Einfluss des Interplanarwinkels der Liganden eines neutralen
(P^P)Cu(N^N)-Komplexes auf dessen spektroskopische Eigenschaften
Bachelorarbeit
vorgelegt von Mathias Putze
Januar 2017 durchgeführt am
Institut für Theoretische Chemie und Computerchemie Mathematisch Naturwissenschaftliche Fakultät
Heinrich-Heine-Universität Düsseldorf
1. Gutachterin: Prof. Dr. Christel M. Marian
Hiermit erkläre ich, dass ich die Bachelorarbeit mit dem Thema
„Einfluss des Interplanarwinkels der Liganden eines neutralen (P^P)Cu(N^N)-Komplexes auf dessen spektroskopische Eigenschaften“
selbstständig und nur mit den angegebenen Quellen und Hilfsmitteln verfasst habe. Wörtliche oder inhaltliche Übernahmen, Abbildungen und Formeln welche aus anderen Quellen übernommen wurden, habe ich als solche gekennzeichnet. Diese Arbeit hat in gleicher oder ähnlicher Form noch keiner Prüfungsbehörde vorgelegt.
Unterschrift (Mathias Putze)
Zusammenfassung
Im Rahmen dieser Arbeit wird der Einfluss des Winkels zwischen den Liganden eines (PyrTet)Cu(DPEPhos) Moleküls untersucht. Hierfür werden die beiden Diastereomere auf Energieminima des Grundzustandes und des ersten angeregten Triplettzustandes untersucht. Die spektroskopischen Eigenschaften des Moleküls werden dabei über die Energieabstände zwischen diesen Zuständen sowie den S1 Zuständen der jeweiligen Geometrie ermittelt. Aufgrund des großen Rechenaufwandes im Rahmen der DFT/MRCI Rechnung und den damit verbundenen wenigen ermittelten Energien, kann jedoch keine Aussage über die Änderungen der Energie bei einer bestimmten Änderung des Winkels gemacht werden. Allerdings steht fest, dass der Winkel die spektroskopischen Eigenschaften beeinflusst.
Abstract
In this Bachelor thesis the influence of the dihedral angle between the bonding atoms of the two ligands of a copper(I) complex. To do so the geometries of the ground state and the first exited triplet state are investigated and the spectroscopic properties are calculated using the information about the energy of those geometries gained by DFT/MRCI calculations. Due to the low number of geometries calculated no pattern of the change of the spectroscopic properties by changing the angle by a number of degrees can be given.
Danksagung
Ich möchte mich bei Prof. Dr. C. M. Marian bedanken für die Möglichkeit, in ihrem Arbeitskreis diese Bachelorarbeit schreiben zu dürfen, sowie für die viele Arbeit und Mühe, die sie in die Betreuung meiner Bachelorarbeit investiert hat.
Bei Prof. Dr. C. Ganter für die Übernahme des Postens als Zweitgutachter.
Bei dem Arbeitskreis der Theoretischen Chemie und Computerchemie für die gute Atmosphäre und die freundliche Aufnahme in den Arbeitskreis. Ganz besonders möchte ich mich hier bei Jelena Föller und Dr. Oliver Weingart bedanken, dafür das ich mich mit meinen Fragen zu jeder Zeit an sie wenden konnte. Für die Hilfe bei technischen Schwierigkeiten möchte ich mich auch noch bei Klaus Eifert bedanken.
Außerdem möchte ich mich bei meinen Eltern und meiner Familie bedanken, die mich finanziell unterstützt haben und wesentlich mehr Geduld mit mir hatten, als ich sie hätte aufbringen können.
Zum Schluss möchte ich mich auch noch bei meinen Freunden und Bekannten bedanken, die während des Studiums oft für mich da waren und mich immer Unterstützt haben. An dieser Stelle möchte ich mich auch noch bei Dr. Rolf Packroff bedanken, der mich überhaupt erst auf diesen Studiengang aufmerksam gemacht hat.
Inhaltsverzeichnis
Abkürzungsverzeichnis
1. Einleitung 1
1.1 Lumineszenz 1
1.2 OLEDS 3
1.2.1 OLEDs 1. Generation 3
1.2.2 OLEDs 2. Generation 4
1.2.3 OLEDS 3. Generation 5
1.3 (bis(2-(Diphenylphosphino)phenyl)ether)Cu
(5-(2-pyridyl)tetrazolat) 8
2. Theorie 9
2.1 Dichtefunktionaltheorie (DFT) 9
2.2 Multireference configuration interaction (MRCI) 11
3. Durchführung 12
4. Darstellung der Ergebnisse 13
4.1 Ergebnisse der Geometrieoptimierung 13
4.2 DFT/MRCI Rechnung 16
4.2.1 Geometrien mit positiven
NNPP-Diederwinkeln 16
4.2.1.1 Grundzustandsgeometrien 16
4.2.1.2 Triplettgeometrien 24
4.2.1.3 Übersicht der berechneten Minima und Vergleich mit dem Experiment 30 4.2.2 Geometrien mit negativen
NNPP-Diederwinkeln 34
4.2.2.1 Grundzustandsgeometrien 34
4.2.1.2 Triplettgeometrien 39
4.2.1.3 Übersicht der berechneten Minima und Vergleich mit dem Experiment 43
5. Zusammenfassung und Ausblick 48
6. Literaturverzeichnis 50
7. Programmverzeichnis 52
8. Anhang 53
8.1 Bilder der SOMOs 53
8.2 Bilder der Orbitale der Grundzustandsgeometrien nach 59 bh-lyp
8.3 Bilder der Orbitale der Triplettgeometrien nach bh-lyp 72 8.4 Bindungslängen- und Winkeländerung der Geometrien 85 8.5 Grundzustandsminimumgeometrien nach PBE0
in Koordinaten 87
8.6 Triplettminimumgeometrien nach PBE0 in Koordinaten 93
Abkürzungsverzeichnis
Abb. Abbildung
CI configuration interaction DFT Dichtefunktionaltheorie
DPEPhos bis(2-(Diphenylphosphino)phenyl)ether (siehe Abb. 6 rechts)
E Energie
GGA generalized gradient expansion HOMO highest occupied molecular orbital
HSOMO highest single occupied molecular orbital IC internal conversion
ILCT inter-ligand-charge-transfer ISC intersystem-crossing
LCD liquid crystal display
LDA local density approximation LSDA local spin-density approximation LUMO lowest unoccupied orbital
LSUMO lowest single unoccupied molecular orbital MLCT metall-to-ligand-charge-transfer
MRCI multireference configuration interaction OLED organic light-emitting diode
PyrTet 5-(2pyridyl)tetrazolat (siehe Abb. 6 links) RISC reverse intersystem-crossing
Sn n. angeregter Singulettzustand (bei n=0 Grundzustand) Tn n. angeregter Triplettzustand
TD-DFT time-dependent density functional theory TADF thermally activated delayed fluorescence ZPE zero point energy
1. Einleitung
OLED Emitter sind für moderne Informationssysteme und Beleuchtungstechnologien von zunehmender Bedeutung. Im Rahmen dieser Arbeit wird der Einfluss des Winkels zwischen den beiden Liganden eines solchen OLED Emitters auf seine spektroskopischen Eigenschaften überprüft.
1.1 Lumineszenz
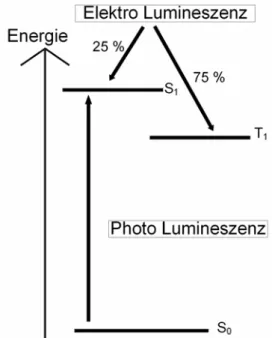
Als Lumineszenz bezeichnet man das Phänomen der strahlenden Abgabe von Energie (Emission), um aus dem ersten angeregten Zustand heraus den tiefer liegenden Grundzustand zu erreichen. Bei diesem Vorgang wird zwischen Phosphoreszenz und Fluoreszenz sowie zwischen elektrisch angeregter Lumineszenz und Photolumineszenz unterschieden.
Unterscheiden sich die Spinmultiplizitäten der beiden an der Emission beteiligten Zustände von einander, so spricht man von Phosphoreszenz. So handelt es sich im Falle des strahlenden Übergangs aus einem Singulett-Zustand heraus in den Singulett- Grundzustand um Fluoreszenz[1]. Ist der Grundzustand jedoch ein Triplett-Zustand, so beobachtet man Phosphoreszenz.
Bei der Photolumineszenz wird ein Elektron des Moleküls durch ein Photon aus dem Grundzustand heraus in ein höher gelegenes Orbital angeregt. Das Photon muss für diese Anregung jedoch genauso viel Energie enthalten, wie der Energieunterschied zwischen dem Grundzustand und dem erreichten angeregten Zustand. Da der Übergang aus einem Singulett-Zustand in den Triplett-Zustand oder der umgekehrte Prozess eine Spinumkehr erfordern würde, ist dieser im Falle einer Photoanregung verboten[2], solange die Spin-Bahn-Kopplung nicht zu groß ist. Es existieren also im Normalfall nur Singulett- Zustände und somit auch nur Fluoreszenz sowie nicht strahlende Effekte. Im Rahmen der Elektrolumineszenz, werden hingegen an der Anode Elektronen aus dem eigentlichen Emittermaterial gelöst und
somit ein Elektronenloch induziert. Zeitgleich wird an der Kathode ein Elektron in das Material eingespeist, sodass Elektron und Elektronenloch aufeinander zuwandern und schließlich als sogenanntes Exciton aufeinander treffen. Aufgrund der Energie des Elektrons in diesem Vorgang, ist das Molekül zunächst in einem angeregten Zustand. Von hier aus relaxiert es schnell bis zum ersten angeregten Singulett-Zustand oder Triplett-Zustand. Von diesem Zustand aus, fällt das Molekül dann in den Grundzustand zurück. Da drei verschiedene Triplett-Zustände sowie ein Singulett-Zustand existieren, macht Fluoreszenz lediglich bis zu 25% der möglichen strahlenden Übergänge aus[3-7].
Das emittierte Licht entspricht bei diesen Vorgängen mit seiner Wellenlänge dem Energieunterschied zwischen dem relaxierten angeregten Zustand und dem (vibronisch angeregten) Grundzustand[3].
Abb. 1) Energieschema elektrische und photochemische Anregung
1.2 OLEDs
Organic light emitting diodes (kurz. OLEDs) sind Bauteile der Elektroindustrie zur Beleuchtung und Darstellung von Informationen.
Ihre Vorteile gegenüber anderen Displaytechnologien sind hohe Energieeffizienz, gute Blickwinkelabhängigkeit, bessere Kontrastdarstellung aber auch eine mögliche bessere Grafikauflösung und eine im Vergleich zur LCD-Technologie bessere Reaktionszeit. Im Bereich der Beleuchtungstechnologie gibt es außerdem den großen Vorteil, dass OLED Leuchtflächen über kein Leuchtzentrum verfügen, sondern über eine gleichbleibend helle Fläche. Dies führt dazu, dass bei Leuchtmitteln mit gleicher Helligkeit OLEDs als wesentlich weniger blendend empfunden werden. Allerdings gibt es hier noch energieeffizientere Technologien, welche aber in ihren Einsatzmöglichkeiten nicht so flexibel sind. Der größte Nachteil der OLED Technologie liegt weiterhin in ihrer Lebensdauer, welche insbesondere im Bereich der blauen OLEDs wesentlich geringer ist[3,8].
1.2.1 OLEDs 1. Generation[6]
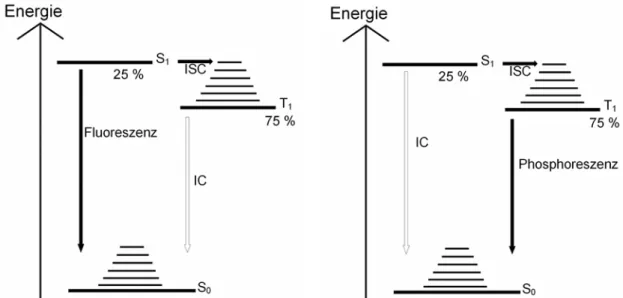
Die erste Generation von OLEDs besteht aus sogenannten Fluoreszenzemittern.
Das bedeutet, dass die Moleküle lediglich 25% der erzeugten angeregten Zustände zur Lichterzeugung verwenden können. Die verbleibenden Triplett-Zustände relaxieren vibronisch unter Intersystem- crossing (ISC) zurück in den Grundzustand, wobei kein Licht sondern Wärme erzeugt wird.
Hierdurch wird also die interne Quantenausbeute der OLEDs erster Generation auf 25% beschränkt. Da es bisher allerdings noch keine besseren stabilen Moleküle gibt, welche blaues Licht emittieren, finden OLEDs erster Generation auch heutzutage Verwendung.
Abb. 2) Energieschema Fluoreszenzemitter (1. Generation) Phosphoreszenzemitter (2.Generation)
1.2.2 OLEDs 2. Generation[6]
Um die Effizienz der OLEDs zu erhöhen wurden später phosphoreszente Emitter entwickelt, welche folglich 75% der angeregten Zustände zur Lichterzeugung nutzen können.
Die schweren Zentralatome haben bei diesen Molekülen zur Folge, dass das Übergangsmoment aufgrund der Spin-Bahn-Kopplung ungleich null wird und somit nicht mehr so streng verboten ist wie bei den OLEDs erster Generation. Dadurch steigt die Wahrscheinlichkeit, dass das Molekül im ersten angeregten Singulett-Zustand mittels Intersystem-crossing zunächst in den ersten angeregten Triplett- Zustand wechseln und von dort aus ebenfalls strahlend in den Grundzustand zurück kehren kann. Die maximal erreichbare Ausbeute der angeregten Zustände beträgt also 100%. Diese Methode zur Erhöhung der Effizienz wird als triplet harvesting bezeichnet. Die Notwendigkeit für das Elektron, seinen Spin zu ändern, um in den Grundzustand zu gelangen, führt allerdings zu einer langen Strahlungslebensdauer des Triplett-Zustands. Da Moleküle im angeregten Zustand wesentlich reaktiver sind, sinkt bei OLEDs der zweiten Generation im Vergleich zu OLEDs der ersten Generation die Lebensdauer der Emittermoleküle.
1.2.3 OLEDs 3. Generation
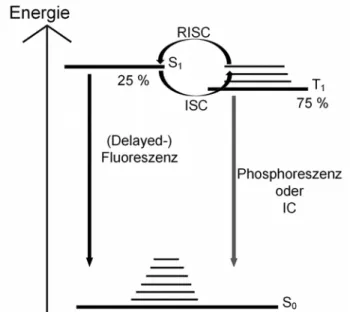
Die Entdeckung des reverse intersystem crossing (RISC), also eines Prozesses, in dessen Folge der T1 Zustand zurück in den energetisch höher liegenden S1 Zustand wechselt, führte zu einem neuen Ansatz im Bereich der OLED Entwicklung. Die bei diesen Molekülen erzeugte Fluoreszenz wird als thermally activated delayed fluorescence (TADF) bezeichnet. Auch bei diesem Vorgehen spricht man von triplet harvesting.
Damit ein Molekül TADF Eigenschaften zeigt, muss es mehrere Bedingungen erfüllen. Zunächst sollte der Energieunterschied zwischen dem ersten angeregten Singulett-Zustand und dem ersten angeregten Triplett-Zustand möglichst gering sein (ΔΕ(S1-T1) ≤ 120 meV)[6]. Hierdurch ist die thermische Energie des Moleküls im Triplett-Zustand bei Umgebungstemperatur hoch genug, um das Energieniveau des S1
Zustands zu erreichen.
Abb. 3) TADF Emitter (3. Generation) ΔΕ(S1-T1) ist deutlich kleiner.
Phosphoreszenz kann zusätzlich möglich sein.
Des weiteren müssen sich die Elektronendichteverteilungen der am S1
→ S0 Übergang beteiligten Orbitale überlappen, um einen möglichst stark erlaubten erlaubten Übergang zu erhalten. Es gilt, dass die Überlappung mit zunehmender Größe einen schnelleren Übergang
erzeugt. Das bedeutet, dass die Strahlungslebensdauer des ersten angeregten Singulett-Zustandes sinkt und die Ratenkonstante somit steigt. Allerdings führt eine starke Überlappung zu einem großen ΔΕ(S1- T1), sodass diese nicht zu stark sein darf, um eine TADF Fähigkeit nicht zu verhindern.
Dieses Phänomen lässt sich damit begründen, dass das Austauschintegral, welches mit steigender Überlappung größer wird auch die (S1-T1) Energiedifferenz in gleicher Weise beeinflusst[5].
Abb. 4) Strahlungsabklingrate (als Indikator für den Überlapp) aufgetragen gegen ΔΕ(S1-T1) für verschiedener Kupfer Komplexe mit TADF Eigenschaften[5]
Natürlich ist auch die Wahl der Liganden von entscheidender Bedeutung für das Lumineszenzverhalten des Emitters, da diese sowohl den Energieunterschied ΔΕ(S1-T1) maßgeblich beeinflussen können als auch das TADF-Verhalten an sich[3,5].
TADF Emitter können in zwei verschiedene Klassen unterteilt werden.
Die klassischen TADF Emitter bestehen aus Cu(I)-Komplexen. Diese zeichnen sich durch eine kürzere Lebensdauer des angeregten Zustandes im Vergleich zu organischen TADF-Emittern aus. Außerdem sind sie im Vergleich zu phosphoreszenten Emittern (2. Generation) deutlich günstiger in der Herstellung, da das verwendete Kupfer
deutlich öfter vorkommt als die Übergangsmetalle Iridium und Platin.
Des weiteren hat dieser Typ von TADF-Emitter normalerweise vergleichsweise hohe Raten bei T1 → S0 Intersystem-crossing Prozessen, sodass die Lebensdauer des angeregten Zustandes im Vergleich zu der Triplett-Lebensdauer von phosphoreszenten Emittern recht niedrig ist[6]. Der zweite Typ der TADF-Emitter besteht aus den organischen Molekülen. Im Vergleich zu den Cu(I)-Komplexen haben diese allerdings häufig einen wesentlich höhere Lebensdauer des angeregten Zustandes und sind somit anfälliger für Reaktionen mit anderen Substanzen[6].
Ein weiterer Nachteil von organischen Molekülen ist eine stärkere Wirkung von Roll-off-Effekten[6]. Also Effekten, die die Effizienz der Emitter bei höherer Helligkeit senken.
Bei Emittern der 3. Generation besteht des weiteren die Möglichkeit, das Molekül so zu entwerfen, dass neben der verzögerten Fluoreszenz auch Phosphoreszenz ermöglicht wird. Dieses Vorgehen ermöglicht es, die Lebensdauer des angeregten Zustandes weiter zu senken und damit die Stabilität der Moleküle weiter zu erhöhen[5].
Abb. 5) Beispiel TADF+Phosphoreszenz für Cu2Cl2(2-Methyl,5-(Diphenylphosphino)pyridin)2[5]
1.3 (bis(2-(Diphenylphosphino)phenyl)ether)Cu (5-(2-pyridyl)tetrazolat)
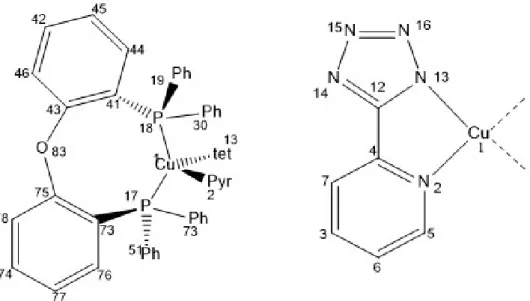
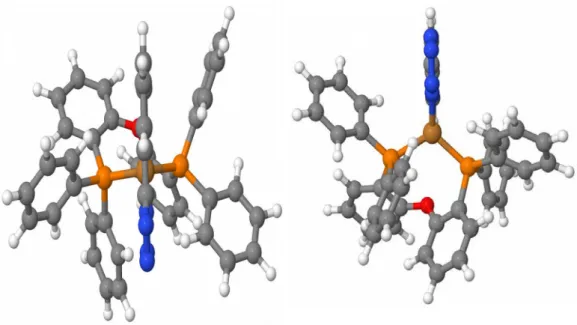
Abb. 6) DPEPhosCu (rechts) und PyrtetCu (links) als Strukturformel. Hier dargestellt, mit einem negativen NNPP-Diederwinkel.
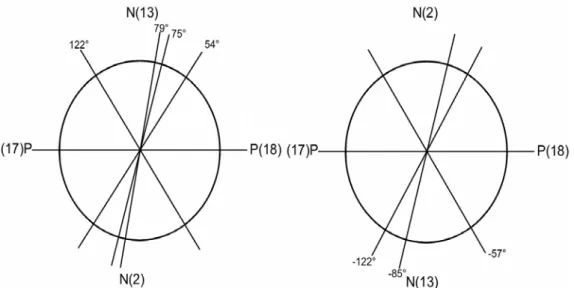
Bei (bis(2-(Diphenylphosphino)phenyl)ether)Cu(5-(2pyridyl)tetrazolat), welches von hier an auch (DPEPhos)Cu(PyrTet) genannt wird, handelt es sich um einen TADF Emitter. Der neutrale Cu(I) Komplex besteht aus zwei verschiedenen zweizähnigen Liganden. Die Winkelangaben für den NNPP-Diederwinkel in dieser Arbeit beziehen sich auf den Diederwinkel P(18)-P(17)-N(13)-N(2). Da es sich bei Diesem um einen Diederwinkel handelt, welcher nicht durch Bindungen zwischen den Atomen beschrieben werden kann, wird er auch als unechter Diederwinkel bezeichnet. Als Film hat die Substanz ein Emissionsspektrum mit einem Maximum bei 535 nm. In Pulverform liegt dieses bei 512 nm. Das emittierte Licht ist also grün oder in der Pulverform cyanblau. Das Spektrum ist jedoch in beiden Fällen sehr breit, was darauf hin deutet, dass die Konformationen einen sehr starken Einfluss auf die Emission haben[9]. Im Rahmen dieser Arbeit soll dieser Einfluss quantifiziert werden, indem der NNPP-Diederwinkel zwischen den beiden Liganden untersucht und variiert wird.
2. Theorie
2.1 Dichtefunktionaltheorie (DFT)[10]
Die klassische Geometrieoptimierung mittels Wellenfunktionen wird Aufgrund der 3N Variablen des Systems sehr schnell aufwändig, sodass eine effizientere Methode gewählt werden muss. Hierfür bietet sich eine Optimierung des Systems mittels DFT an. Der Grundgedanke der DFT basiert darauf, dass das Potential eines jeden Systems mithilfe seiner Elektronendichte beschrieben werden kann. Pierre Claude Hohenberg und Walter Kohn bewiesen dieses gleichnamige Theorem, indem sie die Gegenannahme, das System habe bei einer Dichte zwei verschiedene mögliche Potentiale falsifizierten.
Dieses Theorem bildet also die Grundlage der DFT. Außerdem wurde mithilfe dieses Theorems das zweite Theorem Hohenberg und Kohn hergeleitet, welches besagt, dass die minimale Energie eines Systems nur durch die entsprechende Grundzustandsdichte erreicht werden kann.
Eines der Probleme der DFT ist, dass nicht bekannt ist, wie das Funktional, welches die Energie mit der Elektronendichte verbindet, lautet. Daher haben Walter Kohn und Lu Jeu Sham zunächst im Rahmen ihres Kohn-Sham Theorems die Energie eines beliebigen Systems in unterschiedliche Terme unterteilt und alle einfach lösbaren Teile von den unbekannten Anteilen separiert. Hierzu nahmen Kohn und Sham an, dass die Elektronen nicht wechselwirken und somit mithilfe der Summe ihrer Einelektronenoperatoren im Hamiltonoperator beschreiben lassen, wenn man die Gesamtdichte des dabei erzeugten Systems an die reale Gesamtdichte anpasst. Diese Dichte kann anschließend in der Form von Orbitalen dargestellt werden, um mit diesen weiter zu rechnen.
Aus den unbekannten Termen ergibt sich das Austauschkorrelationsfunktional, welches die unbekannten Größen enthält. Dieses besteht neben den Korrekturen für Selbstwechselwirkungen noch aus den Austauschwechselwirkungen sowie den Korrelationswechselwirkungen, welche mithilfe von Dichtefunktionalen genähert werden. Die einfachsten dieser Dichtefunktionale entstehen mithilfe der sogenannten Lokalen Dichte Näherung (local density approximation, LDA) oder auch der lokalen Spin-Dichte Näherung (local spin-density approximation, LSDA).
Funktionale, die auf Basis dieser Theorie entwickelt werden, hängen lediglich von der Elektronendichte ab, wobei im Falle der LSDA noch zusätzlich der Spin der Elektronen berücksichtigt wird. Wesentlich bessere Ergebnisse werden mithilfe von Dichtefunktionalen auf Basis der generalized gradient expansion (GGA) erzielt. Bei Funktionalen, die auf diese Weise entwickelt werden, wird wie auch bei LDA oder LSDA die Theorie des Elektronengases zur Hilfe genommen. Allerdings wird auch die Änderung der Dichte mithilfe einer Taylorentwicklung und der Parametrisierung des Funktionals berücksichtigt. Zusätzlich können auch Ergebnisse einer Hartree-Fock-Rechnung berücksichtigt werden und mithilfe von Parametern in das Funktional integriert werden. Diese daraus resultierenden Funktionale werden auch als Hybridfunktionale bezeichnet.
Die Vorteile der DFT sind also in ihrer sehr guten Anwendbarkeit sowie in dem geringen Rechenaufwand zu sehen. Allerdings sind die berechneten Bindungsenergien oftmals geringfügig größer als die experimentell ermittelten Werte und die berechneten Energiebarrieren und Schwingungsfrequenzen eher etwas zu klein.
2.2 Multireference configuration interaction (MRCI)[11]
In dieser Theorie bestehen angeregte Zustände aus einer Mischung von verschiedenen möglichen Anregungskonfigurationen. Da dieser Umstand von der DFT Rechnung alleine nicht zu Genüge berücksichtigt wird, nutzt man zur Beschreibung von angeregten Zuständen das MRCI Verfahren. Im Zuge dieser Methode werden die möglichen Zustände linear kombiniert und eine Configuration Interaction (CI) Matrix aufgestellt. Mithilfe dieser Matrix können anschließend die Eigenwerte und die dazu gehörigen Anregungen berechnet werden. Werden alle möglichen Wellenfunktionen in Betracht gezogen, so spricht man von einer Full CI-Rechnung. Allerdings ist dieses mit zunehmender Größe sehr aufwändig, sodass meistens nur Einzelanregungen und Doppelanregungen berücksichtigt werden.
Der Aufwand, die Wellenfunktionen für die Zustände zu erzeugen, limitiert den Anwendungsbereich auf kleine Moleküle, da der Aufwand überproportional mit der Anzahl der Atome eines Moleküls steigt.
Außerdem wächst bei Verbesserung der Basis die Genauigkeit der Rechnung nur langsam.
Um diese Probleme für die MRCI Rechnung zu umgehen und diese Methode auch für große Moleküle nutzbar zu machen, wird in der Praxis auf die DFT/MRCI Methode zurück gegriffen. Bei dieser Methode wird die dynamische Elektronenkorrelation mithilfe der DFT Methode gelöst und nur die übrigen Teile mithilfe der MRCI. Dadurch müssen wesentlich weniger Linearkombinationen berechnet werden, sodass der Aufwand deutlich sinkt. Allerdings kann es passieren, dass bei der DFT/MRCI Rechnung die Elektronenkorrelation doppelt berücksichtigt wird, was zu verfälschten Ergebnissen führen würde. Um diesen Fehler zu vermeiden, wird diese in Abhängigkeit von der Energie skaliert.
3. Durchführung
Als erstes wird das Molekül (siehe Kapitel 1.3) mithilfe des Spartan Programms gebaut und anschließend mittels Turbomol die Geometrie optimiert. Hierfür wird das PBE0[17] Dichtefunktional sowie die Basissätze cc-PVDZ-pp[14] für das Kupferatom und SV(P)[15] für die restlichen Atome verwendet. Als effektives Rumpfpotential wird, für das Kupferatom, das Stuttgart/Köln energy consistant pseudopotential (MCDHF RSC ECP)[16] benutzt. Außerdem werden Grimmes Dispersions Wechselwirkungskorrekturen DFT-D3[19] verwendet.
Von den bei diesem Molekül gefundenen Geometrien mit einem Energieminimum aus wird ein Torsionswinkelscan am unechten NNPP- Diederwinkel durchgeführt. Der gleiche Vorgang wird noch einmal für den Triplett-Zustand wiederholt, allerdings wird hier das Minimum des Singuletts als Ausgangslage für die Geometrieoptimierung genutzt.
Anschließend wird der PyrTet Ligand um 180 Grad, entlang des obigen Diederwinkels, gedreht und die Rechnungen noch einmal wiederholt.
An den gefundenen Minima wird außerdem eine Schwingungsfrequenzanalyse durchgeführt, um die Existenz des Minimums zu bestätigen und die Berechnung der Phosphoreszenzspektren zu ermöglichen.
Außerdem wird an den Minima eine DFT/MRCI Rechnung durchgeführt, diese liefert Informationen über das Dipolmoment der verschiedenen Zustände sowie die Energieunterschiede zwischen diesen. Für diese Rechnung wird im Gegensatz zum Funktional für die Geometrieoptimierung das bh-lyp[18] Funktional verwendet. Die anderen Einstellungen bleiben aber wie oben angegeben.
4. Darstellung der Ergebnisse:
Im folgenden Teil werden die Ergebnisse der Berechnungen dargestellt.
Zuerst werden die Ergebnisse der Rechnungen am Singulett- Grundzustand präsentiert. Anschließend werden die Geometrien des Triplett-Zustand beschrieben.
4.1 Ergebnisse der Geometrieoptimierung
Abb. 7) Schema des Interplanarwinkels bei Zentrierung auf den Kreuzungspunkt des NN-Abstandes mit dem PP-Abstand. Die Geraden repräsentieren den Abstand zwischen den Atomen.
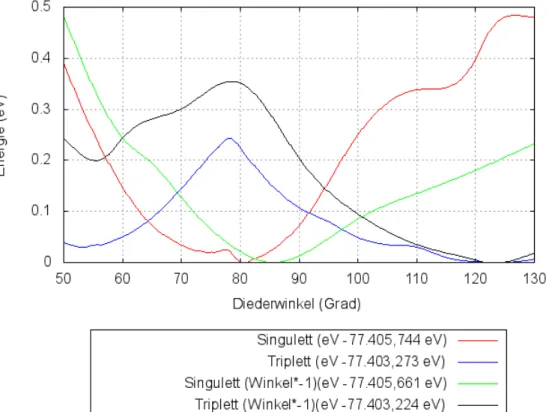
Im Zuge der Geometrieoptimierung konnten 3
Grundzustandsgeometrien in einem Energieminimum gefunden werden sowie 4 Triplettgeometrien in einem solchen. Auf der unten gezeigten Abbildung 8 ist deutlich zu sehen, dass die Singulett- und Triplett- Kurven jeweils einen ähnlichen Verlauf haben. Die Singulettkurve mit einem positiven NNPP-Diederwinkel zeigt deutlich den energetischen Unterschied zwischen den beiden Minima. Natürlich ist ein solcher Sprung in der Kurve nicht normal und eine genauere Untersuchung des Bereichs zeigt, dass hier ab einem Winkel von 72,6°
Dispersionswechselwirkungen zwischen einem Phenylring und dem Pyrtet Liganden auftreten können, die die Energie der Struktur weiter
absenken. Bei der Grundzustandsgeometrie von 74,92° beträgt diese diese mögliche Absenkung 0.01 eV unter den berechneten Zustand.
Abb. 8) Energieverlauf der S0 und T1 Geometrien in Abhängigkeit von dem NNPP-Diederwinkel laut Geometrieoptiomierung.
Auffällig ist außerdem, dass die Anregung jedes mal eine MLCT Anregung mit dem zusätzlichen Charakter einer lokalen Anregung ist.
Das beteiligte HSOMO-X (Highest Single Occupied Molecular Orbital -X) besitzt Elektronendichte auf dem Kupfer und dem Tetrazol.
Lediglich das an der Anregung beteiligte HSOMO-X der Geometrie bei einem NNPP-Diederwinkel von 122° besitzt zusätzlich Elektronendichte auf dem Pyridinring. Bei den anderen beteiligten Orbitalen unterscheidet sich die Elektronendichteverteilung lediglich leicht. Bei allen Geometrien ist das für die Anregung verwendete HSOMO-X energetisch abgesenkt. Bei der Geometrie mit einem NNPP- Diederwinkel von 54° ist das HSOMO-3 das nur einfach besetzte Orbital der Triplettanregung (HOMO-X). Bei den anderen Geometrien sind dies HSOMO-2 (NNPP-Diederwinkel von 122°), HSOMO-5 (-57°) und HSOMO-3 (-122°). Angeregt wird in allen Fällen in das LSUMO
(Lowest Single Unoccupied Molecular Orbital), welches seine Elektronendichte zu großen Teilen auf dem Pyridin aber auch auf dem Tetrazol hat.
Abb. 9) links HSOMO-4 (NNPP-Diederwinkel von 54°), sowie rechts HSOMO-2 (NNPP-Diederwinkel von 122°)
Diese Umlagerung der Eektronendichte passt gut zu der Veränderung der Bindungslängen, da die Bindungen des Kupfers an das Phosphor während der Anregung in den Triplettzustand länger werden. Zeitgleich werden die Bindungen zum Stickstoff kürzer. Im PyrTet Liganden selber werden die Bindungen im Tetrazol minimal länger (0-1 pm), während die Bindungen im Pyridin um etwa 1-2 pm kürzer werden.
Abb 10) links HSOMO-5 (NNPP-Diederwinkel von -57°, sowie rechts HSOMO-3 (NNPP-Diederwinkel von -122°)
Abb. 11) LSUMO (stellvertretend bei NNPP-Diederwinkel von 54°)
4.2 DFT/MRCI Rechnung
4.2.1 Geometrien mit positivem NNPP-Diederwinkeln
4.2.1.1 Grundzustandsgeometrien
Die Geometrieoptimierung des Grundzustandes von (DPEPhos)Cu(PyrTet) hat aufgezeigt, dass es im Singulett-Zustand mindestens zwei verschiedene Energieminima gibt. Diese liegen bei NNPP-Winkeln von 74.92 und 79.43 Grad. Eine Geometrieoptimierung ohne Berücksichtigung der Dispersionswechselwirkungen offenbart dabei, dass die Minima sich insbesondere aufgrund der, bei
Vergrößerung des Diederwinkels, eintretenden
Dispersionswechselwirkungen zwischen einem der Ether-Phenylringe und einem Phenylring des zweiten Phosphoratoms sowie dem anderen nicht verbrückten Phenylring mit dem PyrTet Liganden unterscheiden.
Die Bindungslängen zwischen den einzelnen Atomen sind dabei identisch. Die Ausnahme bilden hier die beiden Bindungen zwischen dem Kupferatom und den Phosphoratomen, sowie die Bindung zwischen dem Phosphoratom P17 und dem Kohlenstoffatom des verbückenden Phenylrings (+1 pm). Bei der Vergößerung des Winkels wächst die Bindung zwischen dem Phosphor mit der Nummer 17 und dem Kupfer um 3 Pikometer, während die Bindung zum anderen
Phosphoratom um die gleiche Distanz schrumpft.
4.2.1.1.1 Minimum des Singulett-Zustands bei 74.92°
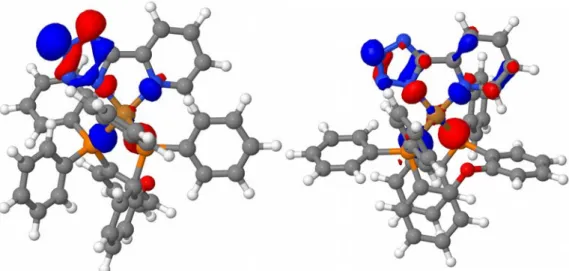
Abb. 12) Minimum bei einem NNPP-Winkel von 74,92° aus zwei verschiedenen Perspektiven.
Bei einem NNPP-Winkel von 74.92° sind die
Dispersionswechselwirkungen noch nicht erkennbar. Die Energie des Moleküls mit diesem Winkel, wird bei der Geometrieoptimierung mit -77.405,724 eV angegeben, womit es etwa 0,02 eV über dem anderen Minimum liegt. Nach der DFT/MRCI Rechnung ist dieses Minimum im Grundzustand bei -77.438,313 eV.
Abb. 13) Absorptionsspektrum bei einem NNPP-Diederwinkel von 74,92° mit Dispersionskorrekturen
Das Absorptionsspektrum zeigt das es mehrere Anregungen mit einer hohen Oszillatorstärke gibt (Abb. 10). Diese liegen bei 326 nm, 308 nm, 286 nm sowie bei 257 nm.
Die Anregungen bei 326 nm und 308 nm sind über die HOMO → LUMO Anregung sowie eine HOMO-1 → LUMO Anregung definiert. Allerdings ist beim ersten angeregten Zustand die Anregung aus dem HOMO dominant, während im Zweiten die HOMO-1 Anregung dominiert.
Abb. 14) Grundzustands-KS Orbitale bei Anwendung der bh-lyp Dichtefunktionale bei einem NNPP- Diederwinkel von 74.92°.
Die HOMO → LUMO Anregung ist ein Metall-to-Ligand-charge-transfer- Übergang (MLCT) mit einer sehr kleinen Überlappung, wie er für die TADF notwendig ist (siehe Kapitel 3.Generation OLEDS S.5). Eine solche kleine Überlappung korreliert in der Regel aber auch mit einer sehr geringen Oszillatorstärke. Im Gegensatz dazu ist die HOMO-1 → LUMO Anregung eine Liganden-zentrierte Anregung. Das heißt, dass die Elektronendichte innerhalb des Liganden umverteilt wird. Dieser Übergang besitzt eine sehr große Überlappung der Elektronendichten
und damit auch eine sehr große Oszillatorstärke.
Der Übergang bei 286 nm besteht im wesentlichen aus einem HOMO
→ LUMO +2 sowie einem HOMO → LUMO +1 Übergang. In beiden Fällen handelt es sich hier um MLCT Übergänge, welche allerdings im Gegensatz zu den anderen beiden Anregungen die Elektronendichte nicht auf den PyrTet Liganden verschiebt, sondern auf den DPEPhos Liganden.
Beim letzten sehr wahrscheinlichen Übergang handelt es sich um den Übergang zwischen dem HOMO -1 und dem LUMO +4; +5 und +3 Orbital sowie den Übergängen HOMO -13; -14 und -16 in das LUMO Orbital. Alle Übergänge haben gemeinsam, dass die Elektronendichte in den Liganden sitzt, während die Ausgangsorbitale HOMO -13; -14 und -16 Elektronendichte im Bereich des Kupferatoms besitzen. Die Anregung besteht also ebenfalls sowohl aus MLCT Anregungen, als auch aus Liganden zentrierten Anregungen.
Die Energie des S0 liegt bei 77.438,353 eV. Der erste angeregte Singulett Zustand (326 nm) liegt 3,80 eV höher. Bei den anderen Zuständen beträgt die Differenz 4,03 eV (S3)(308 nm), 4,33 eV (S6)(286 nm) und 4.83 eV (S20)(257nm) mehr als im Grundzustand.
Der T1 Zustand besteht zum Hauptteil aus der HOMO-1 – LUMO Anregung aber auch aus den HOMO-1 → LUMO+4, LUMO+2 und LUMO+5 Anregungen (Reihenfolge entspricht der Oszillatorstärke). Bei dieser Anregung, wird Elektronendichte im PyrTet-Liganden um verteilt und auf die Phenylringe des DPEPhos verlagert. Hierbei ist das Kupferatom jedoch völlig unbeteiligt und auch die Phosphoratome haben keinen Anteil an diesem Zustand. Erst bei sehr geringen Anteilen an dem Zustand sind HOMO Orbitale zu finden, bei denen das Kupfer einen Anteil hat. Im Zuge dieser Triplettanregung, kann also von einer lokalen Anregung sowie einem ILCT gesprochen werden. Der erste Triplett-Zustand liegt bei -77.434,999 eV.
Die Energielücke zwischen dem ersten angeregten Singulett-Zustand (326 nm) und dem ersten angeregten Triplett-Zustand beträgt 446,27
meV, wenn die Geometrie identisch bleibt. Diese liegt damit deutlich über den angestrebten 120 meV Energielücke für einen guten TADF- Emitter.
4.2.1.1.2 Minimum des Singulett-Zustands bei einem NNPP-Winkel von 79,43°
Abb. 15) Minimum bei einem NNPP-Winkel von 79,43° aus zwei verschiedenen Perspektiven. In Abb. Ist deutlich zu erkennen, dass der PyrTet Ligand aufgrund der Dispersionswechselwirkung zur Seite hin abknickt. Der Ligand selber bleibt aber planar aufgebaut.
Bei diesem Winkel sind deutlich die Dispersionswechselwirkungen zwischen dem PyrTet Liganden und dem DPEPhos zu erkennen. Auch zwischen Zweien der Phenylringe, von denen einer ein verbrückender Ring ist, kommt es zu Dispersionswechselwirkungen. Im folgenden Text wird immer von der Rechnung mit Berücksichtigung dieser Wechselwirkungen ausgegangen, sofern dies nicht explizit anders vermerkt ist.
Abb. 16) Minimum bei 79,43° aus zwei verschiedenen Perspektiven ohne Berücksichtigung der Dispersionswechselwirkungen.
Die Grundzustandsenergie wird von der Geometrieoptimierung mit -77.405,744 eV angegeben. Die DFT/MRCI Rechnung gibt die Energie des Grundzustandes mit -77.438,353 eV. Hier unterscheiden sich die Energien der beiden Singulettminima um 0,04 eV. Beide Zustände sind von der energetischen Lage des Grundzustandes aus beinahe identisch.
Abb. 17) Absorptionsspektrum bei einem NNPP-Winkel von 79,43°
Bei der Geometrie mit diesem Winkel gibt es ebenfalls mehrere Anregungen, die eine hohe Oszillatorstärke besitzen. Die erste liegt bei einer Wellenlänge von 328 nm (S1 (+3,78 eV)) und die zweite bei 303 nm (S4 (+4.09 eV). Die Wellenlänge von 328 nm gehört zu der Anregung von Grundzustand in den ersten angeregten (Singulett-)Zustand. Die 303 nm können der Anregung in den vierten angeregten Zustand zugeordnet werden. Die 328 nm Anregung ist, wie beim 74,92° Minimum von der HOMO → LUMO Anregung definiert.
Auch die HOMO-1 → LUMO Anregung hat hier den zweitgrößten Anteil, auch wenn dieser wesentlich geringer ist, als bei dem anderen Minimum.
Abb. 18) Grundzustands-KS Orbitale bei Anwendung der bh-lyp Dichtefunktionale bei einem NNPP- Diederwinkel von 79,43°.
Die zweite Anregung bei 303 nm unterscheidet sich hingegen sehr stark. Diese entspricht der HOMO → LUMO+1 Anregung. Allerdings sind die weiteren Anteile anderer Anregungen grundlegend unterschiedlich von den Anregungen des 74,92° Minimums. Bei diesem Minimum ist der HOMO-1 → LUMO Übergang zusätzlich enthalten.
Diese IL Charge Transfer Anregung besitzt einen großen Überlapp, wodurch sich erklärt, weshalb der Übergang in den vierten angeregten Zustand, im Gegensatz zum anderen Singulett Minimum, so eine hohe Oszillatorstärke besitzt.
Der T1 Zustand bei dieser Geometrie besteht in erster Linie aus der HOMO-1 – LUMO Anregung, was einer Umverteilung der Elektronendichte im PyrTet Liganden entspricht. Desweiteren sind die Anregungen aus dem HOMO-1 in das LUMO+5 und das LUMO+1 Orbital in höherem Maße an der Anregung beteiligt. Die Anregung in das LUMO+5 Orbital entspricht dabei ebenfalls einer lokalen Anregung im PyrTet Liganden. Das LUMO+1 Orbital hat hingegen seine Elektronendichte zum größten Teil im Bereich des DPEPhos Liganden, sodass dieser Teil der Anregung einer Inter-Ligand-Charge-Transfer (ILCT) Anregung entspricht. Das Kupferatom ist erst bei den kleiner anteiligen Anregungen an diesem Zustand beteiligt. Die Energie des T1
Zustands liegt bei -77.434,964 eV.
Der energetische Abstand zwischen dem S1 Zustand und dem T1 bei identischer Geometrie beträgt 391 meV und liegt damit ebenfalls über der für optimale TADF Eigenschaften benötigte Energielücke von 120 meV. Allerdings zeigt das Energieprofil der Geometrieoptimierung, dass sich die Triplettenergien bei den Singulettminima nahe an einem Energiemaximum liegen. Hier muss also zunächst das Energieminimum der Triplettgeometrien berechnet werden, um bessere Aussagen treffen zu können.
4.2.1.2 Triplettgeometrien
Die Geometrieoptimierung zeigt zwei verschiedene Minima. Das erste Minimum liegt bei einem NNPP-Diederwinkel von 53,887° mit einer Energie von -77.403,235 eV und das zweite Minimum bei einem NNPP- Diederwinkel von 122,068° bei -77.403,273 eV. Diese Minima unterscheiden sich also schon aufgrund ihrer Geometrie deutlich von einander. Bei beiden Minima sind die Bindungen des Kupfers an die Stickstoffe um etwa 10 pm kürzer als bei den Singulettgeometrien, während die Bindungen zu den Phosphoratomen länger werden. Die Bindungswinkel N-Cu-N und P-Cu-P reagieren hingegen umgekehrt.
Außerdem wird der Winkel der Etherbrücke (C43-O-C75) um ein bzw. 2 Grad kleiner.
Minimum des Triplett-Zustands bei einem NNPP-Diederwinkel von 53,89°
Abb. 19) Molekül bei einem NNPP-Diederwinkel von 53,89°.
Dieses Triplettminimum weist der DFT/MRCI Rechnung zufolge im T1
Zustand eine Energie von -77.435.442 eV auf. Der Grundzustand (S0) dieser Geometrie läge bei -77.437,461 eV und der S1 Zustand bei -77.435,288 eV. Somit ergäbe sich eine (S1-T1) Energielücke von 154 meV. Diese liegt bereits deutlich näher an den für die TADF Eigenschaften benötigten 120 eV oder weniger, als dies bei den Grundzustandsgeometrien der Fall war.
Abb. 20) Grundzustands-KS Orbitale bei Anwendung der bh-lyp Dichtefunktionale bei einem NNPP- Diederwinkel von 53,89°.
Der T1 Zustand ist hauptsächlich durch die HOMO-LUMO Anregung definiert und hat somit vorwiegend MLCT Charakter. Zusätzlich beeinflusst noch die Anregung aus dem HOMO-3 und HOMO-1 den T1
Zustand. Die beiden Orbitale verfügen beide über größere Elektronendichte im Bereich der beiden Liganden aber auch im Bereich des Metalls. Da das LUMO nur auf dem PyrTet Liganden, sowie in sehr geringen Maßen auch auf dem Kupfer, über Elektronendichte verfügt, kann man insgesamt sowohl von MLCT Eigenschaften als auch ILCT sowie einer lokalen Anregung sprechen. Bei dieser Geometrie, wechselt das T1 Orbital seine energetische Lage. Bei der S0 Geometrie ist dieses
Orbital das T2 Orbital.
Der S1 Zustand dieser Geometrie besteht zum Großteil aus der HOMO – LUMO Anregung, sowie der HOMO-3 – LUMO Anregung. Erstere hat, wie oben schon beschrieben hauptsächlich MLCT Charakter und verlagert die Elektronendichte rund um das Kupferatom in den Pyridinring, sowie die nächst gelegenen Stickstoffatome. Die HOMO-3 – LUMO Anregung verschiebt ebenfalls Elektronendichte aus dem Kupfer.
Allerdings ist hier ebenfalls viel Dichte auf dem DPEPhos. Diese wird dann in den PyrTet Liganden verschoben. Insgesamt kann also von einer MLCT Anregung mit ILCT Einfluss gesprochen werden. Da alle beteiligten Orbitale Elektronendichte an den am Kupfer gebundenen Stickstoffatomen haben, besitzt diese Anregung einen guten Überlapp.
Minimum des Triplett-Zustands bei einem NNPP-Winkel von 122,07°
Abb. 21) Geometrie bei einem NNPP-Winkel von 122,068°.
Bei einem NNPP-Diederwinkel von 122,068° hat das Molekül im Triplett-Zustand eine Energie von -77.435,333 eV. Somit liegt er laut DFT/MRCI Rechnung 109 meV über dem Minimum bei 53,887°. Dieser Fakt ist deshalb besonders interessant, da die Geometrieoptimierung angibt, dass dieses Minimum etwa 30 meV unterhalb des Triplettminimum mit dem NNPP-Diederwinkel von 53,887° liegt. Die
Energie des Singulett Grundzustands bei dieser Geometrie ist angegeben mit -77.437,153 eV und die des S1 Zustandes mit -77.435,164 eV. Mit 169 meV ist die Energiedifferenz ΔΕ(S1-T1) zwar um 35 meV größer als die des Minimums bei 53,887° aber weiterhin über 200 meV geringer als die Minima der Singulettzustände.
Abb. 22) Grundzustands-KS Orbitale bei Anwendung der bh-lyp Dichtefunktionale bei einem NNPP- Diederwinkel von 122,068°.
Der erste angeregte Triplett-Zustand besteht wiederum zum größten Teil aus der HOMO-LUMO Anregung. Des weiteren beeinflussen die Anregungen vom HOMO-2 und HOMO-3 in das LUMO den Zustand noch relativ stark. Die Elektronendichte der HOMO-2 und HOMO-3 liegt wieder zum großen Teil auf dem DPEPhos Ligand und auf dem Kupfer, während die Dichte des LUMO am PyrTet Liganden liegt. Da aber alle drei beteiligten HOMO-X auch etwas Elektronendichte am PyrTet Liganden haben, ist der gesamte Überlapp groß. Der zweite angeregte Triplett-Zustand setzt sich aus den Anregungen aus dem HOMO in das LUMO+1 sowie das LUMO+2 und das LUMO+14 zusammen. Des weiteren geht auch die Anregung aus dem HOMO-1 in das LUMO+1 in diesen Zustand mit ein. Den größten Anteil an der Anregung stellt die Anregung vom HOMO in das LUMO+1 Orbital.
Danach folgen, in anteiliger Reihenfolge, die Anregungen in das LUMO+14, das LUMO+1 und das LUMO+2. Bei dieser Anregung wird die Elektronendichte von dem PyrTet Liganden und dem Kupfer sowie den Phosphoratomen, vorwiegend in die Phenylringe verlagert. Für den Überlapp sorgt wieder die Anregung in das LUMO+14 Orbital, sodass die hohe Oszillatorstärke gegeben ist. Vom energetischen Standpunkt liegt der T2 Zustand bei -77.438,944 eV und damit 0,911 eV über dem T1
Zustand.
Der S1 Zustand setzt sich aus der HOMO – LUMO Anregung sowie zu geringen Teilen aus den Anregungen HOMO-2 und HOMO-3 in das LUMO Orbital zusammen. Bei der HOMO – LUMO Anregung, handelt es sich um eine klassische MLCT Anregung. Es wird die Elektronendichte rund um das Kupferatom in den PyrTet Liganden um verteilt. Die Überlappung ist hierbei sehr klein. Die Orbitale HOMO-2 und HOMO-3 umfassen jeweils zwei der Phenylringe, sowie das Kupferatom und die Stickstoffatome des PyrTet Liganden. Aufgrund der Elektronendichte auf dem Letzteren sorgen die Anregungen für eine gute Überlappung und somit eine höhere Oszillatorstärke.
4.2.1.3 Übersicht der berechneten Minima und Vergleich mit dem Experiment
Wie in der unten stehenden Tabelle ersichtlich wird, unterscheiden sich die Grundzustandsgeometrien aus energetischer Sicht nur wenig von einander. Da mit den bei dieser Arbeit zur Verfügung stehenden Mitteln keine S1-Geometrien berechnet werden können, muss hier davon ausgegangen werden, dass diese Geometrie und die Triplettgeometrien identisch sind. Da für dieses Molekül bereits TADF Eigenschaften nachgewiesen wurden[9] und diese Art von Lumineszenz auf der energetischen Nähe des S1 und T1 Zustands aufbaut, kann diese Annahme als gültig angesehen werden.
Abb. 23) Die Formel für die Fluoreszenzraten. Als Wert für den Vorfaktor wurde der im Buch genannte Wert von 2.062*10-6 verwendet[13], dazu wurden die Zustandsenergien in Wellenzahlen und das Dipolmoment in atomic units verwendet.
Alle berechneten Fluoreszenzraten liegen deutlich über der gemessenen TADF-Rate von 86.957 s-1 [9]. Dies ist allerdings zu erwarten, da die verzögerte Fluoreszenz zunächst den RISC Vorgang durchlaufen muss. Der gemessene S1 → T1 ISC Vorgang ist hingegen mit einer Rate von 3.7*1010 s-1 deutlich schneller als die Fluoreszenz.
Dieser Umstand erklärt, warum im Experiment keine prompte Fluoreszenz gemessen werden konnte. Die berechneten Raten sind allerdings aufgrund der möglichen Ungenauigkeit des S1 Zustandes ebenfalls recht ungenau. Um eine bessere Genauigkeit zu erzielen, sollte der S1 Zustand mithilfe des TD-DFT Verfahrens untersucht werden.
Abb. 24) Absorptionsspektrum in CH2Cl2 bei Raumtemperatur und Emissionsspektrum des festen Zustands[12].
Leider ist es, vermutlich aufgrund der starken Verzerrung der Triplettgeometrien, im Rahmen dieser Arbeit nicht möglich, ein Emissionsspektrum zu modellieren. Die Tabelle zeigt jedoch auf, dass ein solches Spektrum, im Rahmen des adiabatischen Übergangs, bei 418 nm (S0-Geometrie mit NNPP-Winkel von 74.92°) oder 412 nm (NNPP-Winkel = 79.43°) beginnen würde, wenn die Triplettgeometrie mit einem NNPP-Diederwinkel von 53,89° als Grundlage genommen wird. Bei einer Triplettgeometrie von 122,07° würde das Spektrum bei 402 nm (NNPP-Winkel = 74,92°) bzw. 397 nm beginnen. Das Experiment zeigt, dass die Emission bei einer Wellenlänge von etwas unter 400 nm beginnt.
Das Maximum liegt bei einer Wellenlänge von 571 nm (53,89°) oder 623 nm (122,07°). In beiden Fällen zeigt sich eine (im Falle der vertikalen Anregung deutliche) rot-Verschiebung und damit eine Unterschätzung der S1 → S0 Energiedifferenz. Auch hier gilt, dass der S1 Zustand mithilfe der TD-DFT Methode genauer untersucht werden
sollte. Die in der Abbildung eingezeichnete Messung bei 77K stellt vermutlich die Phosphoreszenz dar, da bei tiefen Temperaturen, wie in Abbildung 5 auf Seite 7 gezeigt, die TADF-Emission nahezu auf null sinkt und lediglich Phosphoreszenz entsteht. Dazu passt auch, dass die 77K Kurve im Vergleich zu der 300K Kurve rot verschoben ist und die emittierten Lichtquanten somit weniger Energie besitzen. Auch hier zeigt sich bei den berechneten Daten eine Rotverschiebung sowohl am Beginn der Emission (Phosphoreszenz mit ZPE) als auch bei dem berechneten Maximum (der vertikale Übergang entspricht ΔΕ(T1-S0) )
Tab. 1) Übersicht über gefundene Minima mit positivem NNPP-Diederwinkel. Es wird angenommen, dass die S1-Geometrie mit der jeweiligen Geometrie der Spalte übereinstimmt.
Grundzustandsgeometrie Triplettminimumgeometrie
NNPP-Winkel (°) 74,92 79,43 53,89 122,07
E(S0) (eV)
-77.438,353 eV 0,040 0,000 0,892 1,200
E(T1) (eV)
-77.438,353 eV 3,354 3,389 2,911 3,020
E(S1) (eV)
-77.438,353 eV 3,800 3,780 3,065 3,189
ΔΕ(S1-T1) (meV) 446 391 134 169
ΔΕ(S1-S0) (eV) 3,760 3,780 2,173 1,989
ΔΕ(S1-S0) (nm) 330 328 571 623
ΔΕ(T1-S0) (nm) 374 366 614 681
Übergangsdipol moment S1-S0
(Debye)
3,087 2,930 1,446 1,293
ZPE (eV) 17,575 17,572 17,516 17,509
Übergänge sind ab hier adiabatisch und bis hier vertikal.
Fluoreszenz S1 → S0(74,92°) (nm)
332 410 394
mit ZPE (nm) 332 418 402
Fluoreszenz S1 → S0(79,43°) (nm)
326 405 389
mit ZPE (nm) 326 412 397
Phosphoreszenz T1 → S0(74,92°) (nm)
370 432 416
mit ZPE (nm) 371 441 420
Phosphoreszenz T1 → S0(79,43°) (nm)
370 426 410
mit ZPE (nm) 369 434 415
Fluoreszenzrate
(s-1) 546.874 335.234
4.2.2 Geometrien mit negativen NNPP-Diederwinkeln
Die Geometrieoptimierung zeigt, dass es nicht möglich ist, den Interplanarwinkel des Moleküls auf die Werte null Grad und 180 Grad zu bringen, ohne die Bindung zwischen dem Kupferatom sowie dem Pyridinring aufzulösen. Dies führt dazu, dass eine Drehung eines Liganden um 180 Grad, zu einem echten Diastereomer führt. Bei diesem Stereomer gibt es eine weitere Singulett Minimum-Geometrie sowie zwei Geometrien mit einem energetischen Minimum des Triplett- Zustands. Diese Geometrien haben einen Diederwinkel von -57,238°;
-85,066° und -121,695°. Offensichtlich, sind diese Winkel denen des gegenüber liegenden Verlaufs sehr ähnlich. Die Singulett-Minimum Geometrie liegt bei -85,066°. Die beiden anderen Geometrien sind Minima der Triplettkurve.
4.2.2.1 Grundzustandsgeometrie
Abb. 25) Aufbau des Moleküls bei einem NNPP-Diederwinkel von -85,066°.
Bei einem NNPP-Winkel von -85,066° liegt die Energie des Moleküls laut Geometrieoptimierung bei -77.405,661 eV. Die DFT/MRCI Rechnung gibt die Energie des Grundzustands hingegen mit -77.438,314 eV an. Diese Energie liegt 0.04 eV über dem niedrigsten Minimum des anderen Torsionswinkelscans und ist somit nahezu identisch mit der Energie des Moleküls bei einem Diederwinkel von
74,92°. Die Energie des ersten Triplettzustandes liegt hier bei -77.434,931 eV und der S1 bei -77.434,446 eV. Die Energielücke beträgt somit 485 meV. Diese ist somit sogar noch größer als bei den anderen beiden Singulett-Zustände.
Auffällig ist, dass es auch bei diesem Winkel zu Dispersionswechselwirkungen wie bei der Geometrie mit einem NNPP- Diederwinkel von 79,43° kommt, was auch hier zu einem Abknicken des Pyrtet Liganden in Richtung des Phenylringes führt.
Abb. 26) Anregungsspektrum der Grundzustandsgeometrie mit dem NNPP-Diederwinkel von -85,066°
Das Spektrum zeigt deutlich, dass die ersten drei Anregungen bei dieser Geometrie die höchste Oszillatorstärke besitzen aber auch die energetisch wesentlich höher liegenden Übergänge in die Zustände S15
und S19 besitzen eine relativ hohe Oszillatorstärke.
Der S0 → S1 Übergang zeichnet sich im wesentlichen durch den Wechsel HOMO → LUMO aus. Hier wird Elektronendichte in den Pyrtet Liganden vom Kupferatom verlagert, sowie aus dem Tetrazolring und den Phenylring, welcher mit dem Pyrtet wechselwirkt. Die Überlappung der beiden beteiligten Orbitale ist relativ gut, da beide Orbitale über
Elektronendichte im Bereich des Tetrazolringes verfügen. Des weiteren trägt auch die Anregung vom HOMO-3 in das LUMO Orbital einen Teil zu der Gesamtkonfiguration des Zustandes bei. Diese Anregung ist beinahe identisch, mit der HOMO → LUMO Anregung. Die zusätzliche Elektronendichte auf dem Stickstoff des Pyridinringes, sorgt allerdings für eine bessere Überlappung des Übergangs und steigert somit die Oszillatorstärke. Insgesamt, kann der S0 → S1 Übergang im wesentlichen als MLCT Übergang betrachtet werden.
Abb. 27) Grundzustands-KS Orbitale bei Anwendung der bh-lyp Dichtefunktionale bei einem NNPP- Diederwinkel von -85,066°.
Der zweite wahrscheinliche Übergang ist der Übergang aus dem Grundzustand in den S2 Zustand. Hierbei sind die Anregungen HOMO-1
→ LUMO; HOMO → LUMO; HOMO-3 → LUMO und HOMO-1 → LUMO+1 die am stärksten beteiligten Übergänge. Das HOMO-1 Orbital besitzt ausschließlich auf dem Pyrtet Liganden Elektronendichte. Diese wird im Zuge der HOMO-1 → LUMO Anregung lediglich im Liganden um verteilt. Zusätzlich wird ein wenig Dichte in einen der nicht verbrückten Phenylringe geschoben. Im wesentlichen kann also von einer lokalen Anregung gesprochen werden. Der HOMO → LUMO Übergang wurde bereits beim S1 Zustand besprochen und hat vorwiegend MLCT Charakter. Das gleiche gilt für den HOMO-3 → LUMO Übergang. Hier ist allerdings auch der Anteil der lokalen Anregung etwas höher als bei der Anregung aus dem HOMO. Im Zuge der Anregung des HOMO-1 Orbitals in das LUMO+1 Orbital, wird Elektronendichte aus dem Pyrtet Liganden in die Phenylringe des DPEPhos um verteilt. Hier muss also ebenfalls von einer lokalen Anregung gesprochen werden.
Die größte Oszillatorstärke hat der Wechsel in den dritten angeregten Singulett-Zustand.
In diesem Fall besteht die Anregung im wesentlichen aus den Übergängen HOMO → LUMO+1; HOMO-1 → LUMO; HOMO → LUMO und HOMO-2 → LUMO. Im Rahmen der HOMO → LUMO+1 Anregung wird Elektronendichte von Kupfer in das DPEPhos verlagert sowie etwas Elektronendichte im PyrTet Liganden um verteilt. Im Wesentlichen ist hier von einem MLCT Effekt zu sprechen. HOMO-1 → LUMO verteilt seine Elektronendichte im Pyrtet Liganden um.
Außerdem wird etwas etwas Elektronendichte in einen der Phenylringe verlagert. Diese Anregung hat eine sehr starke Überlappung und ist als lokale Anregung zu betiteln. Die HOMO → LUMO Anregung ist bereits weiter Oben beschrieben. Sie hat einen weitestgehenden MLCT Übergang. Die Überlappung der beiden Orbitale ist relativ gut. Die letzte verbliebene wichtige Anregung des S0 → S3 Übergangs betrifft HOMO-2
→ LUMO. Hier wird die Elektronendichte aus dem Kupfer und dem DPEPhos Liganden in den Pyrtet Liganden verlagert. Außerdem wird ein Teil der Dichte im DPEPhos Liganden innerhalb dieses um verteilt.
Allerdings findet man bei diesem Übergang die einzige Überlappung im
Bereich des Tetrazols. Trotzdem ist dieser Zustand aufgrund der vielen starken Überlappungen der verschiedenen Anregungen sehr stark erlaubt.
Der erste Triplett-Zustand bei dieser Geometrie wird beinahe ausschließlich aus dem HOMO-1 Orbital heraus angeregt. Die LUMO Orbitale in welche angeregt wird, sind hierbei die Orbitale LUMO;
LUMO+1 und LUMO+5. Die Elektronendichte liegt also vor der Anregung auf dem Pyrtet Liganden und wird im wesentlichen innerhalb dieses neu verteilt (lokale Anregung). Außerdem wird Elektronendichte in den DPEPhos Liganden verlagert (ILCT). Insgesamt ist hier die Überlappung der beteiligten HOMO und LUMO Orbitale sehr gut.
Dieser Zustand sollte also für einen Triplett-Zustand sehr stark erlaubt sein, sofern andere Faktoren die Oszillatorstärke nicht verringern.
4.2.2.2 Triplettgeometrien
4.2.2.2.1 Triplettminimumgeometrie bei einem NNPP-Diederwinkel von -57,238°
Abb. 28) Triplettminimumgeometrie mit einem NNPP-Diederwinkel von -57,238°.
Bei einem NNPP-Diederwinkel von -57,238° liegt die Energie lauf Geometrieoptimierung bei -77.403,017 eV. Das DFT/MRCI gibt die Energie des T1 Zustandes hin gegen mit -77.435.177 eV an. S0 und S1
liegen bei -77.437,286 eV und -77.435,029 eV. Somit liegt die Energielücke bei 148 meV. Dieser Wert liegt nur knapp (14 meV) oberhalb der Energielücke des Minimums bei dem NNPP-Diederwinkel von 53,89°.
Der T1 Zustand wird mithilfe der HOMO-LUMO Anregung gebildet.
Außerdem übt auch die HOMO-2 → LUMO Anregung Einfluss auf diesen aus. Der Übergang in den T1 Zustand ist also ein typischer MLCT Übergang.
Abb. 29) Grundzustands-KS-Orbitale bei Anwendung der bh-lyp Dichtefunktionale bei einem NNPP- Diederwinkel von -57,238°
Der erste angeregte Singulett-Zustand hat bei dieser Geometrie eine Energie von -77.435,029 eV. Da der Grundzustand eine Energie von -77.437,286 eV hat, beläuft sich die S1 → S0 Energiedifferenz auf 2,257 eV, was einer Emission von grünem Licht mit 549nm Wellenlänge entspricht. Der S1 Zustand besteht im wesentlichen aus 2 verschiedenen Anregungen. Die wichtigste Anregung ist die HOMO → LUMO Anregung, bei welcher die Elektronendichte aus dem Kupfer in das Pyrtet verschoben wird. Außerdem hat die Anregung
HOMO-2 → LUMO einen großen Einfluss auf diesen Zustand. Bei dieser Anregung wird die Elektronendichte ebenfalls vom Kupfer aber
auch vom Tetrazolring des Pyrtet Liganden in dem Liganden verteilt.
Dabei liegt der größte Teil der Elektronendichte des LUMO auf dem Pyridinring.
4.2.2.2.2 Triplettminimum bei einer Geometrie mit einem NNPP- Diederwinkel von -121,695°
Abb. 30) Geometrie bei einem NNPP-Diederwinkel von -121,695°.
Diese Geometrie hat eine Grundzustandsenergie von -77.437,237 eV.
Der S1 Zustand liegt mit -77.435,189 eV 2,048 über diesem und der T1
Zustand bei -77.435,353 eV. Damit ergibt sich ein ΔΕ(S1-T1) von 164 meV, was noch einmal 16 meV mehr sind, als bei der Geometrie mit dem NNPP-Diederwinkel von -57,238°. Wäre an der Fluoreszenz lediglich diese Geometrie beteiligt, so hätte das emittierte Licht eine Wellenlänge von 605 nm und wäre somit orange.
Die T1 Anregung ist wieder hauptsächlich durch den HOMO → LUMO Übergang definiert und hat somit MLCT Charakter. Außerdem hat noch der HOMO-2 → LUMO Übergang noch Einfluss auf die auf die Anregung. Auch diese hat MLCT Charakter allerdings hat das HOMO- 2 auch Elektronendichte auf dem DPEPhos und im Pyrtet, sodass auch ILCT und lokale Anregung vorhanden ist.
Abb. 31) Grundzustands-KS Orbitale bei Anwendung der bh-lyp Dichtefunktionale bei einem NNPP- Diederwinkel von -121,695°.
Der erste Angeregte Singulett-Zustand (S1) entsteht ebenfalls über die HOMO → LUMO Anregung. Zusätzlich ist auch noch die HOMO-2 → LUMO Anregung zu berücksichtigen. Das HOMO-2 besitzt Elektronendichte sowohl auf dem DPEPhos Liganden, als auch auf dem Kupfer und dem Pyrtet. Diese Anregung kann also als MLCT Anregung in das Pyrtet bezeichnet werden, mit zusätzlichen lokalen und ILCT Einflüssen.
4.2.2.3 Übersicht der berechneten Minima und Vergleich mit dem Experiment
Für die berechneten Fluoreszenzwellenlängen muss hier von der gleichen Näherung ausgegangen werden, wie bei der Tabelle 1 in Kapitel 4.2.1.3, da auch hier mit den zur Verfügung stehenden Mitteln keine S1 Geometrie berechnet werden kann. Allerdings zeigt hier die Phosphoreszenz bei einem NNPP-Diederwinkel eine weniger starke Rotverschiebung, als dies bei allen anderen Triplettgeometrien der Fall ist.
Abb. 32) Absorptionsspektrum der verschiedenen Grundzustandsgeometrien. In schwarz sind experimentelle Daten aus Abbildung 24 eingezeichnet. Diese wurden in CH2Cl2 bei Raumtemperatur aufgenommen.
Für die berechneten Fluoreszenzraten, gilt auch hier, das diese deutlich größer sind, als die experimentell ermittelte Fluoreszenzrate. Auch die berechnete Fluoreszenz, ist im Vergleich zu den experimentellen Daten rot verschoben. Bei einem Emissionsspektrum mit den, aus den Geometrien mit negativen NNPP-Winkeln, berechneten Werten als Grundlage würde die Emission bei 402 nm bzw. 425 nm (-122°
Geometrie) beginnen. Das Maximum läge dann bei 549 nm bzw. 605 nm.
Tab. 2) Übersicht über gefundene Minima mit negativem NNPP-Diederwinkel. Es wird angenommen, dass die S1-Geometrie mit der jeweiligen Geometrie der Spalte übereinstimmt.
Grundzustandsgeometrie Triplettminimumgeometrie
Winkel (°) -85,066 -57,238 -121,695
E(S0) (eV)
-77.438,353 eV 0,039 1,067 1,116
E(T1) (eV)
-77.438,353 eV 3,422 3,176 3,000
E(S1) (eV)
-77.438,353 eV 3,907 3,324 3,164
ΔΕ(S1-T1) (meV) 485 148 164
ΔΕ(S1-S0) (eV) 3,868 2,257 2,015
ΔΕ(S1-S0) (nm) 321 549 605
ΔΕ(T1-S0) (nm) 366 587 658
Übergangsdipol moment S1-S0
(Debye)
2,434 1,368 2,220
ZPE (eV) 17,570 17,515 17,524
Übergänge sind ab hier adiabatisch und bis hier vertikal.
Fluoreszenz S1
→ S0(-85°) (nm) 377 397
mit ZPE (nm) 384 403
Phosphoreszenz T1 → S0(-85°) (nm)
395 419
mit ZPE 402 425
Fluoreszenzrate
(s-1) 548.215 313.606
Das Absorptionsspektrum zeigt, dass die Struktur des experimentellen Spektrum relativ nahe an den Strukturen der berechneten Spektren liegt. Lediglich im Bereich bis 260nm und ab 330 nm fallen die berechneten Spektren stark ab, da lediglich 20 angeregte Singulettzustände berechnet wurden und Übergänge in Triplettzustände aufgrund der fehlenden Spin-Bahn-Kopplung verboten sind.
Interessanterweise verläuft ein Maximum des experimentellen Spektrums von 260 nm bis 290 nm und deckt damit das erste Maximum aller drei berechneten Spektren ab, sowie das zweite Maximum der Spektren der Geometrien mit dem NNPP-Winkel von 75° und -85°. Es könnten also alle drei Geometrien an diesem experimentellen Spektrum beteiligt sein. Der weitere Verlauf der Spektren bis etwa 330 nm liegt dicht beieinander.
Im Vergleich untereinander ist der Verlauf des Spektrums bei einem NNPP-Winkel von 79° leicht rot verschoben im Vergleich zu dem Spektrum des Geometrieminimums bei einem NNPP-Winkel von 75°.
Zeitgleich fallen die Unterschiede in der Oszillatorstärke bei einem Winkel von 79° deutlich geringer aus, als bei dem 75°-NNPP-Winkel Spektrum und sind gleichmäßiger über das gesamte Spektrum verteilt.
Bei einem NNPP-Diederwinkel von -85° ist der Peak bei 263 nm im Vergleich zur 75° Geometrie leicht rot verschoben, während er verglichen mit dem Peak des 79° NNPP-Winkel Spektrums blau verschoben ist. Ansonsten sind auch hier die Peaks gleichmäßig über das Spektrum verteilt. Im Gegensatz hierzu steht das 75° NNPP-Winkel Spektrum, bei welchem der erste und zweite Peak lediglich etwa 10 nm von einander entfernt sind. Auffällig ist außerdem, dass alle drei berechneten Maxima über genau drei Peaks verfügen.
4.3 Strukturvergleich
Ein Vergleich der Minimumgeometrien mit den Kristalldaten des Electronic Supplementary Material (ESI)[12] zeigt, dass die Geometrie mit einem NNPP-Winkel von 74,92° am ehesten den Bindungslängen und dem NNPP-Winkel entspricht. Die berechneten Bindungslängen unterscheiden sich bei dieser Geometrie um maximal 3 pm (mit Ausnahme der C-H Bindungen) von den experimentellen Daten. Der größte Unterschied betrifft die beiden Bindungen zwischen Kupfer und Stickstoff. Hier ist die Bindung an den Tetrazolring im Vergleich zu den berechneten Werten stärker (3 pm kürzer) und die Bindung zum Pyridin um 3 pm länger. Die dritte Bindung, welche sich bei dieser Geometrie
![Abb. 4) Strahlungsabklingrate (als Indikator für den Überlapp) aufgetragen gegen ΔΕ(S 1 -T 1 ) für verschiedener Kupfer Komplexe mit TADF Eigenschaften [5]](https://thumb-eu.123doks.com/thumbv2/1library_info/4530255.1596175/18.892.169.666.375.725/strahlungsabklingrate-indikator-überlapp-aufgetragen-verschiedener-kupfer-komplexe-eigenschaften.webp)
![Abb. 5) Beispiel TADF+Phosphoreszenz für Cu 2 Cl 2 (2-Methyl,5-(Diphenylphosphino)pyridin) 2 [5]](https://thumb-eu.123doks.com/thumbv2/1library_info/4530255.1596175/19.892.128.763.663.989/abb-beispiel-tadf-phosphoreszenz-für-methyl-diphenylphosphino-pyridin.webp)