Quantenchemische Charakterisierung von Cu(I)-Komplexen gemischter P,N-Liganden auf
[2.2]Paracyclophanbasis: Variation der Pyridyleinheit
Abschlussarbeit zur Erlangung des akademischen Grades des Bachelor of Science im Fach Chemie
Vorgelegt von Andrea Garnica Tapia
März 2018 durchgeführt am
Institut für Theoretische Chemie und Computerchemie Mathematisch Naturwissenschaftliche Fakultät
Heinrich-Heine-Universität Düsseldorf
1. Gutachterin: Prof. Dr. Christel M. Marian 2. Gutachter: Prof. Dr. Christian Ganter
Hiermit erkläre ich, dass ich die vorliegende Bachelorarbeit selbstständig und nur mit den angegebenen Quellen erstellt habe.
Wörtliche oder inhaltliche Stellen, sowie Abbildungen und Formeln, die aus anderen Werken entnommen worden sind, sind als solche gekennzeichnet.
Düsseldorf, den 2018
__________________________
Unterschrift (Andrea Garnica Tapia)
Danksagung:
An dieser Stelle möchte ich mich gerne bei allen Personen bedanken, die mich während der Anfertigung dieser Bachelorarbeit unterstützt und motiviert haben.
Als erstes gebührt mein Dank Frau Prof. Dr. C. Marian, die mir die Möglichkeit gegeben hat meine Bachelorarbeit am Institut der theoretischen Chemie zu schreiben. Für die hilfreichen Anregungen und die konstruktive Kritik bei der Erstellung dieser Arbeit möchte ich mich herzlich bedanken.
Des Weiteren danke ich Herrn Prof. Dr. C. Ganter dafür, dass er sich als Zweitkorrektor für diese Arbeit zur Verfügung gestellt hat.
Ich möchte mich auch beim Arbeitskreis der Theoretischen Chemie und Computerchemie für die nette Atmosphäre und freundliche Aufnahme bedanken. Mein besonderer Dank geht an Jelena Föller, da Sie sich dazu bereit erklärt hat, mich während dieser Arbeit zu betreuen, was sie mit viel Geduld und Herzlichkeit tat.
Ebenfalls möchte ich mich herzlich bei Anna Keller bedanken, die mich sprachlich unermüdlich unterstützt hat.
Außerdem möchte ich mich bei meinen Eltern, Clara Arlene Tapia de Garnica und Luis Alberto Garnica Rosado, bedanken, die an mich geglaubt haben und mir in schwierigen Zeiten immer Mut gegeben haben.
Weiterhin möchte ich der Familie Boog meinen großen Dank zukommen lassen, weil sie mir die Tür öffnete, sodass ich in Deutschland studieren konnte.
Zum Schluss möchte ich mich bei meinem Ehemann, Holger Keller, herzlich bedanken, der mich trotz großer Hürden immer wieder ermutigte weiter zu machen und davon überzeugte, dass ich es schaffen könnte.
Andrea Garnica Tapia de Keller, Düsseldorf, 06.03.2018
Zusammenfassung:
In der vorliegenden Arbeit wurde der Komplex (Sp)-[κ2-N,P-4-(2`-Pyridyl)-1,2- diphenylphosphinyl[2.2]paracyclophan]chlorokupfer(I) quantenchemisch untersucht und charakterisiert, um die Eignung als TADF-Emitter zu ermitteln. Hierfür wurden die S0- und die T1-Geometrie optimiert und mit Hilfe einer DFT/MRCI-Berechnung die Anregungs- und Emissionsenergien erfasst. Anhand der Energieabstände, der Oszillatorstärken und der Anregungsenergien lässt sich ermitteln, dass sich dieser Komplex tendenziell nicht als TADF- Emitter eignet. Darüber hinaus wurden unterschiedliche Substitutionen vorgenommen und ihr Einfluss auf die quantenchemischen Eigenschaften des Komplexes analysiert. Es konnte anhand der Ergebnisse kein deutlicher Trend festgestellt werden. Solche substituierten Komplexe, bei denen die Substitution an der meta-Stellung des Pyridylrings erfolgte, zeigen ebenfalls, dass die Wahrscheinlichkeit für einen TADF-Übergang gering ist. Es deutet sich allerdings an, dass mit zunehmenden negativen I-Effekt die TADF-Eigenschaft positiv beeinflusst wird. Anders verhält es sich bei der Substitution am Paracyclophan, wo die TADF- Fähigkeit verringert wird.
Abstract:
In this thesis, the complex „(Sp)-[κ2-N,P-4-(2`-Pyridyl)-1,2- diphenylphosphinyl[2.2]paracyclophan]chlorokupfer(I)“ was examined and characterized from a quantum chemical perspective in order to find out about its suitability as TADF emitter. Both geometries, S0 and T1, were optimized and based on a DFT/MRCI calculation the excitation energy and emission energy identified. Based on the energy distances, the oscillator strengths, as well as the excitation energies, it can be seen that this complex is most likely not suitable as TADF emitter. Furthermore, a varying set of substitutes was applied and their impact on the quantum chemical properties of the complex was analysed. The results show no specific trend.
Substituted complexes with a substituent at the meta-position of the pyridylring confirm that the probability for a TADF transition is low. Nevertheless, an increasing negative inductive effect indicates a positive influence the TADF quality. In turn, a substitution at paracyclophane reduce the TADF ability.
Inhaltsverzeichnis:
Abkürzungsverzeichnis
1 Einleitung: ... 1
1.1 Lumineszenz: ... 2
1.2 OLEDs: ... 3
1.2.1 Erste OLED Generation: ... 3
1.2.2 Zweite OLED Generation: ... 3
1.2.3 Dritte OLED Generation: ... 4
1.3 TADF: ... 4
2 Theorie: ... 6
2.1 Dichtefunktionaltheorie (DFT) ... 6
2.2 Hybridfunktionale: ... 7
2.2.1 Das PBE0-Funktional ... 7
2.2.2 Das BH-LYP-Funktional... 8
2.3 DFT/MRCI ... 8
2.4 Effektives Kernpotential (ECP) ... 9
3 Durchführung und technische Details zur Rechnung ...10
4 Darstellung der Ergebnisse: ...12
4.1 Standard Cu-Komplex: ...12
4.1.1 Struktur: ...12
4.1.1.1 Vergleich mit den Literaturwerten………15
4.1.1.2 Vergleich mit der T1-Geometrie……….. 15
4.1.2 DFT/MRCI: ...15
4.2 Cu-Komplex mit tert-Butyl-Substituent: ...21
4.2.1 Struktur: ...21
4.2.2 DFT/MRCI: ...22
4.3 Cu-Komplex mit Methyl-Substituent: ...23
4.3.1 Struktur: ...23
4.3.1.1 Vergleich mit den Literaturwerten……….. 23
4.3.1.2 Vergleich mit der T1-Geometrie……….. 24
4.3.2 DFT/MRCI: ...25
4.4 Cu-Komplex mit Fluor-Substituent am Pyridylring: ...26
4.4.1 Struktur: ...26
4.4.2 DFT/MRCI: ...27
4.5 Cu-Komplex mit Fluor-Substituent am Paracyclophan: ...28
4.5.1 Struktur ...28
4.5.2 DFT/MRCI: ...29
4.6 Vergleich zwischen Standard Cu-Komplex und den substituierten Cu-Komplexen: ....31
4.6.1 Struktur: ...31
4.6.2 DFT/MRCI: ...32
5 Zusammenfassung und Ausblick: ...39
6 Literaturverzeichnis: ...41
7 Anhang: ...43
Abkürzungverzeichnis:
LED light emitting diodes
OLED organic light emitting diodes
TADF thermally activated delayed fluorescence
s. siehe
Abb. Abbildung
d.h. das heißt
S Singulett
T Triplett
ISC intersystem crossing
RISC reverse intersytem crossing
PHOLED phosphorescent organic light emitting diode
CT charge transfer
Eng. englisch
BH-LYP Becke Half and Half Lee-Yang-Parr
PBE0 Perdew, Burke and Ernzerhof, hybridisiert durch Adamo
DFT Dichtefunktionaltheorie
UDFT uneingeschränkten Dichtefunktionaltheorie MRCI Multi Reference Configuration Interaction
ECP effective core potential
Cu Kupfer
pm Picometer
Tab. Tabelle
HOMO Highest Occupied Molecular Orbital
LUMO Lowest Occupied Molecular Orbital
Vgl. Vergleich
eV Elektronenvolt, Einheit der Energie
XMLCT Halide/Metal-to-Ligand Charge Transfer
I-Effekt Induktiver Effekt
Pcy Paracyclophan
Pyr Pyridyl
1 1 Einleitung:
Seit mehreren Jahren gewinnt die Anwendung und Weiterentwicklung von neuen Leuchtmitteln an globaler Bedeutung. Die LED (Light Emitting Diode) wird besonders wertgeschätzt, da diese im Gegensatz zu einer herkömmlichen Glühlampe keine Wärme produziert. Sie verspricht außerdem bessere Leistungsfähigkeit, längere Lebensdauer und eine bessere Umweltverträglichkeit. [1]
Im stark wachsenden Sektor der Technologieentwicklung aktueller Displays spielen die organischen Leuchtdioden (OLED) eine zunehmend wichtige Rolle. [2] Es wird dabei auch untersucht, inwiefern man diese in der allgemeinen Lichterzeugung (Beleuchtung) anwenden kann. Die OLEDs sind, anders als LEDs, nicht nur kristalline Punktlichtquellen, sondern eine der wenigen Lichtquellen mit einem flächigen Erscheinungsbild. OLEDs weisen daher einen großen Anwendungsspielraum auf und eignen sich dabei für eine Vielzahl von Produkten. Sie besitzen auch eine starke Flexibilität und eine freie Auswahl der Farben. [3] Bisher sind die Kosten für die Produktion allerdings noch sehr hoch. Deswegen forschen derzeit viele Wissenschaftler, um die Herstellungskosten zu reduzieren und die maximale Ausnutzung dieser organischen Leuchtdioden zu erhöhen. Das Forschungsgebiet verbreitet sich daher seit einigen Jahren zunehmend.
Emitter, die sich die thermisch aktivierte verzögerte Fluoreszenz (eng. thermally activated delayed fluorescence, TADF) zu Nutze machen, sind eine relativ neue Form von OLEDs. Sie versprechen eine noch bessere Effizienz. Zudem wird auf die Anwendung von Schwermetallen verzichtet. [4]
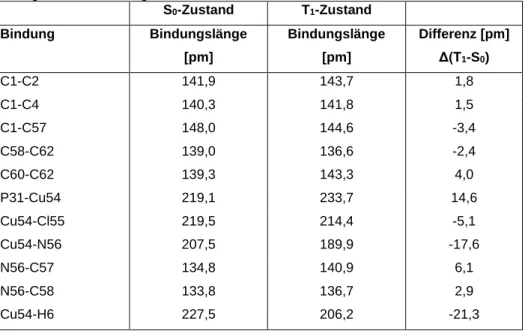
In vielen Fällen zeigen einwertige Kupfer-Komplexe TADF Eigenschaften [5]. In Rahmen dieser Arbeit wird der Cu-Komplex: (Sp)-[κ2-N,P-4-(2`-Pyridyl)-1,2- diphenylphosphinyl[2.2]paracyclophan]chlorokupfer(I) (s. Abb. 1) quantenchemisch untersucht und charakterisiert, um zu ermitteln, ob er sich als TADF-Emitter eignen könnte.
Danach wird die Auswirkung von verschiedenen Substitutionen an diesem Komplex näher betrachtet und ebenfalls quantenchemisch charakterisiert, um zu erkennen, ob diese einen positiven oder negativen Einfluss auf die TADF-Eigenschaften haben.
Abb.1: Darstellung von (Sp)-[κ2-N,P-4-(2`-Pyridyl)-1,2-diphenylphosphinyl[2.2]paracyclophan]chlorokupfer(I)
2 1.1 Lumineszenz:
Das Phänomen, bei dem eine spontane elektromagnetische Strahlung in Form von Licht von einem angeregten Zustand in den energetisch tiefer liegenden Grundzustand erfolgt, wird Lumineszenz genannt. Hierbei handelt es sich um einen elektronischen Übergang. Die Strahlung sollte hierbei nur in dem für das menschliche Auge wahrnehmbaren Spektralbereich zwischen 400 nm bis 780 nm [6] erfolgen. Nur wenn ein Molekül in diesem Bereich emittiert, ist es nutzbar für OLEDs. Um diesen Übergang zu erreichen, braucht das System Energie. Diese Energie wird zuerst elektrisch (über eine Stromquelle) zugeführt und anschließend in Form von Licht emittiert. [7]
Es gibt verschiedene Arten der Lumineszenz. Die Aufteilung hängt davon ab, wie die Anregung der Elektronen erzeugt wird. Bei der Photolumineszenz wird beispielsweise ein Molekül durch ein Photon angeregt. Dabei wird ein Elektron für kurze Zeit vom Grundzustand in einen angeregten Zustand angehoben. Dafür ist es notwendig, dass das Photon dieselbe Energie wie die Energiedifferenz zwischen den beiden Zuständen enthält. Bei der Elektrolumineszenz wird die Anregung durch Elektronen verursacht. [8] Dies beruht auf dem Zerfall von Exzitonen, d.h. von Paaren freier Elektronen und Löcher auf einem Emittermaterial.
Die Energie, die für die Anregung der Elektronen aufgenommen worden ist, wird wieder an die Umgebung abgegeben, wenn das Elektron in den Grundzustand zurückkehrt. Die Abgabe kann in unterschiedlichen Formen erfolgen. Bei der Relaxation wird Wärme abgegeben. Eine andere Form besteht in Photonenstrahlung. Dabei basiert die Abgabe auf zwei unterschiedlichen Prozesse, nämlich Fluoreszenz und Phosphoreszenz.[9]
Bei der Fluoreszenz wird die Lichtemission aus dem niedrigsten Singulett-Zustand S1 in den Grundzustand S0 abgegeben, d.h. die Spin-Multiplizität bleibt erhalten. Die Lebensdauer beträgt zwischen 10-9 und 10-7 s. Im Fall der Phosphoreszenz muss zuerst ein Triplett-Zustand (T1) erreicht werden, von dem dann die Emission in den S0-Zustand abgeht. Es ist jedoch sehr unwahrscheinlich, dass bei der Anregung ein Triplett-Zustand direkt erreicht wird, weil es quantenchemisch verboten ist und dafür einen Spin-Umkehr-Prozess benötigen würde.
Allerdings kann mit Hilfe der Spin-Bahn-Kopplung der Übergang zwischen verschiedenen Multiplizitäten strahlungslos erreicht werden. Unter diesem Spin-Umkehr-Prozess (eng.
Intersystem Crossing, ISC) kann das Molekül aus dem S1-Zustand in einen Triplett-Zustand überführt werden. Vom T1-Zustand aus kann das Molekül wieder strahlungslos in den S1- Zustand zurückkehren (eng. Reverse Intersystem Crossing, RISC) oder durch Emission von Phosphoreszenz-Strahlung in den Grundzustand übergehen.[7] [10] [11] Außerdem kann ISC in den Grundzustand erfolgen.
3 1.2 OLEDs:
Wie bereits eingangs erwähnt, spielen OLEDs in der Entwicklung von neuen und besseren Displays sowie Leuchtmitteln eine große Rolle. Sie verfügen über eine große Quantenausbeute und haben einen geringen Stromverbrauch, weswegen sie sehr wichtig in der Industrie sind. [12] Sie lassen sich in drei Gruppen (Generationen) unterteilen.
1.2.1 Erste OLED Generation:
In der ersten Generation von OLEDs spielen hauptsächlich Fluoreszenz-Emitter eine wichtige Rolle, da das ISC hier noch sehr langsam erfolgt und die Phosphoreszenz-Rate relativ klein ist. Aus diesem Grund können nur die Singulett-Exzitonen geerntet werden, also nur 25% der gesamten Ausbeute können zur Lichterzeugung genutzt werden, da die übrigen 75% zu den Triplett-Anregungen gehören. Die kleine innere Quantenausbeute stellt einen deutlichen Nachteil dar. Von Vorteil ist die Klarheit der erzeugten Farben sowie die gute operationale Stabilität, weswegen OLEDs der ersten Generation heutzutage nach wie vor für stabile blau- Licht Emitter-OLEDs verwendet werden.[5] [13]
1.2.2 Zweite OLED Generation:
Mit der Entwicklung der zweiten Generation von OLEDs, den sogenannten PHOLEDs, wurde eine Effizienzsteigerung der organischen Leuchtdioden erreicht. Durch die starke Spin-Bahn- Kopplung kann eine schnelle Relaxation (ISC-Übergang) vom niedrigsten Singulett-Zustand in einen Triplett-Zustand erfolgen. Durch die effektive Besetzung des Triplett-Zustands wächst die Wahrscheinlichkeit, dass Photonen vom T1-Zustand in den S0-Zustand als Phosphoreszenz abgestrahlt werden.[13] Weil damit neben den Singulett-Exzitonen auch die Triplett-Exzitonen geerntet werden können, wird die Quantenausbeute auf 100% gesteigert.
Allerdings werden hier Organometall-Komplexe mit einem Zentrum von Iridium oder Platin verwendet, welche sehr knappe und damit auch sehr teure Elemente darstellen.[14] Hinzu kommt, dass durch eine lange Strahlungslebensdauer unerwünschte Nebeneffekte auftreten können, beispielsweise Quenching-Prozesse oder auch Bleaching-Reaktionen, welche für blaue PHOLEDS eine große Bedeutung haben. Um die Instabilität der blauen phosphoreszierenden Emitter zu vermeiden, werden Hybrid-OLEDs aus fluoreszierenden und phosphoreszierenden Emittern eingesetzt. Auf diese Weise wird weißes Licht erzeugt, in der die fluoreszierenden Emitter blaues Licht und die phosphoreszierenden Emitter grünes bis rotes Licht abgeben.[5]
4 1.2.3 Dritte OLED Generation:
Die Weiterentwicklung der organischen Leuchtdioden mündete in der dritten Generation.
Diese beruht auf Fluoreszenz. Hier werden organische Donor-Akzeptor Systeme und Übergangsmetall-Komplexe angewendet. Voraussetzung für den Übergang zwischen Singulett- und Triplett-Zustand ist die Spin-Bahn-Kopplung (eng. spin-orbit-coupling, SOC).
Ein kleiner Singulett-Triplett-Energieabstand (∆EST) ist auch in diesem Fall von großer Bedeutung, denn der geringe Abstand ermöglicht ein reverse intersystem crossing (RISC) vom niedrigsten angeregten Triplett-Zustand in den niedrigsten angeregten Singulett-Zustand. Der RISC-Prozess erfolgt unter Umgebungstemperatur. Der Energie-Abstand, der überbrückt werden soll, sollte dabei nicht größer als 1000 cm-1 (≈120 meV) sein.[13] Werden diese Bedingungen erfüllt, kann eine thermisch verzögerte Fluoreszenz, die sogenannte TADF (eng.
thermally activated delayed fluorescence), stattfinden. Es kann also durch TADF und die normale direkte Fluoreszenz eine Ausbeute von 100% erreicht werden. Außerdem können Übergangsmetalle der ersten Reihe eingesetzt werden. Dies ist ein klarer Vorteil gegenüber der zweiten Generation, da man auf Iridium oder Platin verzichten kann, was wiederum die Herstellungskosten von OLEDs deutlich senken würde. Nachteilig wäre, wie auch bei den PHOLEDs, dass für den strahlenden Zerfall eine relativ niedrige Ratenkonstante vorliegt, wodurch die OLEDs anfälliger für nichtstrahlende Zerfälle wie beispielsweise Triplett-Triplett- Auslöschung wären. Außerdem ist bei dieser Art von OLEDs die Emission aus den Zuständen mit CT-Charakter relativ breit, was sich als zusätzlicher Nachteil erweist, da dies nicht günstig für die Anwendung in Displays ist.[5]
1.3 TADF:
Die thermisch aktivierte verzögerte Fluoreszenz wird aufgrund ihrer externen Quanteneffizienz als eine neue Technologie von großer Bedeutung im Bereich von OLEDs betrachtet. Die TADF-basierten OLEDs erweisen sich als eine günstige Alternative zu den Hochleistungs- PHOLEDs. [15] Ihre hohe Effizienz basiert auf einem RISC-Prozess, in dem nichtstrahlende Triplett-Exzitonen (T1) auch für die Fluoreszenz mittels up-conversion vom Triplett-Zustand (T1) in den niedrigsten Singulett-Zustand (S1) durch thermische Energie verwendet werden können. [14] Damit ein Übergang zwischen T1 und S1 mittels RISC erfolgen kann, muss die Energiedifferenz zwischen diesen Zuständen gering sein (maximal 0,12 eV). Mit steigendem Abstand zwischen T1 und S1 vermindert sich die Wahrscheinlichkeit von einem RISC, wodurch die TADF-Fähigkeit negativ beeinflusst wird.[13] Zusätzlich sollen idealerweise eine starke Fluoreszenz bei minimalen nichtstrahlenden Zerfällen in den Grundzustand erfolgen. Alle diese Bedingungen sind allerdings nur schwer gleichzeitig zu erfüllen.
5 Die Energie-Differenz zwischen offenschaligen Konfigurationen mit Singulett-Triplett- Kopplung ist abhängig von der Austauschwechselwirkung der ungepaarten Elektronen. Ist die Überlappung zwischen den Elektronendichten der Orbitale, die an der Anregung teilnehmen, klein, wird die Energiedifferenz ebenfalls klein. Diese Anforderung wird von CT-Zuständen erfüllt. Die Überlappung von Elektronendichten spielen eine entscheidende Rolle bei der Größe der Spin-Bahn-Kopplung (SOC, eng. spin-orbit-coupling) als auch bei der Fluoreszenzrate. Elektronische SOC ist eine Voraussetzung für ein wirksames RISC. Häufig kommt es vor, dass S1 und T1 die gleiche Zusammensetzung der Wellenfunktionen haben.
Das bedeutet, dass die selben Raumorbitale besetzt sind. Die SOC zwischen Singulett- und Triplett-Konfigurationen mit gleicher Besetzung der Raumorbitale verschwindet aufgrund der Symmetrie. Deshalb ist die SOC zwischen Singulett- und Triplett-Zuständen mit CT-Charakter sehr schwach. Allerdings kann sich dieses Verhalten, dank der nahen Entartung von d- Orbitalen mit verschiedener magnetischer Quantenzahl, für Übergangsmetallkomplexe mit angeregten MLCT-Zuständen verbessern.[5]
Es gibt zwei wichtige Materialklassen, die TADF-Eigenschaften potentiell aufweisen können.
Eine davon basiert auf Kupfer(I)-Komplexen. Bei dieser Art von Komplexen ist der ISC- Prozess sehr schnell, weshalb keine signifikante direkte Fluoreszenz beobachtet werden kann.
Diese TADF-Emitter zeigen einen bedeutsamen Vorteil gegenüber Phosphoreszenz-Emittern, da Kupferkosten niedriger als Iridium- und Platinkosten sind. Die zweite Klasse von TADF- Emittern basiert auf reinen organischen Molekülen. In der Vergangenheit haben diese Emitter Nachteile im Vergleich zu Cu(I)-Komplexen gezeigt, weil sie bedingt durch die höhere Lebensdauer von angeregten Zuständen eine stärkere Wirkung von Roll-off-Effekten aufwiesen und dadurch die Effizienz der Emitter bei höherer Helligkeit gesenkt wurde.
Allerdings konnten reine organische Emitter in dieser Hinsicht optimiert werden, indem ihre Lebensdauer verkürzt wurde.[13]
6
2 Theorie:
2.1 Dichtefunktionaltheorie (DFT)[16] [17] [18]:
Mit Hilfe der Dichtefunktionaltheorie kann die Gesamtenergie eines Mehrelektronensystems minimiert werden. Basierend auf dem Hohenberg-Kohn-Theorem wird dabei statt der Wellenfunktion mit der Elektronendichte gearbeitet. Die Elektronendichte gibt die Aufenthaltswahrscheinlichkeit von einem Elektron an einem Punkt im Raum wieder. Die Idee dahinter ist, dass die Energie nicht mehr von 3N Koordinaten (wobei N die Anzahl der Elektronen repräsentiert) sondern nur von 3 Koordinaten, nämlich den Raumrichtungen abhängt. Dies ist ein Vorteil gegenüber der Hartree-Fock-Theorie. Die Anzahl der Elektronen N lässt sich durch die Integration der Elektronendichte über den gesamten Raum berechnen (s. Formel 1):
𝑁 = ∫ 𝜌(𝑟)𝑑𝑟
(1)Die genaue Abhängigkeit der Energie von der Dichte ist unbekannt, weswegen das Dichtefunktional (s. Formel 2) analog zum Hartree-Fock-Formalismus in vier Komponenten entsprechend der Schrödingergleichung aufgeteilt wird:
𝐸[𝜌] = 𝑇[𝜌] + 𝐸
𝑒𝑁[𝜌] + 𝐽[𝜌] + 𝐾[𝜌]
(2)Dafür gilt: T[𝜌] ist die kinetische Energie, EeN[𝜌] ist das Funktional der Coulombwechselwirkung zwischen dem Elektron und dem Kern, J[𝜌] beschreibt die Coulombwechselwirkung zwischen den Elektronen untereinander und K[𝜌] beschreibt die Austauschwechselwirkung. Allerdings lassen sich nur die Coulombwechselwirkungen (Anziehungen bzw. Abstoßungen) über die klassische Physik darstellen, während die Austauschwechselwirkung und die kinetische Energie durch Näherungen beschrieben werden müssen. Für die Herleitung der letzteren Funktionale wird der Kohn-Sham-Formalismus [19] angewendet. Dafür wird das Energiefunktional in einen exakt berechenbaren Term und Korrekturterme aufgeteilt. Für die Beschreibung dieses Systems aus miteinander wechselwirkenden Elektronen wird ein fiktives Referenzsystem verwendet, das die selbe Grundzustandsdichte besitzt und in welchem die Elektronen nicht miteinander interagieren. Aus diesem neuen Kohn-Sham-System, welches durch eine einzelne Determinante beschrieben werden kann, können die Kohn-Sham-Orbitale hergeleitet werden. Die kinetische Energie von nicht wechselwirkenden Teilchen in diesem System Ts[𝜌] wird durch den Ausdruck (3):
𝑇
𝑠[𝜌] = ∑ ∫ 𝜓
𝑘∗(𝑟 ⃗⃗ ) [−
𝑖 ħ22𝑚𝑒
∆
𝑖] 𝜓
𝑘(𝑟 ⃗⃗ )𝑑𝑟
𝑖⃗⃗
𝑖𝑁𝑘=1
(3)
7 beschrieben. Ts[𝜌] stimmt nicht mit T[𝜌] überein, entspricht aber einer guten Näherung. Hinzu kommt der Korrekturterm ∆T[𝜌], der die Wechselwirkung zweier Teilchen repräsentiert. Dieser Term wird mit dem Austausch-Wechselwirkungsterm K[𝜌] zusammengefügt. Das daraus resultierende Kohn-Sham-Austausch-Korrelationsfunktional Exc[𝜌] wird zur Berechnung der Kohn-Sham-Dichtefunktional (4) benötigt:
𝐸
𝐷𝐹𝑇[𝜌] = 𝑇
𝑠[𝜌] + 𝐸
𝑒𝑁[𝜌] + 𝐸
𝑥𝑐[𝜌]
(4)Das Austausch-Korrelation-Funktional Exc[𝜌] umfasst die Austausch-Wechselwirkung zweier Elektronen, die dynamische Elektronenkorrelation und Korrekturterme der kinetischen Energie. Es lässt sich mittels Formel (5) berechnen:
𝐸
𝑥𝑐[𝜌] = (𝑇[𝜌] − 𝑇
𝑠[𝜌]) + (𝐸
𝑒𝑒[𝜌] − 𝐽[𝜌])
(5)Insgesamt benötigt eine Dichtefunktionalrechnung relativ ähnlichen Aufwand wie eine Hartree- Fock-Rechnung, allerdings wird dabei die Elektronenkorrelation mit einbezogen, wenn auch nur näherungsweise über das Austausch-Korrelation-Potenzial. Die Dichtefunktionaltheorie wird daher als äußerst vorteilhaft gegenüber anderen Methoden in der Quantenchemie betrachtet. Voraussetzung ist jedoch, dass man das passende Austausch-Korrelation- Funktional auswählt.
2.2 Hybridfunktionale:
In vorliegender Arbeit werden alle Geometrieoptimierungen mit dem Hybridfunktional PBE0 durchgeführt. Anschließend wird für die DFT/MRCI-Rechnungen das Funktional geändert und anstelle von PBE0 das BH-LYP-Hybridfunktional verwendet.
2.2.1 Das PBE0-Funktional [20] [21]:
Im PBE0-Funktional wird das PBE-Funktional[22] von Perdew, Burke und Ernzerhof mit einem vordefinierten exakten Austauschterm kombiniert. Die Austausch-Korrelations-Energie wird durch Gleichung (6) gegeben. Sie wird zu ¼ durch die Hartree-Fock-Austauschenergie und zu
¾ durch die PBE-Austauschenergie bestimmt:
𝐸
𝑥𝑐𝑃𝐵𝐸0=
14
𝐸
𝑥𝐻𝐹+
34
𝐸
𝑥𝑃𝐵𝐸+ 𝐸
𝑐𝑚𝑃𝑊91 (6)Das Perdew-Wang-Korrelationsfunktional wird mit einem neuen Austauschterm verwendet, woraus sich Gleichung (7) ergibt:
𝐸
𝑥𝑃𝐵𝐸=
𝑏𝑥21+𝑎𝑥2
(7)
8 Die Konstanten haben folgende Werte: b= 0,00336 und a= 0,00449. x wird mittels:
𝑥 =
|𝛻𝜌|𝜌3⁄4
(8)
ermittelt. Hierbei entspricht 𝜌 der Elektronendichte und 𝛻𝜌 die Gradiente der Elektronendichte.
2.2.2 Das BH-LYP-Funktional [23] [21]:
Im Fall des BH-LYP-Funktionals setzt sich die Austauschenergie (s. Gleichung 9) zu 50% aus dem Hartree-Fock-Austauschterm und zu 50% aus einem Becke-Funktional (B88) zusammen.
Hinzu kommt für die Korrelationsenergie ein LYP-Korrelationsfunktional.
𝐸
𝑥𝑐𝐵𝐻−𝐿𝑌𝑃=
12
𝐸
𝑥𝐻𝐹+
12
𝐸
𝑥𝐵88+ 𝐸
𝑐𝐿𝑌𝑃 (9) 2.3 DFT/MRCI[17] [18] [24] [25] [26]:MRCI bezeichnet die Multireferenz-Konfigurationswechselwirkung (eng. multireference configuration interaction) und ist eine Methode, die auf der Konfigurationswechselwirkung (CI, configuration interaction) zur Berechnung der Wellenfunktion unter Berücksichtigung der Elektronenkorrelation beruht. Die Mehrelektronen-Wellenfunktion wird dabei als eine Linearkombination von Konfigurationen zur Darstellung der Korrelationsenergie beschrieben.
|𝛷0⟩ = 𝑐0|𝛹0⟩ + ∑ 𝑐𝑎𝑟 𝑎𝑟|𝛹𝑎𝑟⟩+ ∑𝑎<𝑏,𝑟<𝑠𝑐𝑎𝑏𝑟𝑠|𝛹𝑎𝑏𝑟𝑠⟩+ ∑𝑎<𝑏<𝑐,𝑟<𝑠<𝑡𝑐𝑎𝑏𝑐𝑟𝑠𝑡|𝛹𝑎𝑏𝑐𝑟𝑠𝑡⟩+ ⋯ (10) Hierbei werden die angeregten Zustände, angefangen mit der Wellenfunktion des Grundzustands |𝛹0⟩, als Summanden dargestellt. 𝛹ar beschreibt zum Beispiel eine Einfachanregung aus einem besetzten Orbital a in ein unbesetztes Orbital r. Die anderen Terme beschreiben eine Doppel- bzw. Dreifachanregung. Die Summanden werden über die Koeffizienten ci, die aus der CI-Matrix berechnet wurden, gewichtet.
Für den Fall, dass alle möglichen Konfigurationen berücksichtigt werden können, spricht man von einer Full-CI, welche eine exakte Korrelationsenergie liefern kann. Da die Matrix und der entsprechende Rechenaufwand mit steigender Anzahl von Atomen in einem Molekül an Komplexität zunimmt, bedarf es allerdings der Vereinfachung. Man beschränkt sich zum Beispiel auf Einfach- und Zweifachanregungen, d.h. solche Anregungen, die einen hohen Einfluss auf die Wellenfunktion haben.
Um diese Problematik der MRCI-Rechnung zu umgehen, wird die DFT/MRCI-Methode [24]
angewendet. Sie kombiniert die Dichtefunktionaltheorie und die Konfigurationswechselwirkungsmethode (und profitiert aus ihren Vorteilen), indem die dynamische Elektronenkorrelation aus der DFT und die statische Elektronenkorrelation aus der MRCI-Methode miteinander verbunden werden.
9 Eine doppelte Berücksichtigung der dynamischen Korrelationseffekte ist möglich, wenn sie sowohl in Diagonal- als auch in Außendiagonaltermen der Matrix miteinbezogen werden, was als solches allerdings die Ergebnisse verfälschen würde. Um dies zu vermeiden, werden die Außendiagonalterme durch eine energieabhängige Skalierung exponentiell auf null abgeschwächt (Parametrisierung der Hamilton-Matrix), wodurch der Konfigurationsraum verkleinert und somit die Rechenzeit verkürzt wird.
Insgesamt sind die Ergebnisse der DFT/MRCI-Rechnungen äußerst verlässlich (die Fehlerspanne für die relative Energie liegt zwischen 0,2 und 0,3 eV). [24] Aufgrund dessen ist diese semiempirische Methode für die Berechnung von großen Molekülen und CT-Zustände gut geeignet. [26]
Der ursprüngliche Hamilton-Operator, der von Grimme und Waletzke entwickelt worden ist, lässt sich gut für die Beschreibung von einfach angeregten Zuständen anwenden. Jedoch können für die Beschreibung von Zuständen mit vier offenen Schalen Schwierigkeiten auftreten. Diese Problematik konnte mit Hilfe einer neuen Parametrisierung des DFT/MRCI- Hamiltonoperators (von Igor Lyskov, Martin Kleinschmidt und Christel M. Marian) gelöst werden. [25]
2.4 Effektives Kernpotential (ECP)[16][17] [18] [27]:
Jedes Element verfügt über kernnahe Elektronen (Rumpfelektronen) und Valenzelektronen.
Werden alle Elektronen von Elementen höherer Perioden in die Berechnungen mit einbezogen, kann es zu übermäßig hohen Speicher- und Rechenzeitanforderungen kommen.
Die Verwendung von Rumpfpotentialen (eng. effective core potential, ECP) dient dem Zweck, Rechenzeit und -aufwand zu reduzieren. Zusätzlich können relativistische Effekte in die Berechnungen mit einbezogen werden.
Weil die Rumpfelektronen in schweren Atomen wenig Einfluss auf die chemische Bindung haben, werden die Wechselwirkungen zwischen den Kernelektronen sowie den Valenzelektronen durch ein justiertes Potential (ECP) erfasst und repräsentiert. Dies ermöglicht es, nur die Wechselwirkungen zwischen den Valenzelektronen untereinander für die Berechnungen zu berücksichtigen. Nach der Geometrieoptimierung rechnet man nur mit den Valenzelektronen weiter. Das Rumpfpotential geht als Einelektronenanteil in den Hamilton-Operator ein.[17]
10
3 Durchführung und technische Details zur Rechnung:
Zuerst wurde mit Hilfe des Programms Avogadro [28] die Startstruktur des Kupfer-Komplexes (Sp)-[κ2-N,P-4-(2`-Pyridyl)-1-2diphenylphosphinyl[2.2]paracyclophan]chlorokupfer(I)
konstruiert. Anschließend wurde die Geometrie-Optimierung im elektronischen Grundzustand (S0) mittels des Turbomole-Programms (TURBOMOLE V7.0 2015 [29]) unter Verwendung von DFT optimiert. Dafür wurde das PBE0-Dichtefunktional verwendet. Für die Liganden wurde als Basissatz def-SV(P)[30,31] genutzt, während für das Kupfer-Atom der Basissatz cc-pVDZ-PP[32]
mit dem skalarrelativistischen Rumpfpotential Stuttgart-Koeln MCDHF RSC ECP[33]
ausgewählt wurde. Um zu überprüfen, ob tatsächlich ein Minimum vorliegt, musste eine Schwingungsfrequenzanalyse durchgeführt werden. Ausgehend von der optimierten Struktur des Grundzustandes wurde der angeregte Triplettzustand (T1) mit UDFT auch mittels Turbomole optimiert. Für die Berechnungen wurden dieselben Basissätze und Funktionale wie für die Optimierung des Grundzustands verwendet.
Anschließend wurden DFT/MRCI Berechnungen an den optimierten Strukturen durchgeführt, um die jeweils fünf niedrigsten angeregten Singulett- und Triplettzustände zu ermitteln. Hier wurde, im Gegensatz zur Geometrieoptimierung, das Funktional BH-LYP[20,23] anstelle von PBE0[20] eingesetzt, da DFT/MRCI nur für BH-LYP parametrisiert wurde. Für die Berechnungen wurde der Hamiltonoperator in der Lyskov-Parametrisierung[25] verwendet.
Für beide optimierte Geometrien kamen sowohl standard- (mit esel=1.0 Eh) als auch tight- Parameter (mit esel=0.8 Eh) zur Anwendung. Für alle weiteren Rechnungen wurden nur standard-Parameter genutzt. Zur Vereinfachung der Rechnungen wurden die untersten 24 Orbitale und alle unbesetzten Orbitale, die eine Energie über drei Hartree besaßen, eingefroren, weil diese fast keine Rolle in der Anregung spielen. Anhand der Ergebnisse aus den DFT/MRCI Rechnungen konnten Anregungsspektren erstellt werden. Daraufhin ließ sich die Energielücke zwischen T1 und S1 berechnen. Anhand der Differenzdichten können die wichtigsten Zustände charakterisiert und die Überlappung der Elektronendichten betrachtet werden.
Später wurden alle Vorgänge für drei neue Komplexe, bei denen jeweils die Pyridyleinheit durch Substitution variiert wurde, wiederholt. Die Substitution erfolgte an der Meta-Position im Pyridylring. Dabei wurden drei verschiedene Substituenten (-fluor, -methyl und -t-Butyl) eingesetzt, anschließend ihr jeweiliger Einfluss auf die Eigenschaften des Komplexes untersucht und miteinander verglichen. Zum Schluss wurde eine Substitution an einem Kohlenstoff-Atom des Paracyclophan mittels Fluor durchgeführt und ebenfalls optimiert. Die Ergebnisse dieser Substitution werden hier auch mit den Ergebnissen des Standards des unsubstituierten Cu-Komplexes verglichen, sodass der Einfluss dieser Substitution ermittelt werden kann.
11 Die Darstellung der optimierten Geometrien, der Orbitale und der Differenzdichten wurde mittels des Programms Jmol[34] durchgeführt. Die Tabellen wurden mit Hilfe von Excel angefertigt.
12
4 Darstellung der Ergebnisse:
Im folgenden Teil werden die Ergebnisse der Berechnungen dargestellt und ausgewertet.
Anschließend werden die Moleküle miteinander verglichen.
4.1 Standard Cu-Komplex:
4.1.1 Struktur:
4.1.1.1 Vergleich mit den Literaturwerten:
In der Doktorarbeit von Carolin Braun (2017) [35] im Arbeitskreis Bräse (KIT) wurde eine Reihe von Cu(I)-Komplexen auf [2.2]Paracyclophanbasis synthetisiert und charakterisiert. Aus ihrer Arbeit wurden ein paar Werte entnommen und den hier ermittelten Werten zum Vergleich gegenübergestellt (vgl.Tab. 1 und 2). Die Literaturwerte stammen von einer Kristallstruktur, bei der das Lösungsmittel Dimethylformamid (DMF) mitkristallisiert wurde.
Tabelle 1: Bindungslängen des Cu-Komplexes im Grundzustand
Literaturwerte [pm] [35, S.73] Berechnete Werte [pm] Differenz [pm]
Cu-N 203,1 207,5 4,4
Cu-P 216,8 219,1 2,3
Cu-Cl 222,5 219,5 3,0
Tabelle 2: Bindungswinkel des Cu-Komplexes im Grundzustand
Literaturwerte [°] [35, S. 73] Berechnete Werte [°] Differenz [°]
N-Cu-P 118,97 121,1 2,13
N-Cu-Cl 107,55 107,5 0,05
P-Cu-Cl 132,53 128,1 4,43
Anhand des Vergleichs lässt sich erkennen, dass die Abweichungen nur sehr klein sind. Die Abweichungen lassen sich dadurch erklären, dass die experimentellen Werte der Kristallstruktur entnommen und diese durch Lösungsmitteleffekte beeinflusst sind, während die Berechnungen innerhalb dieser Arbeit im Vakuum durchgeführt wurden.
4.1.1.2 Vergleich mit der T1-Geometrie:
Nachdem das Molekül erfolgreich im Grundzustand und später im Triplettzustand optimiert wurde, werden die beiden Geometrien im Folgenden miteinander verglichen.
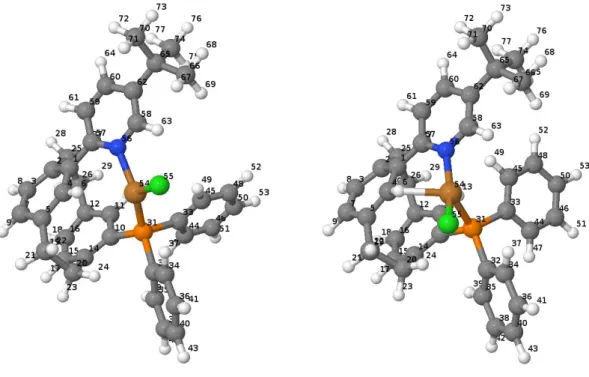
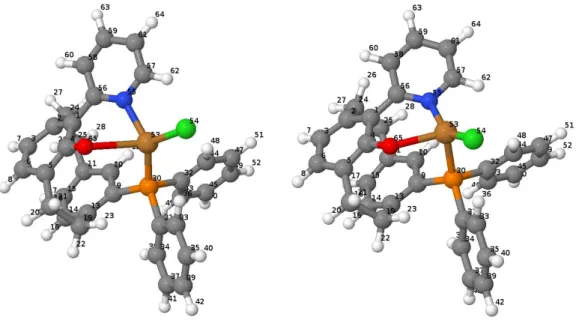
Abbildung 2 zeigt die S0- und T1-Geometrien in visualisierter Form. Die Farben entsprechen folgenden Atomen:
- Weiß: Wasserstoff-Atome
13 - Grau: Kohlenstoff-Atome
- Braun: Kupfer-Atom - Orange: Phosphor-Atom - Blau: Stickstoff-Atom - Grün: Chlor-Atom
Abb. 2: Optimierte Grundzustandsgeometrie (S0) (links) und optimierte Triplettzustandsgeometrie (T1) (rechts) mit Atomnummern.
Anhand der Bindungslängen, Bindungswinkel und Diederwinkel kann man die Änderungen der Geometrien erkennen.
Beim Vergleich der optimierten Strukturen wird sichtbar, dass die Struktur im angeregten Zustand eine deutliche räumliche Abweichung aufweist.
Hinsichtlich der Bindungslängen zeigt sich, dass einige Bindungen länger und einige Bindungen kürzer geworden sind. Die größte Abweichung der Bindungslängen zeigt sich zwischen H6 und Cu. Der Abstand ist so stark verkürzt, dass das Wasserstoff H6 an einem der Phenylringe der Paracyclophan mit dem Kupfer-Zentrum eine Bindung in der angeregten Zustandsoptimierung eingeht. Die größten Unterschiede liegen zwischen Cu und N, wo die Bindung um 17.6 pm kürzer ist und zwischen Cu und P, wo die Bindung um 14,6 pm länger ist. Ansonsten variieren die Bindungslängen um maximal 6 pm. Die größten Abweichungen der Bindungslängen von beiden optimierten Zuständen werden in Tabelle 3 dargestellt. Die übrigen Bindungslängen befinden sich im Anhang in Tabelle A.3.
14
Tabelle 3: Relevante Bindungslängen des Cu(I)-Komplexes im optimierten Grundzustand und Triplettzustand Im Vergleich zueinander gestellt.
S0-Zustand T1-Zustand
Bindung Bindungslänge
[pm]
Bindungslänge [pm]
Differenz [pm]
Δ(T1-S0)
C1-C2 141,9 143,7 1,8
C1-C4 140,3 141,8 1,5
C1-C57 148,0 144,6 -3,4
C58-C62 139,0 136,6 -2,4
C60-C62 139,3 143,3 4,0
P31-Cu54 219,1 233,7 14,6
Cu54-Cl55 219,5 214,4 -5,1
Cu54-N56 207,5 189,9 -17,6
N56-C57 134,8 140,9 6,1
N56-C58 133,8 136,7 2,9
Cu54-H6 227,5 206,2 -21,3
Die relevanten Bindungswinkel und die Diederwinkel werden in den Tabellen 4 und 5 aufgeführt (die übrigen Bindungswinkel befinden sich in Tabelle A.4 im Anhang). Insgesamt wird deutlich, dass sich beim Übergang vom Singulett-Grundzustand in den ersten angeregten Triplettzustand die gesamte Geometrie um das Kupfer-Zentrum verändert hat und eine Verzerrung aufweist.
Tabelle 4: Relevante Bindungswinkeln des S0-Geometrie und T1-Geometrie im Vergleich S0-Zustand T1-Zustand
Bindungswinkel [°] Bindungswinkel [°] Differenz Δ(T1-S0)
C32-P31-C33 101,8 130,8 29,0
C10-P31-Cu54 116,0 106,8 -9,2
P31-Cu54-N56 121,1 106,6 -14,5
P31-Cu54-Cl55 128,1 105,1 -23,0
Cl55-Cu54-N56 107,5 147,9 40,4
Die Änderungen der Bindungswinkel in der Paracyclophan-Einheit sind minimal. Auffallend sind die Änderungen in den Diederwinkeln, was auf eine starke Verzerrung des Gerüsts schließen lässt. Die geometrischen Änderungen sind in Abbildung 3 deutlich zu erkennen.
Tabelle 5: Ausgewählte Diederwinkel des S0-Geometrie und T1-Geometrie.
S0-Zustand T1-Zustand
Diederwinkel [°] Diederwinkel [°] Differenz Δ(T1-S0)
Cl55-Cu54-P31-C32 27,8 -6,7 -34,5
Cl55-Cu54-P31-C33 -87,1 128,9 216,0
N56-Cu54-P31-C32 -175,1 168,3 343,4
N56-Cu54-P31-C33 70,0 46,1 -23,9
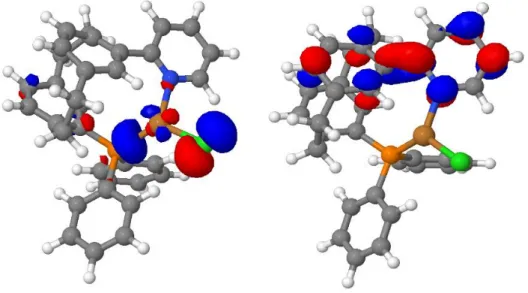
15 Anhand der Molekülorbitale (s. Abb. 3) HOMO (Highest Occupied Molecular Orbital) und LUMO (Lowest Occupied Molecular Orbital) lassen sich einige Änderungen der Struktur zwischen S0- und T1-Geometrien erklären. Durch die Zunahme des bindenden Charakters an den Bindungen zwischen C57-C1, C59-C60 und C62-C58 verkürzen sie sich an der optimierten T1-Geometrie. Die Elektronendichten an C57 und N stoßen sich ab, was die Verlängerung dieser Bindung erklären könnte. Dadurch, dass vom HOMO die Elektronendichte von Cu und Cl auf die Liganden wandert, verringert sich die Abstoßung zwischen Cl-Cu-N und bewirkt eine Verkleinerung des dazwischenliegenden Winkels.
Zwischen Cu und P in HOMO liegt ein bindender Charakter vor, der in LUMO verschwindet (wird nicht bindend), wodurch die Bindung länger wird. In HOMO weist Chlor eine hohe Elektronendichte auf, welche mit der Elektronendichte von Cu zu einer Abstoßung führt. In LUMO sind beide Dichten nicht mehr vorhanden, weswegen die Bindung zwischen Cu und Cl sehr wahrscheinlich kürzer wird. Auch die Phenylringe werden durch diese Verschiebung der Elektronendichten näher einander herangezogen.
Abb. 3: HOMO (links) und LUMO (rechts) der optimierten S0-Geometrie des Cu-Komplexes
4.1.2 DFT/MRCI:
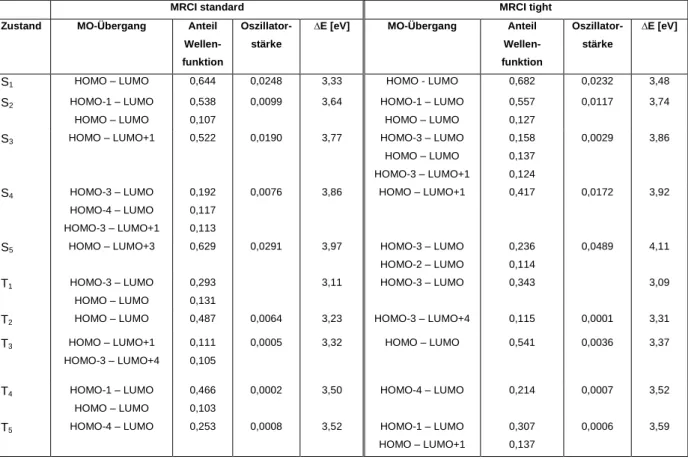
Die MRCI-Berechnungen wurden mit sechs Singulett-Wurzeln und fünf Triplett-Wurzeln berechnet. Für den unsubstituierten Cu-Komplex wurde, wie schon erwähnt, sowohl eine DFT/MRCI-Rechnung mit Lyskov tight-Parametern als auch mit standard-Parametern durchgeführt. Die relevanten Ergebnisse von beiden Rechnungen (s. Tab. 6) ergaben, dass die Unterschiede zu groß sind, um später mit der ressourcen-sparenden tight- Parametrisierung zu rechnen. Dafür wurden die fünf ersten Singulett- und Triplettanregungen genauer untersucht.
16 Bei Betrachtung und Vergleich der Rechenergebnisse mit verschiedenen Parametern im Grundzustand kann man eine deutliche Abweichung erkennen. Die Singulett-Zustände S3 und S4 an der Grundzustandsgeometrie sind vertauscht. Der S3-Zustand mit standard-Parametern entspricht bei der Berechnung mit tight-Parametern S4 und der S4-Zustand mit standard- Parameter S3 mit tight-Parametern.
Tabelle 6: Singulett- und Triplettanregungen des Grundzustandes des Cu-Komplexes mit tight- und Standard- Parametern aus der DFT/MRCI-Rechnung
MRCI standard MRCI tight
Zustand MO-Übergang Anteil Wellen- funktion
Oszillator- stärke
∆E [eV] MO-Übergang Anteil
Wellen- funktion
Oszillator- stärke
∆E [eV]
S1 HOMO – LUMO 0,644 0,0248 3,33 HOMO - LUMO 0,682 0,0232 3,48
S2 HOMO-1 – LUMO
HOMO – LUMO
0,538 0,107
0,0099 3,64 HOMO-1 – LUMO HOMO – LUMO
0,557 0,127
0,0117 3,74
S3 HOMO – LUMO+1 0,522 0,0190 3,77 HOMO-3 – LUMO
HOMO – LUMO HOMO-3 – LUMO+1
0,158 0,137 0,124
0,0029 3,86
S4 HOMO-3 – LUMO
HOMO-4 – LUMO HOMO-3 – LUMO+1
0,192 0,117 0,113
0,0076 3,86 HOMO – LUMO+1 0,417 0,0172 3,92
S5 HOMO – LUMO+3 0,629 0,0291 3,97 HOMO-3 – LUMO
HOMO-2 – LUMO
0,236 0,114
0,0489 4,11
T1 HOMO-3 – LUMO
HOMO – LUMO
0,293 0,131
3,11 HOMO-3 – LUMO 0,343 3,09
T2 HOMO – LUMO 0,487 0,0064 3,23 HOMO-3 – LUMO+4 0,115 0,0001 3,31
T3 HOMO – LUMO+1
HOMO-3 – LUMO+4
0,111 0,105
0,0005 3,32 HOMO – LUMO 0,541 0,0036 3,37
T4 HOMO-1 – LUMO
HOMO – LUMO
0,466 0,103
0,0002 3,50 HOMO-4 – LUMO 0,214 0,0007 3,52
T5 HOMO-4 – LUMO 0,253 0,0008 3,52 HOMO-1 – LUMO
HOMO – LUMO+1
0,307 0,137
0,0006 3,59
Betrachtet man die Anregungen der Zustände (wie aufgeführt in Tab. 6) genauer, kann man erkennen, dass der T1-Zustand an der S0-Geometrie nicht wie erwartet aus einem HOMO → LUMO-Übergang besteht; hingegen besteht der T2 aus dieser HOMO → LUMO-Übergang mit einem Anteil an der Wellenfunktion von 48%. Im Fall der Triplett-Zustände ist deutlich erkennbar, dass die Werte der verschiedenen Zustände bei den Rechnungen mit tight- Parametern sehr stark von den Ergebnissen mit standard-Parametern abweichen. Der T2- Zustand, der dem S1-Zustand entspricht, stimmt in der Berechnung mit den tight- Parametern des T3-Zustands überein.
Im optimierten Triplettzustand sind die Abweichungen zwischen tight- und standard- Parametrisierung nicht mehr so stark ausgeprägt (s. Tab. 7).
Die Oszillatorstärken weichen von den standard-parametrisierten Rechnungen ab. Die größten Abweichungen finden sich an der S0-Geometrie.
17
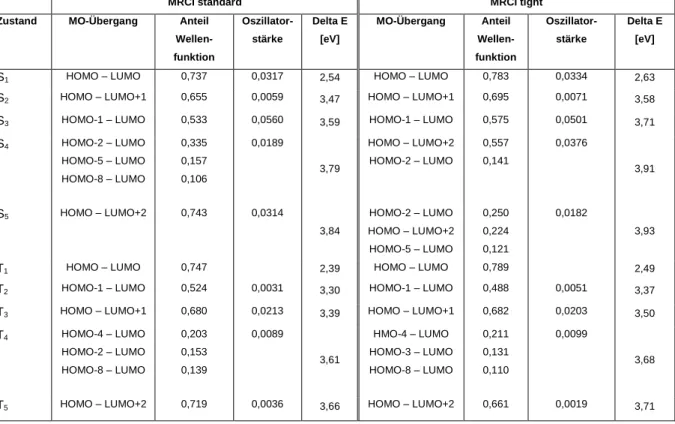
Tabelle 7: Singulett- und Triplettanregungen aus der T1-Geometrie des Cu-Komplex mit tight- und Standard- Parametern aus der DFT/MRCI-Rechnung
MRCI standard MRCI tight
Zustand MO-Übergang Anteil Wellen- funktion
Oszillator- stärke
Delta E [eV]
MO-Übergang Anteil Wellen- funktion
Oszillator- stärke
Delta E [eV]
S1 HOMO – LUMO 0,737 0,0317 2,54 HOMO – LUMO 0,783 0,0334 2,63
S2 HOMO – LUMO+1 0,655 0,0059 3,47 HOMO – LUMO+1 0,695 0,0071 3,58
S3 HOMO-1 – LUMO 0,533 0,0560 3,59 HOMO-1 – LUMO 0,575 0,0501 3,71
S4 HOMO-2 – LUMO HOMO-5 – LUMO HOMO-8 – LUMO
0,335 0,157 0,106
0,0189
3,79
HOMO – LUMO+2 HOMO-2 – LUMO
0,557 0,141
0,0376
3,91
S5 HOMO – LUMO+2 0,743 0,0314
3,84
HOMO-2 – LUMO HOMO – LUMO+2 HOMO-5 – LUMO
0,250 0,224 0,121
0,0182
3,93
T1 HOMO – LUMO 0,747 2,39 HOMO – LUMO 0,789 2,49
T2 HOMO-1 – LUMO 0,524 0,0031 3,30 HOMO-1 – LUMO 0,488 0,0051 3,37
T3 HOMO – LUMO+1 0,680 0,0213 3,39 HOMO – LUMO+1 0,682 0,0203 3,50
T4 HOMO-4 – LUMO HOMO-2 – LUMO HOMO-8 – LUMO
0,203 0,153 0,139
0,0089
3,61
HMO-4 – LUMO HOMO-3 – LUMO HOMO-8 – LUMO
0,211 0,131 0,110
0,0099
3,68
T5 HOMO – LUMO+2 0,719 0,0036 3,66 HOMO – LUMO+2 0,661 0,0019 3,71
Im Folgenden ist es wichtig zu untersuchen, wie groß die Abhängigkeit der vertikalen (aufgelistet in Tab. 6 und 7) und adiabatischen Anregungsenergien der S1- und T1- Zustände vom Selektionsschwellenwert ist. Zum Zwecke der Vereinfachung wird dabei davon ausgegangen, dass die S1-Geometrie die T1-Geometrie ausreichend wiedergibt, weshalb eine weitere Optimierung an S1 nicht notwendig ist.
Hier bestätigt sich, dass die Abweichungen bei den vertikalen Energien mit dem tight- Parametern zu stark sind, nämlich größer als 0,1eV, um mit dieser Parametrisierung weiterrechnen zu können. Auch die adiabatischen Energien sind stark abhängig von der Auswahl des jeweiligen Parameters. Die adiabatische Energie an der S1-Geometrie aus der Berechnung mit standard-Parametern entspricht 2,53 eV und an der T1-Geometrie 2,39 eV, während die Resultate mit tight-Parametern respektive 2,63 eV und 2,49 eV betragen. Dabei ergeben sich Abweichungen von jeweils über 0,1 eV. Insgesamt sind die Abweichungen zwischen den Rechnungen mit standard- und tight-Parametrisierung also sehr groß. Aus diesem Grund werden die weiteren Rechnungen mit den standard-Parametern durchgeführt.
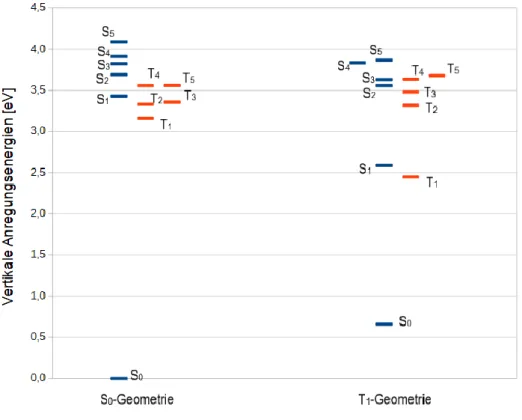
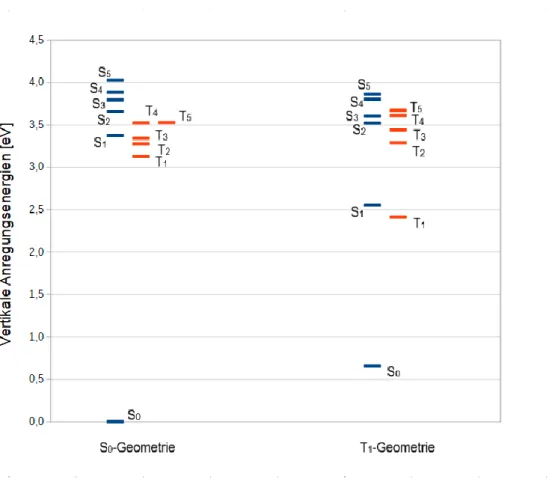
Die vertikalen Anregungsenergien der S0- und T1-Geometrien des Cu-Komplexes (s. Tab. 6 und 7) in Bezug zur Grundzustandsenergie der S0-Geometrie werden in Abbildung 5 graphisch dargestellt. Der S0-Zustand an der T1-Gometrie ist um 0,66 eV gegenüber dem S0-Zustand an der S0-Geometrie angehoben. Im Fall der S0-Geometrie kann man erkennen, dass die ersten
18 zwei Triplettzustände energetisch weiter unten liegen als der erste angeregte Singulettzustand. Der dritte Triplettzustand (T3) liegt fast entartet mit dem S1-Zustand vor. Dies lässt vermuten, dass die Elektronen von S1 aus auf T3, T2 oder T1 übergehen könnten. Die Zustände T4 und T5 liegen fast entartet oberhalb von S1, aber unterhalb von S2. An der T1- Geometrie liegt ausschließlich der T1-Zustand energetisch unterhalb von S1. Im Vergleich zur Grundzustandsgeometrie ist der S1-Zustand sehr stark energetisch abgesenkt. Die Triplett- Anregungen sind mit Ausnahme von T1 energetisch angehoben. Wenn man davon ausgeht, dass die T1-Geometrie der S1-optimierten Geometrie entspricht, kann angenommen werden, dass nur der Übergang zwischen S1 und T1 möglich ist: unterhalb des S1-Zustands befindet sich nur der T1-Zustand. Alle anderen Zustände sind energetisch gestiegen.
Der größte Unterschied liegt in der Änderung der Energie von S1 und T1 an der T1-Geometrie im Vergleich zur S0-Geometrie. Beide haben eine niedrigere Energie und liegen näher bei S0. Die Energielücke zwischen S1 und T1 ändert sich ebenfalls, sie wird kleiner. Die Energiedifferenz bei der T1-Geometrie (0,14 eV) ist geringer als die der S0-Geometrie (0,23 eV).
Abb. 4: Vertikale Anregungsenergien in der S0- und T1-Geometrien
19 Sowohl der Grundzustand als auch die angeregten Zustände (S1 und T1) besitzen ein relativ großes Dipolmoment. Auf den ersten Blick scheint es, als ob sich die Dipolmomente bei der Anregung nicht sonderlich ändern würden, da die Beträge nicht stark voneinander abweichen.
Um die tatsächliche Änderung der Dipolmomente jedoch abschätzen zu können, muss man die Orientierung der Dipolmomentvektoren genauer betrachten.
Tabelle 8: Dipolmomente und Charakterisierung von den Zuständen in der S0- und T1-Geometrie
S0-Geometrie T1-Geometrie
Zustand Dipolmoment [Debye] Charakter Dipolmoment [Debye] Charakter
S0 7,91 7,90
S1 10,31 XMLCT 7,08 XMLCT
S2 8,92 XMLCT 7,07 XMLCT
S3 3,44 XMLCT 5,99 XMLCT
S4 7,64 LC / XMLCT 6,21 XMLCT
S5 2,67 XMLCT / LC 6,10 XMLCT / LC
T1 7,66 LC / XMLCT 7,40 XMLCT
T2 8,96 XMLCT 5,40 XMLCT
T3 6,49 LC / XMLCT 6,75 XMLCT
T4 7,79 XMLCT 5,65 XMLCT
T5 7,47 LC / XMLCT 5,73 XMLCT / LC
Tabelle 9: Vektoranteile und Ausrichtung der Dipolmomente an der S0-Geometrie
Zustand ex ey ez
S0 2.910 0.139 -1.092
S1 1.441 -3.439 1.597
T1 2.819 -0.612 -0.870
T2 1.652 -2.894 1.146
Abb. 5: Dipolmoment Vektoren in der S0-Geometrie, S0 (gelb), S1 (blau), T1 (grün), T2 (rot)
20 Anhand der Ausrichtung der Dipolmomente (s. Tab. 9) kann man erkennen, dass der S1- Zustand genauso wie der T2-Zustand hauptsächlich in -y-Richtung ausgerichtet ist, anders als der T1-Zustand, der genauso wie der S0-Zustand hauptsächlich in +x-Richtung ausgerichtet ist. Dies lässt sich graphisch anhand der Abbildung 5 gut erkennen. Deshalb kann man im Fall einer Solvatation von einer energetischen Erhöhung der Zustände S1 und T2 in der Absorption ausgehen. Wie erwartet bestätigt sich hier, dass der optimierte Triplettzustand an der Grundzustandsgeometrie dem T2- und nicht dem T1-Zustand entspricht.
Mit Hilfe der Differenzdichten kann man die Anregungszustände charakterisieren (s. Tab. 8).
Dabei entspricht die pinke Fläche derjenigen Fläche, von der Elektronendichte abgezogen wird, und die blaue Fläche derjenigen Fläche, zu der Elektronendichte hinzukommt. Die relevanten Anregungen an der T1-Geometrie sind S0→S1 und S0→T1 (s. Abb. 6).
Abb. 6: S0-S1-Differenzdichte (links) und S0-T1-Differenzdichte (rechts) an der optimierten T1-Geometrie.
Den Hauptanteil an beiden Anregungen bildet der Übergang von HOMO auf LUMO. In beiden Fällen deutet der Übergang der Elektronendichte auf einen Charge-Transfer-Übergang hin, spezifischer auf einen XMLCT (engl. Halide/Metal-to-Ligand Charge Transfer) Übergang.
Dieser Charge Transfer Charakter ist für TADF-Moleküle sehr vorteilhaft. Die Ladung überträgt sich hier vom Chlor und Kupfer auf den Liganden, vor allem am Pyridylring. Somit ist die Überlappung der Elektronendichten ziemlich klein. Hauptsächlich überlappen sich die Elektronendichten am Stickstoff. Eine kleine Überlappung korreliert mit einer kleinen Aufspaltung zwischen S1 und T1. Diese wiederum stimmt mit dem auffallenden CT-Charakter des Komplexes überein.
21 4.2 Cu-Komplex mit tert-Butyl-Substituent:
4.2.1 Struktur:
Beim folgenden Schritt wurde am Cu-Komplex die Pyridyleinheit durch Substitution variiert.
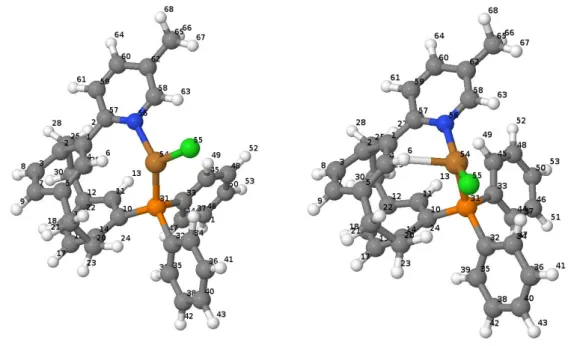
Bei dieser ersten Substitution wurde ein tert-Butyl-Rest an der meta-Position am Pyridinring angebracht. Danach wurde eine Optimierung am Grundzustand und später auch eine Optimierung im angeregten Triplettzustand durchgeführt. Beide Geometrien werden im Folgenden miteinander verglichen (s. Abb. 7).
Abb. 7: optimierte Grundzustandsgeometrie (S0) (links) und optimierte Triplettzustandsgeometrie (T1) (rechts) mit Atomnummern des durch einen tert-Butyl-Rest substituierten Moleküls
Bei der Veränderung der Geometrie von S0 zu T1 gibt es nur wenige Bindungen, die eine Differenz größer als 1 pm aufweisen (genaue Änderungen s. Tab. A.6 im Anhang). Auffällig ist die Bindung zwischen P und Cu, die länger wird, und die Bindung zwischen Cu und N, die kürzer wird. Hinzu kommt, dass sich, wie beim unsubstituierten Molekül, eine neue Bindung zwischen H6 und Cu an der T1-Geometrie bildet. Bei den Bindungswinkeln (s. Tab. A.7 im Anhang) gibt es ebenfalls wenige Änderungen, die über 1° hinausgehen. Die größte und auffälligste Änderung findet um das Cu-Zentralatom statt. Der Winkel zwischen N-Cu-Cl wird um 40° größer. Auch die Diederwinkel (s. Tab. A.8 im Anhang) zeigen, dass das Molekül einer deutlichen Verzerrung an der T1-Geometrie unterliegt: einer der Phenylringe am Phosphor dreht sich um fast 90° um die eigene Achse.
22 4.2.2 DFT/MRCI:
Die MRCI-Berechnungen für dieses Molekül laufen analog zu denen unter Punkt 4.1.2. Die Anregungsenergien für beide Geometrien in Bezug auf den S0 an der S0-Geometrie werden in Abbildung 8 graphisch dargestellt.
Abb. 8: Anregungsenergien von Singulett- und Triplett-Zuständen an der S0- und T1-Geometrie
Die Anregungen (s. Tab. A.9 im Anhang) zeigen, dass S1 an der S0-Geometrie wieder T2
entspricht. Der Übergang von HOMO auf LUMO hat ebenfalls eine große Bedeutung mit dem größten Wellenanteil in S1 und T2 bzw. T1 an beiden Geometrien.
An der S0-Geometrie liegen T1, T2 und T3 unterhalb von S1. Dabei sind T2 und T3 fast energetisch entartet, genauso wie im Fall von T4 und T5. Anders ist es an der T1-Geometrie, wo nur T1 unterhalb von S1 liegt. Der S0-Zustand an der T1-Geometrie ist um 0,66 eV gegenüber dem S0-Zustand an der S0-Geometrie energetisch angehoben (s. Abb. 8). Der T1- Zustand an der T1-Geometrie ist um 0,88 eV energetisch abgesenkt. Die adiabatische Energie des T1-Zustands beträgt 2,45 eV und die des S1-Zustands 2,59 eV. Die Differenzenergie zwischen S1 und T1 an der S0-Geometrie liegt bei 0,1 eV und an der T1-Gometrie, wo der Abstand größer wird, bei 0,14 eV.
23 Die Elektronendichte-Übergänge an den Anregungen S0→S1 und S0→T1 (s. Abb. 9) beschreiben wieder eine XMLCT-Anregung. Die Überlappung ist wieder klein und hauptsächlich am Stickstoff zu verorten.
Abb. 9: S0-S1-Differenzdichte (links) und S0-T1-Differenzdichte (rechts) von der optimierten T1-Geometrie
Die Energiedifferenz zwischen HOMO und LUMO in der S0-Geometrie beträgt 6,004 eV und 4,625 eV in der T1-Geometrie. Die relevanten Orbitalenergien werden in Tabelle A.10 im Anhang aufgelistet.
4.3 Cu-Komplex mit Methyl-Substituent:
4.3.1 Struktur:
4.3.1.1 Vergleich mit den Literaturwerten:
In der zuvor erwähnten Doktorarbeit von Carolin Braun (2017)[35] im Arbeitskreis Bräse (KIT) wurde ebenfalls ein Cu(I)-Komplex mit variierter Pyridyleinheit durch einen Methyl- Substituenten synthetisiert und charakterisiert. Im Folgenden werden die von ihr gemessenen Bindungslängen und -winkel mit den hier ermittelten Werten verglichen (s. Tab. 10 und 11).
Die Literaturwerte stammen von einer Kristallstruktur, bei der das Lösungsmittel Dimethylformamid (DMF) mitkristallisiert wurde.
Tabelle 10: Bindungslängen des Cu-Komplexes im Grundzustand
Literaturwerte [pm] [35, S.75] Berechnete Werte [pm] Differenz [pm]
Cu-N 203,6 207,5 3,9
Cu-P 217,6 219,1 1,5
Cu-Cl 224,5 219,5 5,0