.1 Einfluss von Rydbergfunktionen . . . 61
5.5.2.2 Einfluss des Lösungsmittels . . . 61
5.6 Eingeschränkte Minimumsenergiepfade . . . 61
5.6.1 Pfad T1 . . . 61
5.6.2 Pfad S1 . . . 63
5.7 LIP (Linear interpolierter Pfad) - trans-Stilben . . . 65
5.8 trans-Stilben, S1-Zustand, frühes transientes Spektrum . . . 67
5.9 LIP - cis-Stilben . . . 68
5.10 cis-Stilben, S1-Zustand, frühes transientes Spektrum . . . 70
5.11 Frühes transientes Spektrum des cis-Stilbens, T1-Geometrie . . . 71
5.12 Spätes transientes Spektrum, T1-Geometrie . . . 72
5.13 Spätes transientes Spektrum, trans-Stilben, S1-Geometrie . . . 73
5.14 Zusammenfassung . . . 74
6 Azobenzol 75 6.1 Grundzustand trans-Azobenzol . . . 76
6.1.1 Geometrieoptimierungen . . . 76
6.1.2 Charakterisierung der elektronischen Zustände . . . 76
6.2 S1-Zustand trans-Azobenzol . . . 80
6.2.1 Geometrieoptimierungen . . . 80
6.2.2 Charakterisierung der elektronischen Zustände . . . 81
6.3 S2-Zustand trans-Azobenzol . . . 84
6.3.1 Geometrieoptimierungen . . . 84
6.3.2 Charakterisierung der elektronischen Zustände . . . 85
6.4 T1-Zustand trans-Azobenzol . . . 87
6.4.1 Geometrieoptimierungen . . . 87
6.4.2 Charakterisierung der elektronischen Zustände . . . 88
6.5 Grundzustand cis-Azobenzol . . . 91
6.5.1 Geometrieoptimierungen . . . 91
6.5.2 Charakterisierung der elektronischen Zustände . . . 92
6.6 S1-Zustand cis-Azobenzol . . . 95
6.6.1 Geometrieoptimierungen . . . 95
6.6.2 Linear interpolierter Pfad (LIP) S0 → S1 . . . 96
6.7 T1-Zustand cis-Azobenzol . . . 96
6.7.1 Geometrieoptimierungen . . . 96
6.7.2 Charakterisierung der elektronischen Zustände . . . 97
6.8 Vergleich von Stilben und Azobenzol . . . 99
6.8.1 trans-Form . . . 99
6.8.1.1 S0-Geometrie . . . 99
6.8.1.2 S1-Geometrie . . . 100
6.8.1.3 S2-Geometrie . . . 101
6.8.1.4 T1-Geometrie . . . 102
6.8.2 cis-Form . . . 103
6.8.2.1 S0-Geometrie . . . 103
6.8.2.2 S1-Geometrie . . . 104
6.8.2.3 T1-Geometrie . . . 104
6.9 Zusammenfassung . . . 105
7 Zusammenfassung 107
A Daten der Rydberg-Rechnungen 111
B Daten der COSMO-Rechnungen 115
Tabellenverzeichnis 120
Abbildungsverzeichnis 122
Literaturverzeichnis 126
Danksagung 127
Kapitel 1 Einleitung
Stilben und Azobenzol lassen sich auf Grund ihrer besonderen Eigenschaften als sogenannte Photoschalter für photosensitive Materialien nutzen. Unter Einwirkung von Licht durchlaufen sie eine cis-trans-Isomerisierung. Zur Untersuchung der dabei auftretenden, elektronisch angeregten Zustände sind besonders quantenchemische Methoden geeignet.
Die sich aus dem Photoschalter ergebende Fragestellung ist, wie der Mechanismus hinter dieser photochemischen Umwandlung aussieht. Die Aufklärung könnte zu einer gezielten photochemischen Manipulation der Moleküle führen.
Stilben
Stilben ist auf Grund der Einfachheit seiner Struktur das beste Modellsystem zur Beantwortung dieser Frage.
Cis- und trans-Stilben sind beides thermodynamisch stabile Moleküle. Daher können sie spektroskopisch untersucht werden. Die Moleküle wandeln sich wegen einer zu hohen Energiebarriere nicht im Dunkeln, sondern erst unter Lichteinwirkung in Folge der photochemischen cis-trans-Isomerisierung ineinander um.
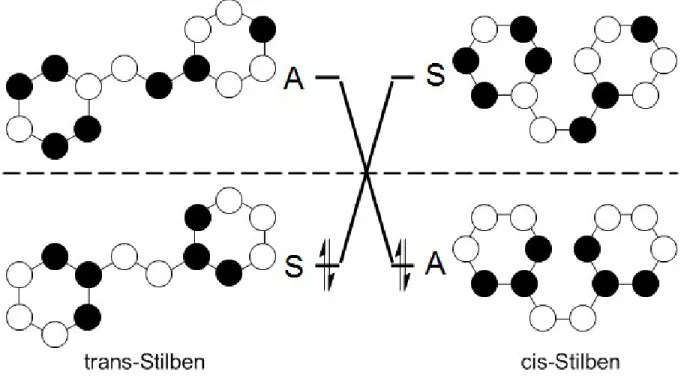
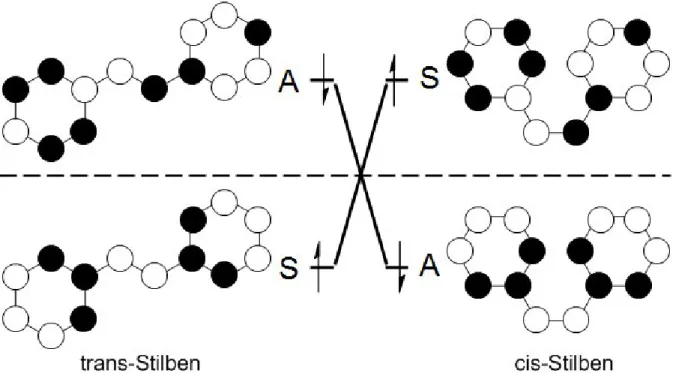
Diese Eigenschaft lässt sich besonders gut anhand eines Orbital- und eines Zustands- korrelationsdiagramms erklären. Es werden nur die Grenzorbitale betrachtet. Das nach den Woodward-Hoffmann-Regeln aufgestellte Orbitalkorrelationsdiagramm zeigt, dass für eine thermische Umwandlung eine große symmetriebedingte Barrriere (Abb. 1.1) existiert. Das zur Aufstellung des Orbitalkorrelationsdiagrammes notwendige Sym- metrieelement ist die C2-Achse und wird in Abb. 1.2 dargestellt. Der Übergang vom trans- zum cis-Stilben ist symmetrieverboten, photochemisch jedoch symmetrieerlaubt (Abb. 1.3). Dies zeigt auch das Zustandskorrelationsdiagramm (Abb. 1.4).
14 KAPITEL 1. EINLEITUNG
Abbildung 1.1: Orbitalkorrelationsdiagramm von Stilben für die symmetrieverbotene Umlagerung im elektronischen Grundzustand
15
Abbildung 1.2: Während der cis-trans-Isomerisierung des Stilbens wird als Symmetrieelement eine C2-Drehachse erhalten. Sie fällt mit der Winkelhalbierenden des Diederwinkels C2-C1-C10-C20 zusammen.
16 KAPITEL 1. EINLEITUNG
Abbildung 1.3: Orbitalkorrelationsdiagramm von Stilben für die symmetrieerlaubte Umwandlung im elektronisch angeregten Zustand
Abbildung 1.4: Zustandskorrelationsdiagramm von Stilben
Die photochemische cis-trans-Isomerisierung ist bereits in der Vergangenheit Gegenstand intensiver Untersuchungen gewesen.
17 Frühere, von Waldeck [1] zusammengefasste Arbeiten zeigen, dass im Wesentlichen zwei Theorien zum Reaktionsverlauf nach elektronischer Anregung diskutiert werden.
Bei tiefen Temperaturen (-196,15◦C) wird sowohl beim cis- als auch beim trans-Stilben im S1-Zustand eine starke Fluoreszenz beobachtet [2, 3].
Bei erhöhten Temperaturen (25◦C) wird die Fluoreszenz gelöscht.
Das unterschiedliche Fluoreszenzverhalten setzt Barrieren auf der S1-Potenzialenergie- hyperfläche voraus. Deren Überwindung ermöglicht eine interne Konversion.
Aus diesen Beobachtungen lässt sich schließen, dass die in der Literatur als wahrscheinlichste dargestellte Theorie eine Drehung um die Ethylenbindung des Stilbens im S1-Zustand, dem ersten angeregten Singulettzustand, zu einer verdrehten Geometrie bei mutmaßlich 90◦ beinhaltet. Zum Singulettgrundzustand besteht voraussichtlich bei einem Winkel von 90◦ eine vermiedene Kreuzung.
Die andere Theorie besagt, dass die Isomerisierung über einen Triplettzustand verläuft.
Diese setzt einen Intersystem-Crossing-Prozess (ISC) gefolgt von einer Isomerisierung entlang der Triplettpotentialfläche voraus.
Beide Annahmen werden mit transienter Absorptions- und Fluoreszenzspektroskopie durch Dr. S. Kovalenko [4] von der Humboldt-Universität zu Berlin experimentell überprüft.
Dazu werden Spektren von cis- und trans-Stilben in Acetonitril und Hexan aufgenommen. Die Spektren beider Moleküle weisen einige Unterschiede auf.
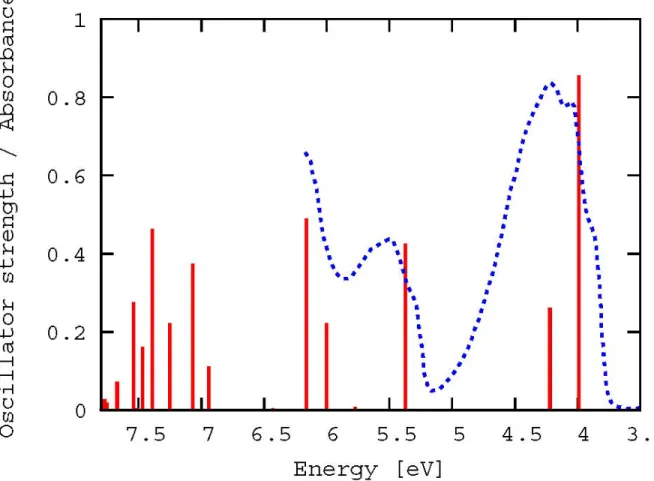
In den sehr frühen (bis 100 fs) und frühen (bis 130 fs) transienten Spektren von cis- Stilben taucht während der Isomerisierung eine Bande auf, die auf einen sogenannten Phantomzustand p∗ (Abb. 1.5) hinweist. Die Bande resultiert aus einer Absorption im angeregten Zustand (ESA - engl.: excited-state absorption) und liegt bei einer Wellenlänge von 340 nm. Sie wächst in dem Maße, wie eine andere ESA-Bande bei einer Wellenlänge von 630 nm verschwindet. Diese gehört zu einem quasistationären Minimum auf der S1-Fläche und wird dem angeregten Zustand c∗ von cis-Stilben zugeordnet (Abb. 1.5). Von diesem geht die Fluoreszenz aus (Abb. 1.5 - grüner Pfeil).
Die Spektren ergeben auch, dass der c∗- und der p∗-Zustand durch eine Barriere voneinander getrennt sind.
18 KAPITEL 1. EINLEITUNG
Abbildung 1.5: Photochemische Isomerisierung von Stilben nach Kovalenko [4]
Die Fluoreszenz des trans-Stilbens entsteht durch den ersten angeregten Zustand t∗ (Abb. 1.5 - hellblauer Pfeil), der ein weiteres lokales Minimum auf der S1-Fläche darstellt. Allerdings sind nach Kovalenkos Modell t∗ und p∗ durch eine höhere Barriere als c∗ und p∗ voneinander getrennt.
Insgesamt werden durch Herrn Kovalenko sowohl im Grundzustand als auch im t∗, c∗ und p∗ quasistationäre Populationen beobachtet. Es werden keine Zwischenstufen erfasst, die einen Weg zwischen den angeregten Zuständen und dem p∗-Zustand aufzeigen würden.
Die Bande bei 340 nm könnte laut Herrn Kovalenko durch eine rechtwinklige Konformation des p∗-Zustands, die zu einer Entkopplung der Phenylringe führen würde, entstehen.
Diese Bande ist nicht im frühen Spektrum des trans-Stilbens vorhanden. Durch die Delokalisation der Elektronen sollte sie keine oder nur eine geringe Oszillatorenstärke haben. Diese würde bei einer Störung der Elektronendelokalisation, wie es beim
19 cis-Stilben auf Grund der Verdrehung der Phenylringe der Fall ist, zunehmen.
Daher ist diese Bande im frühen Spektrum des cis-Stilbens sichtbar. Eine weitere Erklärung für das trans-Stilben wäre wohl, dass dunkle (340 nm) und helle (577 nm) Banden im trans- und cis-Stilben durch die Kopplung von zwei entarteten Zuständen entstehen. Die Zustände sollten benzolähnlich sein und die Kopplung würde über die zentrale Doppelbindung erfolgen. Dadurch könnte auch die abnehmende Absorptionsoszillatorenstärke von cis-Stilben im Vergleich zum trans-Stilben erklärt werden.
Darauf aufbauend werden, neben den Grundzuständen von cis- und trans-Stilben, in diesem Projekt - soweit im zeitlich begrenzten Rahmen einer Diplomarbeit möglich - für die cis-trans-Isomerisierung die photochemischen Reaktionspfade des Singulettzustands S1 sowie des Triplettzustands T1 berechnet. Dabei wird der Winkel für die Einzelpunktrechnungen zwischen cis-Stilben (0◦) und trans-Stilben (180◦) in 10◦-Schritten variiert.
Die quantenchemischen Rechnungen sollen ebenfalls die Theorien für die in den ESA- Spektren vorkommende Bande bei 340 nm für das cis-Stilben erhärten. Dasselbe gilt für die Abwesenheit der Bande im trans-Stilben. Weiterhin könnten die Rechnungen helfen, die anderen auftretenden Banden zuzuordnen.
Azobenzol
Beim Azobenzol liegen ebenfalls zwei stabile cis- und trans-Isomere vor. Azobenzol ist wie Stilben bei höheren Temperaturen (90◦C) nicht fluoreszent [5] und weist zu diesem strukturelle Ähnlichkeiten auf. Daher liegen Vergleiche zur Struktur der elektronisch angeregten Zustände und zur Art des Verlaufs der Potenzialenergiehyperflächen zwischen beiden Molekülen nahe.
Ein besseres Verständnis der bei der Photoschaltung von Azobenzol auftretenden Prozesse würde u.a. Vorhersagen zur Struktur und chemischen Dynamik von Peptiden und Proteinen erleichtern. Die dabei auftretenden lokalen und globalen Änderungen wie eine Proteinfaltung werden unter anderem von Prof. Dr. Karola Rück-Braun [6] an der Technischen Universität Berlin experimentell untersucht.
20 KAPITEL 1. EINLEITUNG
Kapitel 2
Quantenchemie a
Die Quantenchemie untersucht chemische Problemstellungen und befasst sich mit der elektronischen Struktur von Molekülen. Die Grundlage hierfür bildet die Quantenmechanik. Sie beginnt dort, wo die klassische Mechanik versagt und dringt tief in den (sub-)atomaren Bereich vor.
In der Quantenmechanik sind alle Eigenschaften eines Systems in einer Wellenfunktion enthalten. Diese wird durch das Lösen der Schrödingergleichung erhalten.
Die Schrödingergleichung ist der zentrale Bestandteil der nichtrelativistischen Quantenchemie. Es gibt sowohl eine zeitabhängige (Gl. 2.1) als auch eine zeitunabhängige (Gl. 2.2) Gleichung.
Die zeitabhängige Schrödingergleichung
HΨ =ˆ i~∂Ψ
∂t (2.1)
beschreibt die Dynamik, also die zeitliche Änderung eines quantenmechanischen Zustands.
Die zeitunabhängige Schrödingergleichung wird durch einen Separationsansatz erhalten. Voraussetzung hierfür ist die Zeitunabhängigkeit der potentiellen Energie V und damit des Hamiltonoperators. Dabei werden die Variablen für den Ort und die Zeit getrennt.
Die zeitunabhängige Schrödingergleichung ist eine Eigenwertgleichung, wobei Hˆ den Hamiltonperator, Ψdie Wellenfunktion und E die Energie darstellen.
HΨ =ˆ EΨ (2.2)
aGrundlage für dieses Kapitel bilden das Buch von Szabo/Ostlund [7] sowie das Vorlesungsmanusskript zur Quantenchemie II von Prof. C. M. Marian
22 KAPITEL 2. QUANTENCHEMIE
2.1 Born-Oppenheimer-Näherung
Im Allgemeinen treten in der Quantenchemie Vielteilchenprobleme auf. Die betrachteten Moleküle besitzen M Kerne und N Elektronen. Sie werden durch einen nichtrelativistischen, molekularen Hamiltonoperator (Gl. 2.3) beschrieben.
Dieser lautet in atomaren Einheiten:
Hˆ =−
N
X
i=1
1 2∇2i −
M
X
A=1
1
2MA∇2A−
N
X
i=1 M
X
A=1
ZA riA +
N
X
i=1 N
X
j>i
1 rij +
M
X
A=1 M
X
B>A
ZAZB
RAB (2.3) mit
MA: Quotient aus der Masse des Kerns A zu der Masse eines Elektrons ZA: Atomnummer des Kerns
∇i2/∇A2: Ableitungen in Bezug auf die Koordinaten des i-ten Elektrons und des A-ten Kerns
Der erste und der zweite Term beschreiben die Operatoren für die kinetische Energie der Elektronen sowie die der Kerne, der dritte den für die Coulombanziehung zwischen den Elektronen und den Kernen, der vierte und fünfte die für die Abstoßung zwischen den Elektronen sowie den Kernen.
Da sowohl die kinetische Energie der Elektronen als auch die der Kerne berücksichtigt werden, erschwert dies die Lösung der Schrödingergleichung deutlich.
Es wird versucht, dieses Problem mit der Born-Oppenheimer-Näherung zu lösen. In dieser werden die räumlichen Positionen der Kerne in einem Molekül festgehalten, wohingegen sich die Elektronen frei im elektrostatischen Feld der Kerne bewegen können. Dies ist auf Grund des Masse- und damit Geschwindigkeitsunterschiedes zwischen beiden möglich, da die Elektronen sehr viel leichter sind als die Kerne.
Im molekularen Hamiltonoperator können daher näherungsweise der Term für die Bewegung der Kerne vernachlässigt und der für die Abstoßung zwischen den Kernen konstant gehalten werden. Daraus resultiert der elektronische Hamiltonoperator (Gl.
2.4).
Hˆelec =−
N
X
i=1
1 2∇2i −
N
X
i=1 M
X
A=1
ZA riA +
N
X
i=1 N
X
j>i
1
rij (2.4)
Für festgehaltene Kerne gilt
Hˆ0 = ˆHelec+const. (2.5)
2.1. BORN-OPPENHEIMER-NÄHERUNG 23 Für die Lösung der molekularen Schrödingergleichung werden die Kern- und Elektronenbewegung durch einen Produktansatz (Gl. 2.6) separiert.
(−
M
X
A=1
1
2MA∇2A+ ˆHmol−Emol)
∞
X
n
χn(R)~ ·ϕn(~r; ¯R) = 0 (2.6) Die Bewegung der Elektronen hängt sowohl von deren Koordinaten als auch parametrisch von den Kernkoordinaten ab. Die Multiplikation mit ϕ∗k(~r; R) und¯ Integration über r ergibt als Lösung drei Terme. Der erste lautet
Z
ϕ∗k(~r; ¯R)(−
M
X
A=1
1 2MA
∇2A+ ˆHmol−Emol)
∞
X
n
χn(R)~ ·ϕn(~r; ¯R)d~r = (2.7)
−
M
X
A=1
1
2MA∇2Aχn(R)~ (2.8)
−
M
X
A=1
1 MA
∞
X
n
Z
(ϕ∗k(~r; ¯R)∇Aϕn(~r; ¯R)d~r)∇Aχn(R)~ (2.9)
−
M
X
A=1
1 2MA
∞
X
n
Z
ϕ∗k(~r; ¯R)χn(R)∇~ 2Aϕn(~r; ¯R) (2.10) Dabei werden die Integrale von Gl. 2.9und Gl. 2.10 mit Akn und Bkn bezeichnet. Akn beschreibt Außerdiagonalterme, Bkn Diagonal- und Außerdiagonalterme.
Der zweite Term der Lösung gestaltet sich folgendermaßen:
X
n
( Z
ϕ∗k(~r; ¯R) ˆHelec0 ϕn(~r; ¯R)d~r) = Ek,elec0 ( ¯R)χk(R)~ (2.11) Der dritte und letzte Term beschreibt die Anwendung von ϕ∗k auf Emol
X
n
( Z
ϕ∗k(~r; ¯R)Emolϕn(~r; ¯R)d~r) = Emolχk(R)~ (2.12) Insgesamt ergibt sich somit
(−
M
X
A=1
1 2MA
∇2A+Ek,elec0 ( ¯R)−Emol)χk(R)−~ X
n M
X
A=1
1 2MA
(2Akn+Bkn)χn(R) = 0~ (2.13) Der erste Term stellt die Schrödingergleichung für die Kernbewegung dar, der zweite die Kopplungsterme.
24 KAPITEL 2. QUANTENCHEMIE
Uk( ¯R) =Ek,elec0 ( ¯R)−
M
X
A=1
1
2MABkk (2.14)
Das Ergebnis wird inGl. 2.13 eingesetzt. Der erste Term beschreibt die Kernbewegung unter der Wirkung des Potentials Uk, der zweite die Kopplung der Kernbewegung in verschiedenen Elektronenzuständen.
(−
M
X
A=1
1
2MA∇2A+Uk( ¯R)−Emol)χk(R)~ −X
n6=k M
X
A=1
1
2MA(2Akn+Bkn)χn(R) = 0~ (2.15) In der Born-Oppenheimer-Näherung werden alle Diagonalterme Bkk und Außerdiagonalterme Akn und Bkn gleich null gesetzt. Übrig bleibt Gl. 2.16.
(−
M
X
A=1
1
2MA∇2A+Ek,elec0 ( ¯R)−Emol)χk(R) = 0~ (2.16) Die Born-Oppenheimer-Näherung wird im Allgemeinen gut erfüllt. Die elektronische Wellenfunktion ändert sich nur langsam mit der Kerngeometrie. Die Kerne selbst bewegen sich im Potential der Elektronen. Die Auftragung der Energien in Abhängigkeit von den Kernkoordinaten führt zum Konzept der Potenzialhyperflächen.
Diese können sich kreuzen oder vermiedene Kreuzungen ausbilden. In diesen Bereichen bricht die Born-Oppenheimer-Näherung komplett zusammen, da die Kopplung zwischen der Bewegung von Kernen und Elektronen zu stark wird. Die Wellenfunktionen verändern sich sehr deutlich mit R, so dass die erste und zweite¯ Ableitung nicht mehr vernachlässigbar sind.
2.2 Variationsprinzip
Das Variationsprinzip [8] besagt, dass die Berechnung des Erwartungswertes mit einer willkürlichen Wellenfunktion Φ0, die ungleich Ψ0 ist, einen größeren Wert als E0 (Gl.
2.17) ergibt. Dabei sind E0 die exakte Energie, Ψ0 die exakte Wellenfunktion eines Systems im Grundzustand und E0 der Erwartungswert der Energie für jede andere Funktion.
E0 = hΦ0|H|Φˆ 0i
hΦ0|Φ0i ≥ E0 (2.17)
Die exakte Energie E0 ist also eine untere Grenze für beliebige Testwellenfunktionen.
Je niedriger demnach die berechnete Energie ist, desto besser ist die zugehörige Wellenfunktion.
2.3. HARTREE-FOCK 25
2.3 Hartree-Fock
Das Hartree-Fock-Verfahren (HF) ist eine „ab initio“ Methode. Es ist ein Näherungsverfahren zur Lösung der elektronischen Schrödingergleichung eines Vielteilchensystems. Dafür wird ein gemitteltes Feld für die Elektron-Elektron- Abstoßung angenommen.
Die einfachste Wellenfunktion wird durch eine Slaterdeterminante |Ψ0i für ein N- Elektronen-System beschrieben (Gl. 2.18). Diese gehorcht dem Antisymmetrieprinzip.
|Ψ0i=|χ1χ2. . . χNi (2.18) Über das Variationsprinzip wird die Wellenfunktion mit der kleinsten Energie gesucht (Gl. 2.19). Dafür werden die Spinorbitale in einer Orthonormalbasis verändert.
E0 =hΨ0|H|Ψˆ 0i (2.19)
H ist hierbei der elektronische Hamiltonoperator, E0 der Erwartungswert der elektronischen Energie.
Durch die Antisymmetrisierung der Wellenfunktion gibt es neben der Coulombwechselwirkung noch eine Austauschwechselwirkung. Diese entsteht durch die Korrelation von Elektronen mit gleichem Spin und verringert die Coulombabstoßung. Dadurch haben Singulett- und Triplettzustände unterschiedliche Energien. In der Hartree-Fock-Näherung wird die Korrelation von Elektronen, die einen entgegengesetzten Spin besitzen, nicht berüksichtigt.
E0 =X
a
haa+ 1 2
X
ab
(Jab−Kab) (2.20)
haa =hχa(1)| − 1 2
∇~21−X
A
Za
r1A|χa(1)i (2.21) Jab =hχa(1)χb(2)| 1
r12|χa(1)χb(2)i (2.22) Kab=hχa(1)χb(2)| 1
r12|χb(1)χa(2)|i (2.23) Der Erwartungswert setzt sich also aus einem Einelektronenterm haa und zwei Zweielektronentermen Jab und Kab zusammen. In Kab sind von Teilchen 1 und 2 die letzten beiden Orbitalfunktionen vertauscht.
Die Minimierung des Erwartungswertes E0 führt zur Hartree-Fock-Gleichung. Sie ist eine Pseudoeigenwertgleichung, da die Operatoren von den Lösungen abhängen. Sie
26 KAPITEL 2. QUANTENCHEMIE eine kanonische HF-Gleichung umgewandelt. Allerdings ist die HF-Energie ungleich dem Erwartungswert, da in der Summe der Orbitalenergien die Elektron-Elektron- Wechselwirkung doppelt gezählt wird.
faχa=aχa (2.24)
fa=ha+
N
X
b
(Jb−Kb) (2.25)
fa ist der sogenannte Fock-Operator. Dieser ist ein effektiver Einelektronenoperator.
Die HF-Gleichung ist eine gekoppelte Integrodifferentialgleichung, die iterativ gelöst werden muss. Dafür wird die Methode des selbstkonsistenten Feldes (SCF - engl.:
self-consistent field) verwendet. Es wird mit einem willkürlichen Wert gestartet und die Gleichung bis zur Selbstkonsistenz gelöst. Mit einem größer werdenden Basissatz wird die Energie in Richtung des HF-Grenzwertes erniedrigt. Allerdings wird diese nicht erreicht.
2.4 Elektronenkorrelation
Die Elektronenkorrelation stellt die Abweichung der Wechselwirkung zwischen Elektronen vom mittleren Potenzial dar. Dabei wird zwischen der statischen und der dynamischen unterschieden. Die statische beschreibt langreichweitige, die dynamische hingegen kurzreichweitige Korrelationen. Für den statischen Teil werden mehrere, energetisch vergleichbare Determinanten gebraucht, die z.B. durch ein Multireferenzwechselwirkungsverfahren (MRCI - Kap. 2.6) beschrieben werden können. Die Dichtefunktionaltheorie (DFT - Kap. 2.5) ist hierfür nicht geeignet.
Beim dynamischen Teil nimmt die Stärke der Korrelation mit zunehmender Nähe der Elektronen zu. Von der DFT werden im Gegensatz zum MRCI große Teile der dynamischen Elektronenkorrelation erfasst.
Die Energie der Elektronenkorrelation Ecorr berechnet sich zu
Ecorr =E0−EHF (2.26)
Sie ist die Differenz zwischen der Hartree-Fock-Energie EHF und der exakten, nichtrelativistischen Energie E0 in einer gegebenen Basis. Sie ist immer negativ, da die HF-Energie wegen des Variationsprinzips eine obere Grenze zur exakten Energie darstellt. Letztere lässt sich über eine volle Konfigurationswechselwirkungsrechnung (full-CI) bestimmen.
Der Fehler von Ecorr liegt im Bereich von einem Prozent. Das entspricht in etwa der Größenordnung einer chemischen Bindung.
2.5. DFT 27
2.5 DFT
Für die Minimierung der Gesamtenergie wird in der Dichtefunktionaltheorie [9]
anstelle der Wellenfunktion die Elektronendichte (Gl. 2.27) zur Lösung des Vielteilchenproblems genutzt. Dadurch wird die Lösung der Schrödingergleichung vermieden.
Die Dichtefunktionaltheorie geht auf das Theorem von Hohenberg und Kohn [10]
zurück. Demnach ist das exakte Dichtefunktional gleich der Grundzustandsenergie, für jedes andere Funktional hingegen ist die Energie größer. Es gehorcht somit dem Variationsprinzip.
E0[ρ0]≥E0[ρ] (2.27)
Das exakte Dichtefunktional ist allerdings nicht bekannt. Daher wird das Funktional im Rahmen der Born-Oppenheimer-Näherung aufgestellt.
EDF T[ρ] =ET[ρ] +EV[ρ] +EJ[ρ] +EX[ρ] +EC[ρ] (2.28) Es setzt sich aus den Funktionalen für die kinetische Energie ET[ρ]der Elektronen, die Coulomb-Wechselwirkung zwischen den Kernen und den Elektronen EV[ρ], die Coulombabstoßung der Elektronen J[ρ]sowie den Austausch Ex[ρ]und die Korrelation EC[ρ]zusammen.
Die Coulomb-Terme werden durch klassische Ausdrücke formuliert. Die anderen basieren auf dem Modell des homogenen Elektronengases, bei dem zwischen den Elektronen keine Wechselwirkung besteht. Da durch das nicht wechselwirkende System die korrespondierende Wellenfunktion als einzelne Slaterdeterminante betrachtet werden kann, ist eine exakte Berechnung möglich.
Zur Berechnung der Elektronendichte
ρ(r) =
n
X
i=1
|φ(r)|2 (2.29) werden nach Kohn und Sham [11] für die Konstruktion der Dichte Orbitale eingeführt.
Da jedoch in dieser Theorie die Elektronen wechselwirken, werden Korrekturterme mit einbezogen. Diese entsprechen der Elektronenkorrelation und werden mit dem Austauschterm zum sogenannten Austausch-Korrelationsfunktional zusammengefasst.
Änderungen in der Dichte des Elektronengases werden zunächst nicht berücksichtigt.
Die einfachste Näherung ist die lokale Dichtenäherung (LDA - engl.: local density approximation). Sie beruht auf der Grundzustandsenergie des homogenen Elektronengases. Für das inhomogene Elektronengas wird lokal die gleiche Energie wie für das homogene angenommen. Örtliche Dichteschwankungen werden mit Hilfe
28 KAPITEL 2. QUANTENCHEMIE approximation) berechnet. Eine noch bessere Genauigkeit wird durch die Verwendung von Hybridfunktionalen wie B3-LYP [12, 13] und BH-LYP [13, 14] erreicht. Diese setzen sich aus dem exakten Austauschfunktional der Hartree-Fock-Methode und den Dichtefunktionalen zusammen.
2.6 MRCI
MRCI steht für Multireferenzkonfigurationswechselwirkung (engl.: multireference configuration interaction).
Die Konfigurationswechselwirkung (CI) ist konzeptionell eine einfache Methode zur Berechnung der exakten Wellenfunktion und damit der Energie eines Vielteilchenproblems. Die gesamte CI-Wellenfunktion (Gl. 2.30) besteht aus einer Linearkombination von N-Elektron-Funktionen (Konfigurationszustandsfunktionen), deren Koeffizienten variationell bestimmt werden.
|Φ0i=c0|Ψ0i+X
ar
cra|Ψrai+X
a<b r<s
crsab|Ψrsabi+ X
a<b<c r<s<t
crstabc|Ψrstabci+· · · (2.30)
Dabei wird die Determinante mit der tiefsten Energie |Ψ0i durch eine Hartree-Fock- Rechnung bestimmt. Die anderen Determinanten beschreiben angeregte Zustände.
Dabei bedeutet |Ψrai, dass eine Einfachanregung aus dem besetzten Spinorbital χa in das unbesetzte Spinorbital χb auftritt/stattfindet. Der letzte Beitrag zur exakten Determinante stellt die Anregung aller Elektronen aus den besetzten Spinorbitalen in alle unbesetzten Spinorbitale dar. Die Koeffizenten c0 und so weiter sind die Koeffizienten der Eigenvektoren. Die zugehörigen Eigenwerte entsprechen den Energien der Zustände im CI-Ansatz. Die Eigenwerte werden durch die Diagonalisierung der HamiltonmatrixHij =hΨi|H|Ψˆ ji erhalten.
Allerdings ist die Anzahl der möglichen Determinanten für die Rechnungen meist zu groß, sodass die CI-Entwicklung in der Regel abgebrochen werden muss.
MRCI ist eine CI-Entwicklung, bei der mehrere Vielelektronenkonfigurationen als Referenzen anstelle von einzelnen Hartree-Fock-Referenzen genutzt werden.
Die endgültige Entwicklung ist häufig eine Linearkombination, die aus Einfach- und Doppelanregungsdeterminanten (MRSDCI - SD steht für „Singles Doubles“) besteht. Die verschiedenen Konfigurationen werden durch die Anregung aus internen Raumorbitalen erzeugt (Abb. 2.1).
2.6. MRCI 29
Abbildung 2.1: Aufteilung der Orbitalbasis einer MRCI-Entwicklung
Die MRCI-Methode ist sehr genau, sofern alle wichtigen Konfigurationen mit in die Entwicklung einbezogen werden. Dies kann für kleine Systeme erfüllt werden. Bei zunehmender Größe wird die Entwicklung unerschwinglich und muss abgebrochen werden. Dies hat allerdings eine mangelnde Größenkonsistenz zur Folge. Das bedeutet, dass die Energie eines Systems von n nicht miteinander wechselwirkenden Teilchen nicht gleich der n-fachen Energie eines einzelnen Teilsystems ist. Die Konfigurationen können selektiv nach verschiedenen Kriterien abgeschnitten werden.
Dies kann nicht bei der Betrachtung angeregter Zustände über einen Geometriebereich
30 KAPITEL 2. QUANTENCHEMIE
2.7 DFT/MRCI
Die Kombination aus den Methoden DFT und MRCI [15] vereint die Vorteile von beiden. Dies betrifft insbesondere die Elektronenkorrelation, da bei der Berechnung der Grundzustände und der angeregten Zustände sowohl die statische (MRCI) als auch die dynamische (DFT) Elektronenkorrelation benötigt werden.
Diese Methode zielt insbesondere auf Multireferenzzustände ab, die mit dem MRCI-Verfahren abgedeckt werden. Für die Entwicklung der CI-Matrixelemente werden aus Kohn-Sham-Orbitalen aufgebaute spin- und raumsymmetrisierte Konfigurationszustandsfunktionen verwendet. Außerdiagonalelemente der Matrix werden durch eine energieabhängige Skalierung exponentiell auf Null gedämpft.
Energetisch höher liegende Zustandskonfigurationen werden dadurch abgeschwächt.
Somit wird eine Doppeltzählung der Elektronenkorrelation verhindert.
Die fünf dafür verwendeten, semiempirischen Parameter für Singulett- und Triplettzustände werden durch eine Anpassung an experimentelle Anregungsenergien erhalten. Als Hybridaustauschkorrelationsfunktional wird das BH-LYP-Funktional (Becke’s „half-and-half“) benutzt.
Es können sehr große Systeme (mehrere hundert Elektronen) behandelt werden, deren Ergebnisse bei gleichzeitig geringen Kosten sehr genau sind. Der relative Fehler für die berechneten Energien liegt unter 0,2 eV.
Kapitel 3
Intramolekulare Relaxationsprozesse
Das dynamische Verhalten von Atomen oder Molekülen nach einer elektronischen Anregung kann sehr komplex sein. Es reicht von intramolekularen Relaxationsprozessen über photochemische Reaktionen bis hin zum photochemischen Zerfall.
Intramolekulare Relaxationsprozesse sind Prozesse, bei denen energetisch angeregte Atome oder Moleküle in ihren Grundzustand zurückkehren. Dies erfolgt entweder strahlend oder strahlungslos. Zu den strahlenden Prozessen gehören die Fluoreszenz und die Phosphoreszenz.
Fluoreszenz ist eine kurzlebige, spontane Emission von Licht. Die Fluoreszenzlebensdauer liegt typischerweise im Bereich von Nanosekunden. Die Emission eines Photons erfolgt hierbei aus einem angeregten Singulettzustand.
Phosphoreszenz ist eine zeitlich stark verzögerte, spontane Emission von Licht. Die Verzögerung kann mehrere Stunden betragen, da die Emission eines Photons hierbei aus einem angeregten Triplettzustand erfolgt. Die Zeitverzögerung entsteht durch eine Spinumkehrung des Elektrons, die während eines sogenannten Interkombinationsprozesses (ISC - engl.: intersystem crossing) erfolgt.
Der ISC-Prozess ist ein strahlungsloser Übergang zwischen zwei Zuständen mit unterschiedlicher Multiplizität. Er ist nur durch eine Spin-Bahn-Wechselwirkung möglich. Bei Zuständen gleicher Multiplizität findet ein anderer strahlungslose Prozess statt. Dies ist die interne Konversion (IC - engl.: internal conversion). Sie wird durch eine vibronische, also nichtadiabatische Kopplung bewirkt.
Bei beiden sind die Übergänge der Elektronen schneller, wenn sich die Potenzialflächen kreuzen.
Vor der Emission eines Photons wird durch die Wechselwirkung der Teilchen mit der Umgebung auch überschüssige, strahlungslose Schwingungsenergie abgegeben. Der Prozess wird Schwingungsrelaxation (VR - engl.: Vibrational Relaxation) genannt.
Eine weitere wichtige Form der strahlungslosen Prozesse ist die intramolekulare Schwingungsenergieumverteilung (IVR - engl.: Internal Vibrational Redistribution).
Dabei wird Schwingungsenergie, die anfänglich in einer Mode lokalisiert ist, auf andere Schwingungsfreiheitsgrade des Moleküls umverteilt. Voraussetzung ist allerdings derselbe elektronische Zustand. Je mehr Schwingungsfreiheitsgrade vorhanden sind, desto effektiver funktioniert die Umverteilung.
Alle Prozesse werden in einem Jablonski-Diagramm zusammengefasst.
32 KAPITEL 3. INTRAMOLEKULARE RELAXATIONSPROZESSE
Abbildung 3.1: Jablonski-Diagramm: Schematische Darstellung der Potentialkurven des Grundzustands S0, des ersten angeregten Singulettzustands S1 und des ersten angeregten Triplettzustands T1. A: Absorption, F: Fluoreszenz, P: Phosphoreszenz, VR:
Vibrational Relaxation, IC: Internal Conversion, ISC: Intersystem Crossing (Quelle:
C. M. Marian)
Kapitel 4
Theoretische Methoden
Die Berechnungen der Gleichgewichtsgeometrien von trans- und cis-Stilben sowie trans- und cis-Azobenzol werden mit dem Programm TURBOMOLE [16] durchgeführt.
Aus dessen Bibliothek werden sowohl das B3-LYP-Funktional als auch der Basissatz TZVPP benutzt.
TZVPP ist ein triple zeta Valenzbasissatz. Für die Valenzschale werden dreimal so viele Gaußfunktionen wie für einen minimalen Basissatz sowie zwei Sätze an Polarisationsfunktionen für alle Atome verwendet.
Die Geometrien der Singulettgrundzustände S0 werden mit der Dichtefunktionaltheorie (DFT), die angeregten Singulettzustände Sn mit der zeitabhängigen Dichtefunktionaltheorie [17] (TDDFT - engl.: time-dependent density functional theory) und die Triplettzustände mit der uneingeschränkten Dichtefunktionaltheorie (UDFT) optimiert. An diesen stationären Punkten werden Schwingungsfrequenzen ausgerechnet, um zu bestimmen, ob es sich um Minima handelt. Für die Singulettgrundzustände und die Triplettzustände werden diese mit dem AOFORCE-Modul [18] des TURBOMOLE-Pakets analytisch bestimmt. Die Schwingungsfrequenzen der angeregten Singulettzuände werden numerisch mit dem SNF-Programm [19] des TURBOMOLE’s berechnet.
Mit Hilfe der Programme TURBOMOLE und MRCI werden die Anregungsenergien der optimierten Geometrien bestimmt.
Die Berechnung der Singulett- und Triplettzustände erfolgt anhand eines auf einem Dichtefunktional basierendem Multireferenz-Konfigurationswechselwirkungs-Verfahren (DFT/MRCI - s. Abschnitt 2.7). Die Basis für die MRCI-Entwicklung bilden Kohn- Sham-BH-LYP-Orbitale, welche mit TURBOMOLE erzeugt werden.
Die Berechnung spektroskopischer Daten wie der Oszillatorenstärke werden mit dem Programm PROPER [20] gemacht. Dafür werden die Dichtematrizen aus der DFT/MRCI-Wellenfunktion gebraucht.
Bei den eingeschränkten Geometrioptimierungen für das Stilben wird EF.X [21, 22] als externer Treiber für das TURBOMOLE verwendet. Dies sind Geometrieoptimierungen mit einer festgehaltenen internen Koordinate, in diesem Fall θ (englisch: CMEP -
34 KAPITEL 4. THEORETISCHE METHODEN constrained minimum energy path). Die Berechnungen der Gleichgewichtsgeometrien erfolgen ebenfalls mit dem B3-LYP-Funktional.
Bei den Berechnungen für den CMEP-S1 wird auf den Treiber EF.X verzichtet.
Stattdessen werden im TURBOMOLE die kartesischen Koordinaten in interne Koordinaten umgewandelt. Dies hat den Vorteil, dass die Symmetrie nicht auf eine C1-Symmetrie beschränkt ist. Anschließend werden wieder kartesische Koordinaten erzeugt.
Die beschränkten Geometrien der Singulettgrundzustände S0 werden ebenfalls mit DFT, die der angeregten Singulettzustände Sn mit TDDFT und die der Triplettzustände mit UDFT berechnet.
Die Geometrieoptimierungen des Stilbens in einer Lösemittelumgebung werden mit COSMO [23, 24] simuliert. Das Lösmittel ist hierbei Acetonitril. Die Dielektrizitätskonstante wird auf 36 [25] festgesetzt. Die Geometrien der Singulettgrundzustände S0 werden auch hier mit DFT und die Triplettzustände mit UDFT optimiert, wobei der Basissatz TZVPP und das Funktional erneut B3-LYP sind.
Weiterhin werden im Stilben Rydbergzustände mit dem TZVPP+Rydberg-Basissatz berechnet. Hierfür ist das Funktional ebenfalls B3-LYP. Bei der Berechnung der Rydbergzustände werden jedem gewünschten Zentrum zusätzliche Funktionen angefügt. Die Exponenten [26] werden in der folgenden Tabelle aufgelistet.
Tabelle 4.1: Rydbergexponenten Orbital Atom C Atom N
3s 0,023 0,0280
3p 0,021 0,0250
3d 0,015 0,0150
4s 0,0055 0,0066
4p 0,0049 0,0051
4d 0,0032 0,0032
Kapitel 5 Stilben
Stilben besteht aus zwei Phenylringen, die über eine Ethylenbindung miteinander verbunden sind. Daraus resultiert ein konjugiertes π-System. Bezüglich der zentralen Doppelbindung können die Phenylringe cis- oder trans-ständig angeordnet sein. Beide stereoisomeren Formen wandeln sich unter Lichteinwirkung ineinander um. Die dabei zu überwindende Barrierenhöhe ist bereits in einem früheren Forschungsprojekt [27]
bestimmt worden. Sie beträgt, ausgehend vom trans-Stilben, in diesem Modell 1,41 eV.
Abbildung 5.1: Isomerisierung Stilben
Für die eingeschränkten Geometrieoptimierungen erfolgt die Drehung der beiden Phenylringe im Stilben um die Ethylenbindung. Der Diederwinkel θ, den die Atome C2-C1-C10-C20 einnehmen, wird für die einzelnen Rechnungen festgehalten. Dieser wird in 10◦-Schritten im Berich von 0◦ bis 180◦ variiert (Abb. 5.2).
Bei den eingeschränkten Geometrieoptimierungen wird hierbei sowohl auf der Potenzialenergiehyperfläche des ersten angeregten Singulettzustands S1 (CMEP S1
- engl.: constrained minimum energy path) als auch der des Triplettzustandes T1 (CMEP T1) gerechnet.
36 KAPITEL 5. STILBEN
Abbildung 5.2: trans-Stilben mit Atomnummerierung
5.1 Grundzustand trans-Stilben
5.1.1 Geometrieoptimierungen
Aus der Geometrieoptimierung ergibt sich eine planare, C2h-symmetrische Struktur, welche durch eine anschließende Schwingungsanalyse als Minimum identifiziert worden ist. Dies stimmt sowohl mit experimentellen Ergebnissen von Jet-Spektren isolierter Moleküle [28,29] als auch mit denen von Fluoreszenzanregungsspektren [30,31] überein.
Die Bindungslängen der beiden Phenylringe im trans-Stilben entsprechen mit 1,39 Å sehr gut denen des Benzols mit 1,40 Å. Die Ethylenbindung hat mit 1,34 Å die exakte Länge einer Doppelbindung. Die beiden Einfachbindungen C2-C1 und C1’-C2’ mit einer Länge von 1,46 Å stimmen sehr gut mit der von 1,47 Å zwischen zwei sp2-hybridisierten Kohlenstoffatomen überein [32].
Die theoretisch berechneten Daten und die aus der Röntgenstrukturanalyse [33]
erhaltenen sind sich sehr ähnlich (Tab. 5.1). Sie weichen nichtmals um 0,02 Å voneinander ab.
5.1. GRUNDZUSTAND TRANS-STILBEN 37 Tabelle 5.1: Vergleich der Bindungslängen in [Å]
Bindung theo. exp. Diff.
C1-C10 1,342 1,326 0,016 C1-C2 1,463 1,471 -0,008 C2-C3 1,402 1,392 0,010 C3-C4 1,389 1,384 0,005 C4-C5 1,390 1,381 0,009 C5-C6 1,394 1,383 0,011 C6-C7 1,386 1,381 0,003 C7-C2 1,404 1,397 0,007
5.1.2 Charakterisierung der elektronischen Zustände
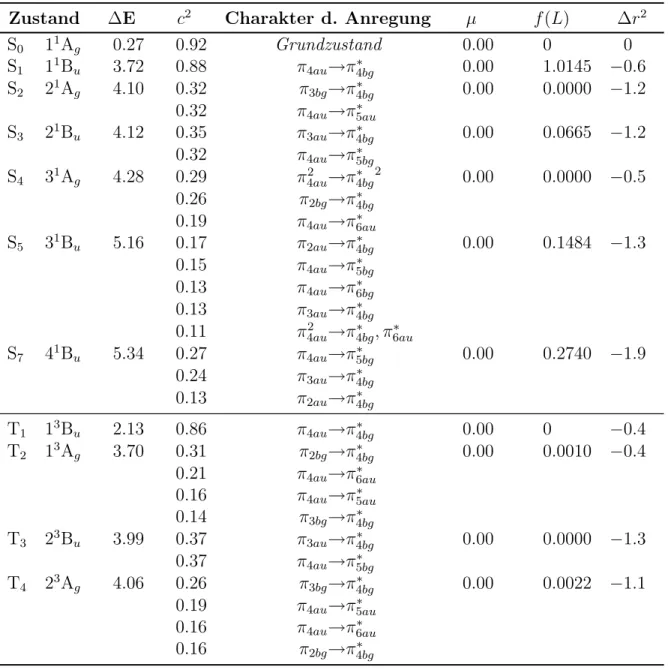
An der optimierten Gleichgewichtsgeometrie sind DFT/MRCI-Rechnungen mit insgesamt 47 Wurzeln (16 Ag - 8 Bg - 8 Au - 15 Bu) für den Singulettzustand und 46 Wurzeln (15 Ag - 8 Bg - 8 Au - 15 Bu) für den Triplettzustand durchgeführt worden, um einen Energiebereich bis etwa 8 eV abzudecken. Ziel ist es, die Ergebnisse mit den experimentellen Daten von Herrn Kovalenko [4] zu vergleichen, die sich in diesem Energiebereich erstrecken.
In Tab. 5.2 wird die elektronische Struktur der Zustände dargestellt. Der darin vorkommende c2-Wert beschreibt in der CI-Entwicklung den quadrierten Koeffizienten der jeweiligen Konfiguration. Er ist somit ein Maß für das Gewicht dieser Konfiguration, da die Normierungsbedingung lautet:
conf
X
i
c2i = 1 (5.1)
Die Oszillatorstärken werden durch f(L) angegeben. Sie beschreiben die Oszillation der Elektronendichte zweier Zustände während eines Elektronenübergangs. Je größer die Differenz zwischen der Verteilung der Elektronendichte ist, desto höher ist die Übergangswahrscheinlichkeit und desto größer ist die Oszillatorstärke [34].
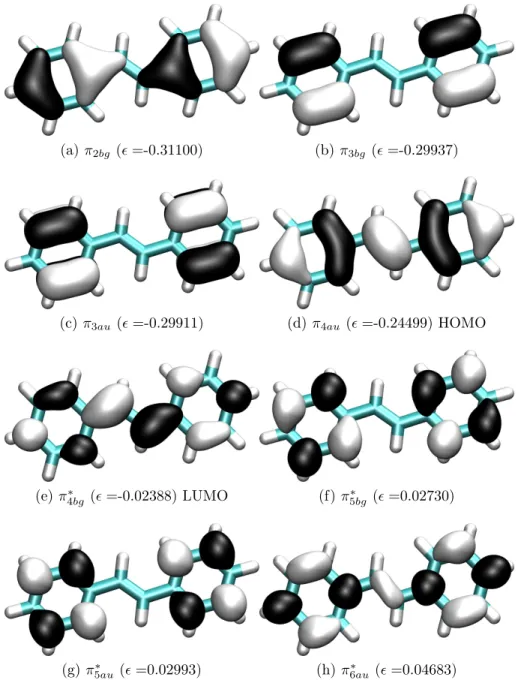
In Abb. 5.3 wird die Verteilung der Elektronendichte relevanter Orbitale im TZVPP- Basissatz dargestellt.
38 KAPITEL 5. STILBEN Tabelle 5.2: Stilben: Vertikales Anregungsspektrum an der trans-S0-Geometrie.
Aufgelistet sind der jeweilige Zustand, der Charakter der Anregung mit dem zugehörigen c2-Wert, die DFT/MRCI-Anregungsenergie ∆E [eV] und die Wellenlänge λ [nm], Dipolmomente µ [D], Oszillatorenstärken f(L) und ∆r2-Werte [bohr2].
Zustand ∆E λ c2 Charakter d. Anregung µ f(L) ∆r2
S0 11Ag 0.00 0.94 Grundzustand 0.00 0 0
S1 11Bu 3.99 310 0.75 π4au→π∗4bg 0.00 0.8550 −1.0 S2 21Ag 4.20 295 0.35 π3bg→π∗4bg 0.00 0.0000 −1.4
0.33 π4au→π∗5au
S3 21Bu 4.22 294 0.32 π3au→π∗4bg 0.00 0.2604 −1.6 0.28 π4au→π∗5bg
0.16 π4au→π∗4bg
S4 31Ag 4.83 257 0.32 π2bg→π∗4bg 0.00 0.0000 −0.7 0.22 π24au→π∗4bg2
0.21 π4au→π∗6au
T1 13Bu 2.53 490 0.83 π4au→π∗4bg 0.00 0 −0.8
T2 13Ag 3.69 336 0.33 π2bg→π∗4bg 0.00 0.0003 −0.6 0.25 π4au→π∗6au
T3 23Bu 3.96 313 0.36 π3au→π∗4bg 0.00 0.0000 −1.3 0.34 π4au→π∗5bg
T4 23Ag 4.01 310 0.32 π3bg→π∗4bg 0.00 0.0006 −1.5 0.28 π4au→π∗5au
T5 33Bu 4.12 301 0.37 π3au→π∗5bg 0.00 0.0000 −1.9 0.34 π3bg→π∗5au
T6 33Ag 4.25 292 0.34 π3bg→π∗5bg 0.00 0.0001 −1.6 0.33 π3au→π∗5au
5.1. GRUNDZUSTAND TRANS-STILBEN 39
(a)π2bg (=-0.31100) (b)π3bg (=-0.29937)
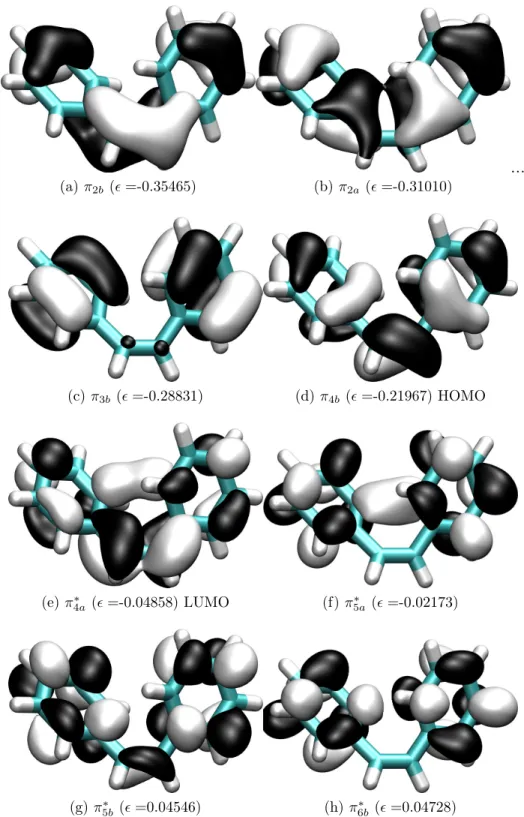
(c)π3au(=-0.29911) (d)π4au(=-0.24499) HOMO
(e)π4bg∗ (=-0.02388) LUMO (f)π5bg∗ (=0.02730)
(g)π∗5au(=0.02993) (h)π6au∗ (=0.04683)
Abbildung 5.3: trans-Stilben-S0-Geometrie: Form und energetische Reihenfolge wichtiger Grenzorbitale. Die Orbitalenergie ist in a.u. angegeben.
Bei allen inTab. 5.2 gezeigten, angeregten Singulett- und Triplettzuständen handelt es sich wie erwartet um vertikale (π → π∗)-Übergänge. Der HOMO →LUMO-Übergang erfolgt im S1-Zustand und weist eine Anregungsenergie von 3,99 eV auf. Dieser ist gleichzeitig optisch hell. Die Oszillatorenstärke liegt bei 0,8550. In der Literatur wird einstimmig der HOMO → LUMO-Übergang dem S1-Zustand mit einer Bu-Symmetrie und einer großen Oszillatorstärke zugeordnet [35, 36]. Champagne et al. [30] finden für diesen Zustand ebenfalls eine experimentelle Anregungsenergie von 4,00 eV im
40 KAPITEL 5. STILBEN Die Intensität des S0 → S3-Übergangs ist mit einer Oszillatorenstärke von 0,2604 deutlich geringer als die des S0 → S1-Übergangs, während die Anregung des S2- Zustands auf Grund seiner Symmetrie dipolverboten ist. Beide Zustände liegen mit 4,22 eV (S3) und 4,20 eV (S2) energetisch sehr nahe zusammen.
Die beiden Zustände nehmen auch in der Literatur eine besondere Stellung ein.
Gemeinsam ist den Papern von Gagliardi et al. [35], Angeli et al. [36] und Molina et al. [37] eine Voraussage für die Symmetrie des S2-Zustands. Sie bestimmen diese zu Bu. Darüber hinaus gehen die Meinungen jedoch weit auseinander. Gagliardi nimmt an, dass der berechnete 21Bu-Zustand eine geringere Intensität besitzt und im Experiment von der starken Absorptionsbande des 11Bu-Zustands verdeckt wird. Angeli ist ebenfalls der Auffassung, dass der 21Bu-Zustand dunkler ist. Beide stellen fest, dass er sich aus einer HOMO → (LUMO+1)- sowie einer (HOMO-1) → LUMO-Anregung zusammensetzt. Molinas Ergebnisse hingegen ergeben eine Vertauschung der 11Bu- und 21Bu-Zustände und der damit verbundenen Eigenschaften.
Der 21Ag-Zustand wird durch Einfachanregungen, der 31Ag-Zustand durch eine HOMO2 → LUMO2-Anregung dominiert. Die letzte Bande wird daher in der Zwei- Photonenspektroskopie beobachtet. Der 21Ag-Zustand und die beiden 1Bu-Zustände liegen energtisch eng zusammen, wobei der 21Ag-Zustand dem S3-Zustand zugeordnet wird.
Orlandi und Siebrand [38] gehen davon aus, dass sich der 31Ag-Zustand bei einer Verdrehung der Phenylringe zum energetisch günstigsten Zustand entwickeln wird.
Die Anordnung der Zustände ist sehr wichtig, wie es in Kap. 5.4 noch aufgezeigt werden wird.
In der Triplettmannigfaltigkeit werden 6 Zustände unterhalb oder in energetischer Nähe zu den niedrigliegenden Singulettzuständen gefunden, welche als Endzustände für einen möglichen Intersystem-Crossing (ISC)-Prozess dienen könnten.
5.2 S
1-Zustand trans-Stilben
5.2.1 Geometrieoptimierungen
Die Berechnungen der Geometrieoptimierungen des ersten angeregten Singulettzustandes S1 im TZVPP-Basissatz ergeben für das trans-Stilben eine planare, C2h-symmetrische Geometrie, welche durch die anschließende Schwingungsanalyse mit SNF als Minimum identifiziert worden ist. Champagne [30] erhält experimentell das gleiche Resultat.
Die Ethylenbindung C1-C10 weitet sich im Vergleich zu der Grundzustandsgeometrie S0 des trans-Stilbens um 0,07 Å auf, während die Bindungen zwischen C2-C1 und C10-C20 um 0,052 Å verkürzt werden. Die hierzu benachbarten Bindungen in den Phenylringen weiten sich deutlich - zwischen C2(0)-C3(0) um 0,034 Å und C2(0)-C7(0) um
5.2. S1-ZUSTAND TRANS-STILBEN 41 0,025 Å. Die restlichen Bingungslängen verändern sich nur geringfügig.
Dies wird anhand der Veränderung der Elektronendichte, die sich in einer anderen Orbitalform widerspiegelt, anschaulich dargestellt.
Da der S1-Zustand durch die HOMO → LUMO-Anregung repräsentiert wird, werden die Trends der Orbitaldichten miteinander in Verbindung gebracht. Es ist gut zu erkennen, dass die Ethylenbindung ihren Orbitalcharakter von bindend zu antibindend wechselt und eine Bindungsverlängerung damit einhergeht. Dies gilt genauso für die Bindungen zwischen C2(0)-C3(0) und C2(0)-C7(0). Bei den Bindungen zwischen C2-C1 und C10-C20 verhält es sich genau umgekehrt.
Tabelle 5.3: Bindungslängen in [Å]
Bindung S1 S0 Diff.
C1-C10 1,413 1,342 0,071 C1-C2 1,411 1,463 -0,052 C2-C3 1,428 1,402 0,026 C3-C4 1,374 1,389 -0,015 C4-C5 1,408 1,390 0,018 C5-C6 1,396 1,394 0,002 C6-C7 1,381 1,386 -0,005 C7-C2 1,438 1,404 0,034
Die Bindungslängen der beiden Einfachbindungen sowie der Ethylenbindung sind jetzt gleich lang. Ihre Längen liegen zwischen der einer Einfachbindung (1,47 Å) und der einer Doppelbindung (1,34 Å).
Die beiden Phenylringe weisen nicht mehr einen benzolartigen, sondern einen stärkeren Einfach- und Doppelbindungscharakter auf.
In Abb. 5.4 werden die Verteilungen der Elektronendichte relevanter Orbitale im TZVPP-Basissatz veranschaulicht.
Ein Vergleich der Orbitale des S1-Zustands mit denen des Grundzustand zeigt, dass sie eine sehr ähnliche Struktur aufweisen. Einzig ihre energetische Reihenfolge hat sich teilweise verändert.
42 KAPITEL 5. STILBEN
5.2.2 Charakterisierung der elektronischen Zustände
In Tab. 5.4 wird die elektronische Struktur der Zustände dargestellt.
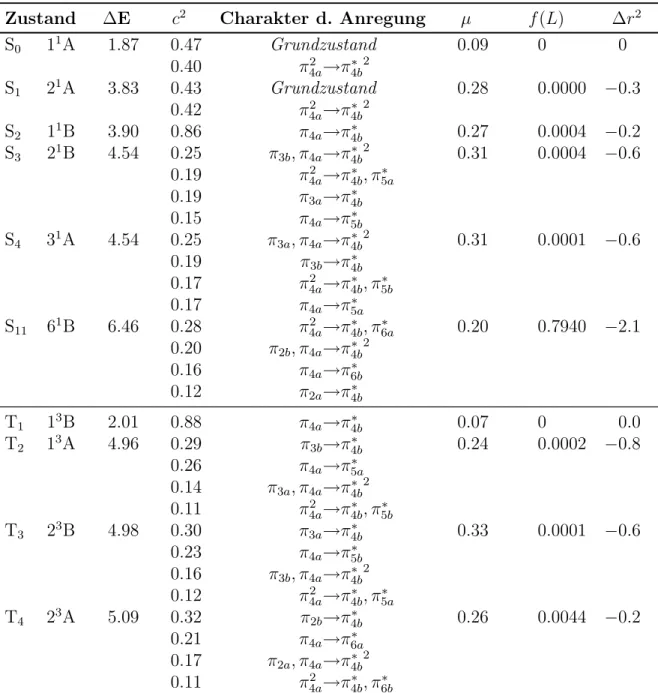
Tabelle 5.4: Stilben: Vertikales Anregungsspektrum an der trans-S1-Geometrie.
Aufgelistet sind der jeweilige Zustand, der Charakter der Anregung mit dem zugehörigenc2-Wert, die DFT/MRCI-Anregungsenergie∆E [eV], Dipolmomenteµ[D], Oszillatorenstärkenf(L)und∆r2-Werte[bohr2]. Als Nullpunkt der Energieskala ist die DFT/MRCI-Energie des Grundzustandsminimums gewählt worden.
Zustand ∆E c2 Charakter d. Anregung µ f(L) ∆r2
S0 11Ag 0.27 0.92 Grundzustand 0.00 0 0
S1 11Bu 3.72 0.88 π4au→π4bg∗ 0.00 1.0145 −0.6 S2 21Ag 4.10 0.32 π3bg→π4bg∗ 0.00 0.0000 −1.2
0.32 π4au→π5au∗
S3 21Bu 4.12 0.35 π3au→π4bg∗ 0.00 0.0665 −1.2 0.32 π4au→π5bg∗
S4 31Ag 4.28 0.29 π4au2 →π4bg∗ 2 0.00 0.0000 −0.5 0.26 π2bg→π4bg∗
0.19 π4au→π6au∗
S5 31Bu 5.16 0.17 π2au→π4bg∗ 0.00 0.1484 −1.3 0.15 π4au→π5bg∗
0.13 π4au→π6bg∗ 0.13 π3au→π4bg∗ 0.11 π4au2 →π4bg∗ , π6au∗
S7 41Bu 5.34 0.27 π4au→π5bg∗ 0.00 0.2740 −1.9 0.24 π3au→π4bg∗
0.13 π2au→π4bg∗
T1 13Bu 2.13 0.86 π4au→π4bg∗ 0.00 0 −0.4
T2 13Ag 3.70 0.31 π2bg→π4bg∗ 0.00 0.0010 −0.4 0.21 π4au→π6au∗
0.16 π4au→π5au∗ 0.14 π3bg→π4bg∗
T3 23Bu 3.99 0.37 π3au→π4bg∗ 0.00 0.0000 −1.3 0.37 π4au→π5bg∗
T4 23Ag 4.06 0.26 π3bg→π4bg∗ 0.00 0.0022 −1.1 0.19 π4au→π5au∗
0.16 π4au→π6au∗ 0.16 π2bg→π4bg∗
5.2. S1-ZUSTAND TRANS-STILBEN 43
(a)π2au(=-0.36538) (b)π2bg (=-0.31022)
(c)π3au(=-0.30184) (d)π3bg (=-0.30169)
(e) π4au(=-0.22929) HOMO (f) π4bg∗ (=-0.04005) LUMO
(g)π∗5bg (=0.02912) (h)π5au∗ (=0.03083)
(i)π∗6au(=0.04692)
...
(j)π∗6bg (=0.10554)
Abbildung 5.4: trans-Stilben-S1-Geometrie: Form und energetische Reihenfolge wichtiger Grenzorbitale. Die Orbitalenergie ist in a.u. angegeben.
Bei allen in Tab. 5.4, angeregten Singulett- und Triplettzuständen handelt es sich um (π → π∗)-Übergänge. Der HOMO-LUMO-Übergang erfolgt im S1-Zustand. Die stimulierte Emission hat eine Oszillatorenstärke von 1,0145. Auch hier liegen der S2- und der S3-Zustand energetisch sehr nahe zusammen, wobei der S2-Zustand ebenfalls optisch dunkel ist. Die optische Helligkeit des S3-Zustands beträgt allerdings nur ein Viertel von der des korrespondierende Zustands in der S -Geometrie. Die weiteren
44 KAPITEL 5. STILBEN
5.2.2.1 Einfluss von Rydbergfunktionen
Manche ∆r2-Werte, die das zweite Moment und damit die Diffusität der Orbitale beschreiben, sind in den eben analysierten MRCI-Werten relativ groß. Mit zunehmender Diffusität nimmt der sogenannte Rydbergcharakter zu. Diese Rechnungen untersuchen die Folgen des Rydbergeinflusses.
Für die Betrachtung der Rydberg-Orbitale werden weitere Zustände mit in die Berechnung hereingenommen. Dazu wird die Atomorbitalbasis erweitert. Durch die Rydbergfunktionen in dieser Atomorbitalbasis werden noch zusätzliche Zustände mit diffuser Ladungsverteilung beschrieben, die in den DFT/MRCI-Rechnungen in der TZVPP-Basis nicht auftreten. Um das gleiche Energiespektrum abzudecken, müssen mehr Wurzeln betrachtet werden.
Durch die Betrachtung zusätzlicher Wurzeln können die Zustände besser beschrieben werden.
Werden lediglich die optisch hellen Zustände betrachtet, so ähneln die Ergebnisse des DFT/MRCI in der erweiterten Basis denen der TZVPP-Basis. Einzig bei einer Energie von 2,53 mit einer Oszillatorenstärke von 0,90953 verteilt sich ein Valenzübergang auf zwei Wurzeln. Diese liegen bei 2,51 und 2,56 eV. Zusammen erreichen ihre Oszillatorenstärken mit 0,11673 und 0,49778 fast den Ausgangswert.
Die berechneten Daten weichen kaum voneinander ab. Daher werden die Rydberg- Ergebnisse auf Grund zu hoher Kosten für einen zu geringen Nutzen nicht weiter beachtet (Tabelle s. Anhang).
5.2.2.2 Einfluss des Lösungsmittels
Herr Kovalenko [4] hat seine Experimente in unterschiedlichen Lösemitteln durchgeführt. Die Messergebnisse weichen für diese nur geringfügig voneinander ab (Abb. 5.5).
Die Berechnungen mit COSMO simulieren eine Acetonitrilumgebung. Dies ist eines der Lösemittel, welches Kovalenko verwendet hat. Die Ergebnisse stimmen mit denen in der Vakuumumgebung gut überein, weshalb sie nicht weiter berücksichtigt werden (Tabelle s. Anhang).