am J 2 - Molek¨ ul des Fortgeschrittenen Praktikums
Wissenschaftliche Arbeit f¨ur das Staatsexamen im Fach Physik
Martine Meyer Physikalisches Institut
Albert-Ludwig-Universit¨at Freiburg
07.Oktober 2014
Inhaltsverzeichnis
1 Vorwort 1
2 Theoretische Grundlagen 1
2.1 Energieniveaus und Wellenfunktionen zweiatomiger Molek¨ule . . 1
2.1.1 Die Born-Oppenheimer-N¨aherung. . . 1
2.1.2 Berechnung und Klassifikation von Elektronenwellenfunk- tionen ΨE . . . 3
2.1.3 Beschreibung der Kernbewegung . . . 7
2.1.4 Wechselwirkung zwischen Elektronenbewegung und Kern- bewegung . . . 14
2.2 Zur Interpretation der beobachteten Absorptions¨uberg¨ange bei Jod 17 2.2.1 Zu welchem angeregten elektronischen Zust¨anden sind Ab- sorptions¨uberg¨ange m¨oglich (Auswahlregeln)? . . . 17
2.2.2 Welche Schwingungs- und Rotationsstruktur besitzt das Absorptionsspektrum? . . . 19
2.3 Zur Interpretation der beobachteten Intensit¨aten der Absorpti- onsbanden . . . 23
2.3.1 Wie sind die Jod- Molek¨ule auf die Schwingungs- und Ro- tationsniveaus des Grundzustands verteilt? . . . 24
2.3.2 Betrachtung der ¨Ubergangsmomente f¨ur Schwingungs¨uberg¨ange 27 2.3.3 Franck-Condon-Faktor . . . 28
3 Aufgabenstellung 29 4 Die alte Versuchsanordnung 29 4.1 Ziel der Arbeit . . . 29
4.2 Versuchsaufbau und Durchf¨uhrung . . . 30
4.3 Fazit . . . 32
5 Verbesserungsprozess 32 6 Versuchsaufbau 35 6.1 Absorptionsmessung . . . 35
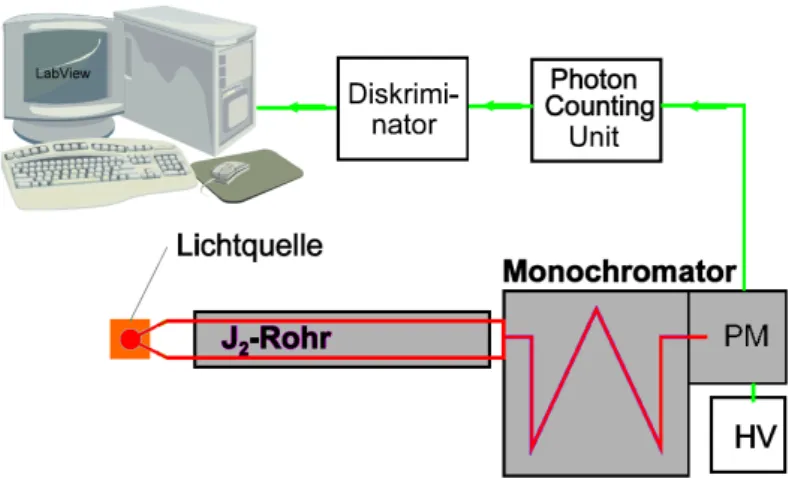
6.2 Emissionsmessung . . . 35
6.3 Ger¨atebeschreibung. . . 37
6.3.1 Lampen . . . 37
6.3.2 Laser . . . 37
6.3.3 Jod-Rohr . . . 38
6.3.4 Spektrometer . . . 39
6.3.5 Monochromator. . . 39
6.3.6 Photomultiplier. . . 41
6.3.7 Photon Counting Unit . . . 42
6.3.8 Diskriminator . . . 42
6.4 Software . . . 43
6.4.1 SpectraSuite . . . 43
6.4.2 JodAnalog . . . 43
7 Aufgabenstellung 44 7.1 Absorption . . . 44
7.2 Emission. . . 45
8 Musterversuch 46 8.1 Durchf¨uhrung . . . 46
8.1.1 Absorption . . . 46
8.1.2 Emission . . . 47
8.2 Auswertung . . . 49
8.2.1 Absorption . . . 49
8.2.2 Emission . . . 56
8.3 Ergebnisse . . . 61
8.3.1 Absorption . . . 61
8.3.2 Emission . . . 62
9 Zusammenfassung 62 10 Literatur 63 11 Anlagen 64 Versuchsanleitung f¨ur den vorl¨aufigen Versuch im Sommersemester 2014 64 Poster . . . 78
Identifizierte Schwingungsbanden, Wellenzahlen und Termdifferenzen mit Fehlern . . . 79
Beispielhafte Aufstellung der berechneten Wellenzahlen . . . 80
Versuchsanleitung . . . 81
1 Vorwort
Die Spektroskopie ist ein wichtiges Hilfsmittel in der Physik und der Chemie, um mehr ¨uber die Struktur von Molek¨ulen und Atomen zu erfahren. Durch die Spek- troskopie lassen sich vor allem Energieniveaus, ¨Ubergangswahrscheinlichkeiten, Symmetrie und Kopplung der Zust¨ande und deren Lebensdauern bestimmen.
Schon 1868 gelang es Angstr¨om die Emissionslinien des Sonnenspektrums auf- zulisten, die erste quantitative Spektralanalyse fand allerdings erst 1925 statt.
Wurde Spektroskopie anfangs mit Gitterspektralapparaten und Photoplatten durchgef¨uhrt so werden aufgrund der hohen Aufl¨osung heute meistens laser- spektroskopische Verfahren verwendet [2], [14].
Dieser Versuch soll eine Einf¨uhrung in die Molek¨ulspektroskopie sein, bei dem wichtige Molek¨ulkonstanten, wie die Schwingungskonstanten und Dissoziations- energien eines Molek¨ulzustands bestimmt werden. Eine Emissions- und eine Absorptionsmessung, jeweils durchgef¨uhrt mit verschiedenen Messanordnungen, sollen die Vielfalt spektroskopischer M¨oglichkeiten andeuten.
Als zu untersuchendes Molek¨ul wird in diesem Versuch das Jod-Molek¨ul verwen- det. Jod steht im Periodensystem der Elemente an 53. Stelle und z¨ahlt zu den Halogenen. Es hat eine relative Atommasse von 126,9u und eine Dichte von 4,93 gcm−3. Die Siedetemperatur von Jod liegt bei 184,4 ˆA°C. Diese Molek¨ul eignet sich sehr gut f¨ur spektroskopische Untersuchungen, da es eine sehr ausgepr¨agte Bandenstruktur besitzt.
2 Theoretische Grundlagen
In diesem Abschnitt werden einige Elemente zur Theorie zweiatomiger Molek¨ule, die f¨ur diesen Versuch relevant sind, eingef¨uhrt. Die theoretischen Grundlagen sind zum Großteil aus [1] entnommen worden. An einigen Stellen wurden sie pas- send zum neuen Versuchsaufbau gek¨urzt oder erweitert und es wurden teilweise neue Quellen und Literaturverweise verwendet.
2.1 Energieniveaus und Wellenfunktionen zweiatomiger Mo- lek¨ ule
2.1.1 Die Born-Oppenheimer-N¨aherung
Exakte Eigenfunktionen und Energieeigenwerte f¨ur ein zweiatomiges Molek¨ul ergeben sich aus der Eigenwertgleichung:
HΨ =EΨ (1)
Der Hamilton-Operator f¨ur zwei durch Coulomb-Wechselwirkung wechsel- wirkende Teilchen A und B ist folgendermaßen definiert (vgl. [4], Kap. 11.1.1):
H =−~2
2m∆ +Epot(qA, qB, r) Epot(qA, qB, r) = qA·qB
r
(2)
m: Masse
Epot: potentielle Energie der Wechselwirkung zweier Objekte (hier: Elek- tron oder Kern)
qA: Ladung eines beteiligten Objekts
qB: Ladung des anderen beteiligten Objekts
r: Abstand der beiden Teilchen
So ergibt sich durch Einsetzen f¨ur die Eigenwertgleichung eines zweiatomigen- Molek¨uls:
H =− ~2 2me
X
i
∆i
| {z }
TE
− ~2 2MA
∆A− ~2 2MB
∆B
| {z }
TN
+X
i>j
e2 rij
| {z }
VEE
−X
i
Z1·e2 rAi −X
i
ZB·e2 rBi
| {z }
VEN
+ZAZB·e2 rAB
| {z }
VN N
(3)
TE ist der Operator der kinetischen Energie aller Elektroneni(xi, yi, zi).
TN ist der Operator der kinetischen Energie der beiden Kerne A und B (xA, yA, zA;xB, yB, zB)
VEE, VEN, VN N sind die potentiellen Energien der wechselseitigen elektro- statischen Abstoßung bzw. Anziehung der Elektronen und Kerne.
Der L¨osungsansatz
Ψ = ΨE(..., xi, yi, zi, ...)·ΨN(xA, yA, zA, xB, yB, zB), (4) wobei ΨE und ΨN L¨osungen der abseparierten Gleichungen
HEΨE= (TE+VEE+VEN)ΨE =EE·ΨE (5) HNΨN = (TN +VN N+EE)ΨN =EN·ΨN (6) sind, bildet dann eine befriedigende N¨aherungsl¨osung von (1), wenn die Dif- ferentiale nach den Kernkoordinaten ∂Ψ∂xE
A, . . . ,∂∂x2Ψ2E
A , . . . klein bleiben, d.h. wenn sich die Elektroneneigenfunktion ΨE nur wenig mit dem Kernabstand ¨andert.
Dies ist gew¨ohnlich erf¨ullt, da sich die Elektronen sehr viel schneller bewegen, als die durch ihre sehr viel gr¨oßere Masse tr¨ageren Kerne. Gezeigt wurde dieser Sachverhalt 1927 von M. Born und R. Oppenheimer.
(5) stellt die Schr¨odinger-Gleichung der Elektronenbewegung f¨ur festgehaltenen Kernabstand rAB =r dar. F¨ur verschiedene Kernabst¨ande ist auchVEN ver- schieden, deshalb h¨angen ΨE undEE vonrals Parameter ab.
(6) ist die Schr¨odinger-Gleichung der Bewegung der Kerne im Potential VN N+ EE.
Die wesentliche Aussage der Born-Oppenheimer-N¨aherung besteht also darin, dass die Bewegung der Elektronen und die der Kerne unabh¨angig voneinander behandelt werden k¨onnen. Die Wechselwirkung zwischen den Elektronen und dem Kern wird dadurch ber¨ucksichtigt, dass einerseits ΨE undEE vom Kern- abstand r abh¨angen, andererseits die elektronischen Eigenwerte EE(r) einen Teil des Potentials bilden, in dem sich die Kerne bewegen. (vgl. [2], Kap. 9.5.1;
[4, Kap. 11.2)
2.1.2 Berechnung und Klassifikation von Elektronenwellenfunktio- nenΨE
Da Gleichung (5) f¨ur das Jod-Molek¨ul nicht exakt l¨osbar ist, muss die L¨osung angen¨ahert werden. Ein N¨aherungsweg besteht darin, N¨aherungsl¨osungen Φ von ΨE aus Einelektronen- Molek¨ulorbitalen (MO’s) zu konstruieren, die ihrerseits als Linearkombinationen von Einelektronen- Atomorbitalen (AO’s) angesetzt werden (vgl. [4], S.42-43). In einer so erhaltenen
”LCAO/MO- Wellenfunktion
“(LCAO: linear combination of atomic orbitals) Φ sind Parametercienthalten, die es erlauben, Φ so zu variieren, dass Φ eine m¨oglichst niedrige Energieliefert:
= P
i
P
kc∗ickR
Φ∗iHEΦkd3~r P
i
P
kc∗ickR
Φ∗iΦkd3~r (7)
Durch Berechnung des Minimums vonbekommt man einen Wert, der dem tats¨achlichen (gemessenen) EigenwertEE am n¨achsten kommt.
Aufbau von Elektronenwellenfunktionen durch Einelektronen- Mo- lek¨ulorbitale am Beispiel des Jod- Molek¨uls
Ein Molek¨ulorbital eines Elektrons wird durch eine Wellenfunktion beschrieben, die nur von den Koordinaten dieses Elektrons abh¨angt. Dieses”Modell der un- abh¨angigen Teilchen“ impliziert, dass die Wechselwirkung eines Elektrons mit den anderen Elektronen nur pauschal dadurch ber¨ucksichtigt wird, dass jene das Axialfeld der beiden Kerne teilweise abschirmen und zu einem resultieren- den Axialfeld beitragen (vgl. [2], Kap. 6.1.1)
In einem axialen Kraftfeld ist nur noch die Komponente lz (Molek¨ulachse z) des Bahndrehimpulses~leines Elektrons Konstante der Bewegung (vgl. [4], Kap.
11.6.1).
Es gilt:
lz=ml·~ (8) mit m¨oglichen Wertenml=l, l−1, . . . ,−l. Hierbei stelltl die Bahndrehim- pulsquantenzahl gem¨aß|~l|=p
l(l+ 1)~dar.
Das Vorzeichen von ml beschreibt den Drehsinn der Bewegung des Elektrons um die z- Achse. Da die Energie des Elektrons aber unabh¨angig von diesem Drehsinn ist, gen¨ugt es, die Quantenzahlλ=|ml|= 0, . . . , lzu verwenden, um den Zustand des Elektrons zu spezifizieren. Der Bahndrehimpulszustand eines Elektrons wird f¨urλ= 0,1,2,3, . . . alsσ, π, δ, φ, . . .- Zustand bezeichnet.
Um anzuzeigen, welche Atomorbitale nl-linear zu einem Molek¨ulorbital kom- biniert werden, notiert man das betreffende Molek¨ulorbital meist in der Form λnl (vgl. [6], S. 121 ff). (Diese Bezeichnung ist bei homonuklearen zweiatomi- gen Molk¨ulen deshalb m¨oglich, weil hier die Bindung auf der ¨Uberlappung von gleichnamigen Atomorbitalen beruht, z.B. zwei 1s- Orbitalen.) In jedem Orbital sind zwei verschiedene Spinzust¨ande m¨oglich, außerdem ist jedes Orbital mit λ≥1 zweifach entartet (die beiden Zust¨ande haben gleich großes, aber entge- gengesetzt gerichtetesml). Deshalb haben in einemσ- Orbital zwei Elektronen Platz, inπ, δ, φ- Orbitalen jeweils vier.
Bei homonuklearen Molek¨ulen kann die Symmetrieoperation i
”Punktspiegelung am Symmetriezentrum“ auf die Orbitalwellenfunktion angewendet werden. Je nach seinem Verhalten kann dann das Orbital als eines mit gerader oder unge- rader Parit¨at identifiziert werden:
iλg= (+1)·λg (9)
iλu= (−1)·λu (10)
Die Parit¨atseigenschaft gibt Auskunft ¨uber die Bindef¨ahigkeit eines Orbitals:
Ist das Symmetrieverhalten der Orbitale so, dass zwischen den beiden Kernen eine große Aufenthaltswahrscheinlichkeit f¨ur das Elektron besteht, so ist das Orbital bindend. (vgl. [4] S.46)
Dazu geh¨oren z.B. die Orbitaleσgns, σgnp, πunp.
Ist das Symmetrieverhalten aber so, dass zwischen den beiden Kernen eine Kno- tenstelle der Aufenthaltswahrscheinlichkeit liegt, so ¨uberwiegt die Abstoßung der Kerne, das Orbital ist antibindend und wird dazu oft mit einem * verse- hen, z.B.σ∗uns, σu∗np, π∗gnp. Die Energieniveaus der Molek¨ulorbitale, die von den Atomorbitalen ns und np ausgehen, liegen i.A. (bei schweren Molek¨ulen) in der in Abb. 1dargestellten Anordnung.
Wie beim Atom kann man die Elektronenkonfiguration eines Molek¨uls mit vielen Elektronen feststellen, indem man
”von unten her“ die Orbitale gem¨aß dem Pauli- Prinzip auff¨ullt (d.h. maximal zwei Elektronen inσ- Orbitalen, ma- ximal vier Elektronen inπ, δ, . . . - Orbitalen)(vgl. [2, Kap. 9.3.1).
Im Falle des Jod- Molek¨uls, das durch die Bindung zweier Jodatome (Elektro- nenkonfiguration des Grundzustands KLM 4d105s25p5) entsteht, erh¨alt man als
Abbildung 1: Veranschaulichung der Energieniveaus der Molek¨ulorbi- tale
Elektronenkonfiguration des Grundzustand:
. . .(σg5s)2(σu∗5s)2(σg5p)2(πu5p)4(π∗g5p)4 (11) Da im Wesentlichen nur die Elektronen der ¨außeren Schale der Atome zur Molek¨ulbindung beitragen, sind nur die Orbitale angeschrieben, die von den Elektronen der Atomschalen= 5 gebildet werden.
Die Elektronenkonfiguration des ersten angeregten Zustands betr¨agt:
. . .(σg5s)2(σ∗u5s)2(σg5p)2(πu5p)4(πg∗5p)3(σ∗u5p) (12) Kennzeichnung der Elektronenzust¨ande durch Drehimpulsquan- tenzahlen und Angabe des Symmetrieverhaltens am Beispiel des Jod- Molek¨uls
Es ist ¨ublich, einen elektronischen Zustand durch Angabe der vorliegenden Ge- samtdrehimpulsquantenzahlen (wie beim Atom) und durch Angabe des Sym- metrieverhaltens der ihm zugeordneten Wellenfunktion zu kennzeichnen.
Wie bei Atomen vom LS- Kopplungstyp bilden in den meisten Molek¨ulen die ein- zelnen Elektronenbahndrehimpulse einen Gesamt- Elektronenbahndrehimpuls L~ = P
i~li und die einzelnen Elektronenspins einen Gesamt- Elektronenspin S~=P
i~si.
Die Komponente des Gesamt- Bahndrehimpulses der Elektronen entlang der Molek¨ulachse betr¨agt:
Lz=ML·~ (13)
mitML=P
imli undML=L, L−1, . . . ,−L(L aus|~L|=p
L(L+ 1)~).
Wieder h¨angt die Energie eines Zustands nur vom Wert Λ =|ML|ab.
Der Bahndrehimpulszustand eines Molek¨uls wird f¨ur Λ = 0,1,2,3, . . . als Σ,Π,∆,Φ, . . .- Zustand bezeichnet. Jeder Zustand mit Λ ≥ 1 ist wiederum zweifach entartet. (Feinere Wechselwirkungen heben diese Entartung auf, vgl.
Abschnitt
”Zur Interpretation der beobachteten Absorptions¨uberg¨ange“) Die Bahnbewegung der Elektronen in Zust¨anden mit Λ≥1 erzeugt ein Magnet- feld entlang der Molek¨ulachse und zwingt den GesamtspinS~ zur Pr¨azession um diese Achse.
Erhalten bleibt wieder nur die Komponente:
Sz= Σ~, (14)
wobei Σ die (2S+ 1) m¨oglichen WerteS, S−1, . . . ,−Sbesitzt (Saus|S~|= pS(S+ 1)~).
Der Betrag der Komponente des Gesamtdrehimpulses entlang der Achse betr¨agt:
|Λ~+ Σ~|=|Λ + Σ|~=: Ω~ (15) Die Spin-Bahn-Wechselwirkung ergibt einen Beitrag in der Elektronenener- gie, der verschieden groß ausf¨allt f¨ur jede Stellung von S~ zur Molek¨ulachse, d.h. f¨ur jeden Wert von Λ + Σ. Dies f¨uhrt zu einer Multiplettaufspaltung der Molek¨ulterme in (2S+ 1) verschiedene Niveaus.
Deshalb benennt man jeden Λ - Zustand mit seiner Multiplizit¨at 2S+1 (links oben) und dem gerade vorliegenden Wert von Λ + Σ (rechts unten).
Elektronenwellenfunktionen werden mit Φ+Eoder Ψ−Ebezeichnet, je nachdem ob die Symmetrieoperation σ
” Spiegelung an einer Ebene durch die beiden Kerne“ das Vorzeichen der Wellenfunktion ¨andert oder nicht; z.B.:
σΣ+= (+1)·Σ+ (16)
σΣ−= (−1)·Σ− (17)
Elektronenwellenfunktionen homonuklearer Molek¨ule k¨onnen außerdem mit ΨEg oder ΨEu (gerade oder ungerade Parit¨at)
bezeichnet werden, je nachdem ob die Operation i
”Punktspiegelung am Symmetriezentrum“ das Vorzeichen der Wellenfunktion ¨andert oder nicht, z.B.:
iΠg= (+1)·Πg (18)
iΠu= (−1)·Πu (19)
(vgl. [2], S.290) Zust¨ande mit gleichen Drehimpulsquantenzahlen Λ,2S+ 1 und Λ+Σ, aber unterschiedlichem Symmetrieverhalten, k¨onnen sich in der Elek- tronenenergie unterscheiden. Beispielsweise kann jedem der beiden - bei Λ≥1 auftretenden - in Λ entarteten Zust¨ande eine Wellenfunktion mit unterschiedli- chem Symmetrieverhalten Ψ+E bzw. Ψ−E zugeordnet werden.
Aufgrund der Wechselwirkung zwischen Elektronenbewegung und Molek¨ulro- tation wird diese Entartung aufgehoben, es bilden sich zwei nur im Symmetrie- verhalten der zugeordneten Wellenfunktionen unterschiedene Energiezust¨ande.
Ein und derselben Elektronenkonfiguration kann entweder genau ein Molek¨ulterm
oder auch mehrere - in den Drehimpulsen und dem Symmetrieverhalten verschie- dene - zugeordnet werden. (vgl. [6], 130 ff.)
So ergibt sich aus der Konfiguration (11) der Term
X1Σ+g (20)
als resultierender elektronischer Grundzustand des Jod- Molek¨uls, w¨ahrend f¨ur die Konfiguration (12) die Terme
1Πu,3Π2u, A3Π1u, B3Π+0u,3Π−0u (21) m¨oglich sind (vgl. [9], S. 755).
2.1.3 Beschreibung der Kernbewegung
Im Rahmen der Born-Oppenheimer-N¨aherung bildet die ElektronenenergieEE(r) einen Teil der potentiellen Energie der Kernbewegung:
V(r) =VN N(r) +EE(r) = ZI2·e2
r +EE(r) (22)
(Die KernladungszahlZI des Jodatoms betr¨agt 53.)
Das Jodmolek¨ul ist ein so schweres (d.h. kompliziert aufgebautes) Molek¨ul, dass eine approximative L¨osung der elektronischen Schr¨odinger- Gleichung (5) (z.B.
durch das LCAO/MO- Verfahren) keine guten EE(r)- Werte ergibt (vgl. [6], S.147).
Deshalb bestimmt man in der PraxisV(r) auf einem anderen Weg:
Die Form der potentiellen Energie bestimmt die Kernbewegung. Deshalb kann man auch umgekehrt aus der Kernbewegung (d.h. aus den Schwingungs- und Rotationskonstanten, siehe unten) den Verlauf der potentiellen Energie bestim- men. N¨aheres dazu im AbschnittDie Morse-Funktion.
Die potentielle Energie V innerhalb eines elektronischen Zustands h¨angt nur vom Kernabstand r ab. Deshalb kann die Schr¨odinger-Gleichung (6) in Schwerpunktsystemen in der Form
∇2ΨN +2µ
~2(E−V(r)) = 0 (23)
geschrieben werden, mit der reduzierten Masseµ=MMA·MB
A+MB.
Schreibt man ∇2 in Kugelkoordinaten und separiert die Wellenfunktion gem¨aß:
ΨN(r, θ, φ) = 1
rS(r)Y(θ, φ) (24)
so gelten f¨ur den RadialanteilS und WinkelanteilY die Gleichungen:
1 sinθ
∂
∂θ
sinθ∂Y
∂θ
+ 1
sin2θ
∂2Y
∂φ2 +J(J+ 1)Y = 0 (25) (J = 0,1, . . . Quantenzahl des Gesamtdrehimpulses) und
d2S dr2 +2µ
~2
E−V(r)−J(J+ 1)~2 2µr2
S= 0 (26)
(vgl. [2], S.302 links). (26) beschreibt die Schwingung der Kerne, genauer des Teilchensµ, im PotentialV(r) +J(J+1)2µr2~2, wobei der zweite Summand die Rota- tion des Molek¨uls im (variierenden) Abstandrber¨ucksichtigt. Die L¨osung ergibt f¨ur jeden Wert vonJ einen Satz von Schwingungseigenfunktionen und Energie- eigenwertenE=EvJ, die durch eine Quantenzahlvunterschieden werden. Um diese Energieeigenwerte des”schwingenden Rotators“ approximativ anzugeben, ist es sinnvoll, zuerst die Quantisierung der Rotations- und Schwingungsenergie anhand zweier Grenzmodelle zu untersuchen:
Der starre Rotator(r=const.)
MitV(r) =: 0 und S(r)r =: 1 wird Gleichung (23) zu Gleichung (25) unter der Bedingung, dass
E=J(J+ 1)~2
2µr2 =B∗J(J+ 1) mit B∗= ~2
2µrE2
(27)
Da in der Molek¨ulphysik die Energien spektroskopischer ¨Uberg¨ange gew¨ohn- lich in cm−1- Einheiten gemessen werden, ist es ¨ublich, alle Energiewerte in cm−1 (Wellenzahlen) auszudr¨ucken. Z.B. lauten dann die Rotationsterme:
F(J) :=E(J)
hc = J(J + 1~2)
4πµcr2E =:BJ(J+ 1) mitB= ~2
4πµcr2E
(28)
Der rotationslose Oszillator (J = 0)
Die Schwingungsbewegung, die nur eine Funktion des Kernabstands rist, wird durch Gleichung (26) beschrieben. F¨urJ = 0 wird sie zu
d2S dr2 +2µ
~2(E−V(r)) = 0 (29)
Ihre Eigenfunktionen und Eigenwerte h¨angen von der potentiellen Energie V(r) ab. Wenn V(r) ein stabiles Molek¨ul beschreiben soll, muss es folgende Eigenschaften besitzen:
r→0 :V(r)→ ∞ (30a)
r→ ∞:V(∞)→const. (30b)
f¨ur ein remit 0< re<∞nimmtV(r)ein Minimum an. (30c)
Ganz allgemein kannV(r) als Taylor- Reihe umreentwickelt werden:
V(r) =V(re) +V0(re)(r−re) +V00(re)
2! (r−re)2+V000(re)
3! (r−re)3+. . . (31) DaV(re) gleich Null gesetzt und außerdem das Minimum der potentiellen Ener- gie ist, fallen die beiden ersten Summanden weg.
Befindet man sich nun im Bereich kleiner Auslenkungen um die Gleichge- wichtslage re (d.h. unten im Potentialtopf), so sind die h¨oheren Potenzen klein gegen (r−re)2, und die potentielle Energie des harmonischen Oszillators (vgl.
[4], Kap. 11.2.4)
Vharm(r) = V00(re)
2 (r−re)2=: ke
2 (r−re)2 (32) n¨ahert dort das reale Potential an (siehe Abb.37).keist die Kraftkonstante f¨ur kleine Schwingungen im realen PotentialV(r).
Setzt manVharm(r) in (29) ein, so erh¨alt man als Schwingungsenergieterme die
¨aquidistante Termleiter des harmonischen Oszillators Gharm(v) := E
hc =ωe(v+1
2) mit v= 0,1,2, . . . , (33) ωe=νe
c = 1 2πc
ske
µ (34)
(νeist die klassische Schwingungsfrequenz)
Mit zunehmender Schwingungsauslenkung n¨ahertVharm V(r) immer schlechter an, insbesondere wird Bedingung (30)b verletzt (vgl. Abb.37).
Setzt man nun die anharmonische potentielle Energie (31) in (29) ein und ste- hen die Vorfaktoren der Anharmonizit¨atsterme im Verh¨altnis V002(re V0003!(re) . . . zueinander, was bei den meisten Molek¨ulen richtig ist, so erh¨alt man nach einer St¨orungsrechnung (vgl. [5], S. 93) die Energieterme des anharmonischen Oszillators
G(v) =ωe(v+1
2)−ωexe(v+1
2)2+ωeye(v+1
2)3+. . . (35) wobei die Schwingungskonstanten im Verh¨altnisωeωexeωeye>> . . . zueinander stehen.
Der Oszillator besitzt eine Nullpunktsenergie, deren Termwert G(0) = 1
2ωe−1
4ωexe+1
8ωeye+. . . (36) lautet.
Der Abstand benachbarter Schwingungsterme betr¨agt:
∆G(v+1
2) :=G(v+1)−G(v) =ωe−ωexe(2v+2)+ωeye(3v2+6v+13
4 )+. . . (37) Bei den meisten molekularen Zust¨anden (so auch bei den beiden betrachte- ten des Jod- Molek¨uls) istωexe positiv undωexeωeye, so dass der Abstand der Schwingungsterme mit zunehmendemv abnimmt.
Da das Potential f¨ur große r mit −r16 geht (van- der- Waals- Wechselwirkung zwischen den beiden molek¨ulbildenden Atomen), ist die Zahl der Schwingungs- terme im Potentialtopf endlich. (Dies gilt f¨ur alle potentiellen Energien mit
−r1s, s > 2.) F¨ur ein endliches vdiss wird ∆G(v+ 12) zu Null. Oberhalb von G(vdiss) befindet sich ein Energiekontinuum, in dem das Molek¨ul dissoziiert vorliegt.
Die Dissoziationsenergie D0, gemessen vom tiefsten Niveau v = 0 aus, be- tr¨agt:
D0=
vXdiss
v=0
∆G(v+1
2) (38)
Die DissoziationsenergieDe, gemessen vom Potentialminimum aus, betr¨agt dann:
De=G(0) +D0 (39)
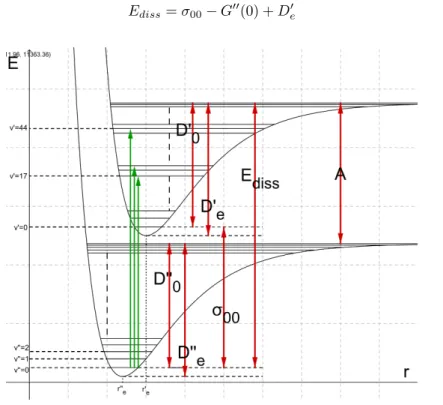
Abbildung 2: Veranschaulichung der Energieniveaus mit Kontinuum und Dissoziationsenergien
Diese Art der Berechnung der Dissoziationsenergie wurde von Raymond Bir- ge und Hertha Sponer entwickelt und nach ihnenBirge- Sponer- Plot genannt.
Zur Bestimmung vonvdiss, tr¨agt man ∆G(v+12) gegenv+12 auf und bestimmt vdissaus dem Schnittpunkt der mithilfe von (37) angelegten Funktion mit der x- Achse. Die Schwingungskonstantenωe,ωexeundωeyekann man dann ebenfalls
aus dieser Funktion ablesen. (vgl. [8]) Die Morse- Funktion
Um mit Hilfe gemessener Molek¨ulkonstanten den Verlauf der potentiellen Ener- gie zu bestimmen, lassen sich zwei Methoden unterscheiden:
Erstens gibt es eine Reihe von analytischen Funktionen, die die Eigenschaf- ten (30) besitzen und zur Approximation der potentiellen Energien realer Mo- lek¨ulzust¨ande verwendet werden. Sie enthalten zun¨achst einige freie Parameter, die dann mittels der gemessenen Molek¨ulkonstanten festgelegt werden.
Die bekannteste ist die Morse- Funktion (vgl. [8]):
V(r) =De(1−e−a(r−re))2
mit re= 2,979 ˚A (40)
Setzt man sie in (31) ein, so erh¨alt man als Schwingungsenergieterme G(v) =a
s~De
πcµ(v+1
2)− ~a2 4πcµ(v+1
2)2 (41)
Aus dem Vergleich mit (35) erh¨alt man ωe=a
s~De
πcµ (42)
ωexe= ~a2
4πcµ (43)
µ= 1,053·10−25 kg entspricht der reduzierten Masse von Jod.
Durch Eliminieren vonaergibt sich De= ωe2
4ωexe
(44) Je besser die berechneten und gemessenen Werte von ωexe und De uber-¨ einstimmen, umso besser approximiert die Morse-Funktion die reale potentielle Energie des betrachteten Zustands.
Zweitenskann man mit der Rydberg-Klein-Rees-Methode (RKR- Methode) die tats¨achliche Potentialfunktion eines Zustands graphisch aus den gemessenen Molek¨ulkonstanten konstruieren (vgl. [12]).
Der schwingende Rotator
Ein reales Molek¨ul vollf¨uhrt gleichzeitig Schwingungen und Rotationen.
Zur Bestimmung der Energieterme des schwingenden Rotators muss man als ers- tes das Modell des starren Rotators aufgeben: Die Tatsache der Molek¨ulschwin- gungen legt nahe, die Rotation besser durch ein Modell des
”nichtstarren Rota-
Abbildung 3:Veranschaulichung der Approximation des realen Poten- tials durch das des harmonischen Oszillator und der Morse-Funktion [2]
tors“ zu beschreiben.
Die elastische Dehnung des Kernabstands w¨ahrend der Rotation wird dabei durch Zuf¨ugung eines ”Dehnungsterms“ im RotationstermF(J) ber¨ucksichtigt (vgl. [5], S. 103 ff).
F(J) =B·J(J+ 1)−D·J2(J+ 1)2+. . . , DB (45) L¨asst man Schwingung und Rotation gleichzeitig zu, so muss man die Ro- tationskonstanteB = 4πcµr~ 2 eines Molek¨uls mit konstantem Kernabstandrso korrigieren, dass sie dessen ¨Anderung w¨ahrend der Schwingung ber¨ucksichtigt.
Da die Schwingungsfrequenz allgemein ca. zwei Zehnerpotenzen gr¨oßer ist als die Rotationsfrequenz, kann man ¨uber den Kernabstandrmitteln (vgl. [5], S.106 ff):
Bv:= ~ 4πcµh1
r2iv (46)
und erh¨alt so f¨ur jedes Schwingungsniveauv einen mittleren WertBv. Rechnungen ergeben (vgl. [5], S. 106 ff), dassBvals Potenzreihe von (v+12) darstellbar ist:
Bv =Be−αe(v+1
2) +γe(v+1 2)2+. . . mitBe= ~
4πcµ 1
r2e, Beaere. . .
(47)
Auf ¨ahnliche Weise erh¨ahlt man einen mittleren Wert der Dehnungskonstan- ten D zu:
Dv :=De+βe(v+1 2) +. . . mitDe= 4B2e
ω2e , Deβe. . .
(48)
Die Termwerte des schwingenden Rotators lauten somit:
T(v, J) =G(v)+Fv(J) =ωe(v+1
2)−ωexe(v+1
2)2+ωeye(v+1
2)3+· · ·+BvJ(J+1)−DvJ2(J+1)2+. . . (49)
Weil die Rotationskonstanten viel kleiner als die Schwingungskonstanten sind, liegen ¨uber jedem SchwingungsniveauG(v) die RotationsniveausG(v) + Fv(J) angeordnet als
”Rotationsleitern “.
Abbildung 4: Veranschaulichung der Struktur der verschiedenen Uberg¨ange [15]¨
Eine sorgf¨altige Studie der feineren Wechselwirkungen zwischen Schwingung und Rotation durch Dunham (vgl. [5], S. 109; [4], Kap. 11.3.6) ergab die Term- werte des schwingenden Rotators, ausgedr¨uckt als Doppelreihe:
T(v, J) =X
lj
Ylj(v+1
2)l·Jj(J+ 1)j (50) Mit großer Genauigkeit gelten z.B. die Entsprechungen:
Y10=ωe, Y20=ωexe, Y30=ωeye, . . . , Y01=Be, . . . (51) so dass man in der Literatur weitgehend dieωe, ωexe, . . . , Be, . . . statt der Ylj weiter verwendet, um die Terme und Spektrallinien zu beschreiben.
Die Schwingungs-RotationstermeT(v, J) eines gegebenen elektronischen Zu- stands haben das Minimum seiner Potentialkurve als Bezugspunkt (Nullpunkt).
Betrachtet man die Gesamtheit aller elektronischen Zust¨ande eines Molek¨uls, so werden die Energieterme ¨ublicherweise auf das Minimum des tiefsten Zustands bezogen. Die Termformel eines angeregten Zustands lautet dann:
T(v, J) =Te+G(v) +Fv(J) (52) wobei Te die Termwertdifferenz zwischen dem Minimum des betrachteten angeregten Zustands und des Grundzustands angibt.
2.1.4 Wechselwirkung zwischen Elektronenbewegung und Kernbe- wegung
Bei der Herleitung der Termformel (?? wurde zwar die Wechselwirkung zwi- schen der Schwingungs- und der Rotationsbewegung der Kerne ber¨ucksichtigt, noch nicht aber die Wechselwirkung zwischen der Kernrotation und der Elek- tronenbewegung. Wechselwirkungen zwischen verschiedenen Bewegungsformen eines atomaren Teilchens k¨onnen qualitativ durch Drehimpuls-Vektormodelle beschrieben werden (vgl. [6], S. 116 ff).
Man geht zun¨achst davon aus, dass gewisse Drehimpulse, von denen jeder mit einer bestimmten Bewegungsform (z.B. der Bahnbewegung der Elektronen, dem Elektronenspin oder der Kernrotation) verkn¨upft ist, unabh¨angig voneinander erhalten bleiben.
Will man dann die Wechselwirkung zwischen bestimmten Bewegungsformen ber¨ucksichtigen, so wird dies im Rahmen des Vektormodells dadurch beschrie- ben, dass die betreffenden Drehimpulsvektoren nicht mehr streng erhalten wer- den, sondern um gemeinsame Feldachsen pr¨azedieren, so dass nur noch ihre quantisierten Komponenten entlang dieser Achsen erhalten bleiben.
Durch die Angabe der Quantenzahlen solcher quantisierter Drehimpulskom- ponenten entlang der Molek¨ulachse wurden z.B. die Elektronenzust¨ande eines zweiatomigen Molek¨uls gekennzeichnet.
Die Schreibweise1Σ0oder etwa3Π0 setzte dabei aber voraus, dass die Kompo- nenten Λ,Σ,Ω einzeln definiert sind, dass also das elektrostatische Kernfeld so groß ist, dassL~ undS~ unabh¨angig voneinander r¨aumlich quantisiert werden.
Wird aber die Wechselwirkung zwischenL~ und S~ relativ st¨arker als deren Wechselwirkung mit dem Kernfeld, koppeln~LundS~ zu einem Elektronen- Ge- samtdrehimpulsJ~e, und nur noch dessen Achsenkomponente Ω ist definiert.
Entsprechend werden die elektronischen Zust¨ande nur noch mit dem Wert von Ω bezeichnet, die beiden Zust¨ande1Σ0und3Π0z.B. gehen also in Zust¨ande Ω = 0 ¨uber
(eventuell noch charakterisiert durch die Bezeichnung des Symmetrieverhal- tens).
Will man nun auch die Wechselwirkung zwischen der Elektronenbewegung und der Kernbewegung ber¨ucksichtigen, so koppelt zu den genannten Elektronen- drehimpulsen der Drehimpuls der Kernrotation, und alle zusammen bilden den GesamtdrehimpulsF~.
Je nach den dominierenden Wechselwirkungen sind verschiedene Kopplungsf¨alle als Grenzf¨alle formulierbar: die Hundschen Kopplungsf¨alle (vgl. [4], Kap. 11.6.4):
Die meisten Molek¨ule zeigen aber ein ¨Ubergangsverhalten zwischen zwei- en (oder mehreren) Kopplungsf¨allen. So liegen die Zust¨ande des Jod- Molek¨uls
”zwischen“ den beiden Kopplungsf¨allen (a) und (c):
Kopplungsfall (a):
Abbildung 5: Hundscher Kopplungsfall (a) Quelle: [4], S.62
Die Kopplung zwischen der Rotation der AtomkerneN und den elektroni- schen Drehimpulsen L und S ist sehr schwach. Dagegen ist die Kopplung des Bahndrehimpulses an die internukleare Achse stark und der Spin koppelt mit dem parallel zur Molek¨ulachse ausgerichteten Magnetfeld.
Daraus folgt:
Ω =|Λ + Σ| J~=Ω +~ N~ mitJ <Ω
(53)
Kopplungsfall (c):
Abbildung 6:Hundscher Kopplungsfall (c) Quelle: [4], S.63
Die Kopplung des BahndrehimpulsesLan die internukleare Achse ist schwach.
Es bildet sich ein elektronischer GesamtdrehimpulsJ~e=~L+S~, der seinerseits an die internukleare Achse koppelt. Die Projektion des Gesamtdrehimpulses Ω koppelt wieder mit der KernrotationN zum GesamtdrehimpulsJ:
J~=Ω +~ N~
mitJ <Ω (54)
Man sieht, dass sich die beiden Kopplungsf¨alle nur gering unterscheiden.
Den Bezeichnungen1Σ0,3Π0, . . . entspricht der Kopplungsfall (a), wo Λ,Σ,Ω und J =|J~| =p
J(J+ 1)~ definiert sind, den Bezeichnungen Ω = 0, . . . ent- spricht der Kopplungsfall (c), wobei Ω und J unabh¨angig quantisiert sind.
Eine St¨orungsrechnung, die die Wechselwirkung zwischen der Elektronen- und der Kernbewegung ber¨ucksichtigt, ergibt kleine additive Beitr¨age zum Schwingungs- und Rotationsterm:
Als Beitrag zum Schwingungsterm erg¨abe sichBv(−Ω2+L2p), wobeiL2p den
zeitlichen Mittelwert der Komponente des elektronischen Drehimpulses senk- recht zur Molek¨ulachse angibt (vgl. [6], S. 201 ff).
Dieser Term soll hier unber¨ucksichtigt bleiben, da Bv ωe. Außerdem ist f¨ur die beiden beteiligten Zust¨ande (siehe unten) Ω = 0 undL2pauch f¨ur den Π- Zustand sehr klein.
F¨ur die Rotationsterme erh¨alt man einen Beitrag Φi(J), der sehr klein ist und auch deshalb hier nicht weiter ber¨ucksichtigt wird, weil die Rotationsstruktur im Detail sowieso nicht aufgel¨ost werden kann (siehe unten).
F¨ur Λ>0 hat Φi f¨ur jedesJ zwei verschiedene Werte, d.h. die Λ-Entartung wird durch die Wechselwirkung zwischen der Elektronen- und der Kernbewegung aufgehoben.
2.2 Zur Interpretation der beobachteten Absorptions¨ uberg¨ ange bei Jod
2.2.1 Zu welchem angeregten elektronischen Zust¨anden sind Ab- sorptions¨uberg¨ange m¨oglich (Auswahlregeln)?
Ein atomares System befinde sich im ZustandEm, es besitze noch weitere (dis- krete) Energieeigenwerte, z. B.En. Ber¨ucksichtigt man nun in der Schr¨odinger- Gleichung dieses Systems die Wechselwirkung mit einem elektromagnetischen Feld, das Strahlung der Wellenzahl En−Ehc m =:σmnenth¨alt, so findet man, dass das System mit einer gewissen Wahrscheinlichkeit in den ZustandEn ubergeht¨ (vgl. [5], S. 18 ff). Dies wird als Absorption oder Emission von Lichtquanten der Wellenzahlσmn interpretiert.
Die Zahl der bei einem Absorptions¨ubergang absorbierten LichtquantenNabsmn der Wellenzahl σmn ist proportional zu (vgl. [5], S. 19 ff)
1. der Zahl der auf das Fl¨achenelement einfallenden Lichtquanten der Wel- lenzahlσmn. (Dabei wird vorausgesetzt, dass die einfallende Strahlung in einem Wellenzahlintervall umσmn, das die ganze Linienbreite ¨uberdeckt, konstante Intensit¨at besitzt. Dies ist bei einer kontinuierlichen Lichtquelle erf¨ullt.)
2. der Zahl der sich im unteren Zustand befindlichen Teilchen in einem der Strahlung ausgesetzten Volumenelement.
3. einer quantenmechanischen Gr¨oße |R~mn|2, die die ¨Ubergangswahrschein- lichkeit misst. R~mn heißt ¨Ubergangsmoment und hat f¨ur elektrische Di- pol¨uberg¨ange (das sind die bei weitem h¨aufigsten ¨Uberg¨ange) die Form
R~mn= Z
Ψ∗nP~Ψmdτ (55)
wobei P~ das elektrische ¨Ubergangs-Dipolmoment des atomaren Systems und Ψmund Ψn die Wellenfunktionen der beteiligten Zust¨ande sind.
Bei homonuklearen Molek¨ulen ist das elektrische Dipolmoment in jedem Au- genblick einer Schwingung oder Rotation aus Symmetriegr¨unden gleich Null.
Deshalb sind alle ¨UbergangsmomenteR~mnf¨ur Schwingungs-Rotations- ¨Uberg¨ange innerhalb eines bestimmten elektronischen Zustands gleich Null. Das Jod-Molek¨ul macht also keine reinen Schwinguns-Rotations- ¨Uberg¨ange.
Andererseits sind bei homonuklearen Molek¨ulen elektronische ¨Uberg¨ange (mit Schwingungs- und Rotationsstruktur) sehr wohl m¨oglich, da sich das momen- tane Dipolmoment w¨ahrend der Neuverteilung der elektronischen Ladung, die der elektronische ¨Ubergang mit sich bringt, ¨andert (vgl. [6], S. 220).
Welche elektronischen ¨Uberg¨ange sind beim Jod-Molek¨ul m¨oglich?
Auskunft dar¨uber geben die Auswahlregeln f¨ur elektronische ¨Uberg¨ange. Man erh¨alt durch Berechnung der ¨Ubergangsmomente R~mn, die nur dann ungleich Null sind, wenn die Quantenzahlen und Symmetrieeigenschaften der beiden je- weils betrachteten Zust¨ande bestimmten Beziehungen gen¨ugen, d.h. wenn sie
”Auswahlregeln“ erf¨ullen.
Man unterscheidet Auswahlregeln, die allgemein gelten, und solche, die nur f¨ur einen bestimmten Drehimpuls-Kopplungsfall gelten (beide betrachteten Zust¨ande m¨ussen zum gleichen Kopplungsfall geh¨oren).
Folgende Auswahlregeln kommen beim Jod-Molek¨ul in Betracht (vgl. [5], S.
240):
g↔u, g=g, u=u(allgemeing¨ultig)
∆Ω = 0,±1
∆Λ = 0,±1 (Kopplungsfall a)
0+↔0+,0−↔0−,0+=0− (Kopplungsfall c)
(56)
Der Grundzustand des Jod- Molek¨uls heißt1Σ+0g.
Als angeregte Zust¨ande (der ersten angeregten Elektronenkonfiguration) kom- men die Zust¨ande1Πu,3Π2u,A3Π1u,B3Π+0u,3Π−0u in Betracht.
Nach den obigen Auswahlregeln sind nun davon die ¨Uberg¨ange1Σ+0g↔3Π−0u und1Σ+0g↔3Π2unicht erlaubt.
Nach [9] wurde der Absorptions¨ubergang zum Zustand 1Πu nicht beob- achtet und ist deshalb vermutlich sehr schwach. Beobachtbar sind Absorpti- ons¨uberg¨ange zu den Zust¨andenA3Π1u undB3Π+0u. Dabei findet der ¨Ubergang X → B ¨uber den ganzen sichtbaren Spektralbereich verteilt (von ca. 500 nm an) recht kr¨aftig statt, w¨ahrend auf dem ¨UbergangX →Anur zu etwa 3−4%
des ¨UbergangsX →B - und zwar vorwiegend im Roten - absorbiert wird (vgl.
[9]).
Das Auftreten und die Auspr¨agung weiterer Bandensysteme im Ultravio- letten (z.B. zwischen 180−200 nm) h¨angen teilweise stark von Temperatur und Druck ab, so dass die experimentelle Realisierung mit großem Aufwand
verbunden ist. Außerdem liegen in diesem Bereich mehrere angeregte Zust¨ande energetisch eng beieinander, so dass die theoretische Analyse und Zuordnung der Banden schwierig wird (vgl. [9]). Es bietet sich also zur spektroskopischen Analyse der ¨UbergangX1Σ+g →B3Π+0u an.
2.2.2 Welche Schwingungs- und Rotationsstruktur besitzt das Ab- sorptionsspektrum?
Durch Lichtabsorption geht das Jodmolek¨ul von einem bestimmten Schwingungs- Rotations- Zustand des GrundzustandsT00=G00(v) +Fv0000(J00) in einen anderen des angeregten ZustandsT0=Te0+G0(v0) +Fv00(J0) ¨uber.
Dementsprechend besitzt das absorbierte Licht die Wellenzahl
σ:=T0−T00=Te0+G0(v0)−G00(v00) +Fv00(J0)−Fv0000(J00) (57) Rotationsstruktur
Zun¨achst soll nur die Rotationsstruktur Fv00(J0)−Fv0000(J00) betrachtet werden.
Mit Formel (49) erh¨alt man (Te0+G0(v0)−G00(v00) =:σ0):
σ=σ0+B0vJ0(J0+1)−Dv0J02(J0+1)2+· · ·−(Bv00J00(J00+1)−D00vJ002(J00+1)2+. . .) (58) Dabei sind f¨ur die Betrachtung der Rotationsstruktur folgende Auswahlre- geln wichtig (vgl. [6], S. 220 ff):
∆J = 0,±1 (J = 0=J = 0) (59)
∆J = 0 verboten bei ¨Uberg¨angen mit Ω = 0↔Ω = 0 (60)
+↔ −,+=+,−=− (61)
s↔s, a↔a, s=a(f¨ur homonukleare Molek¨ule) (62) Bemerkung:
Die beiden letzten Auswahlregeln beziehen sich auf das Symmetrieverhalten der Gesamtwellenfunktion Ψ = ΨE ·ΨN des Molek¨uls (nicht ber¨ucksichtigt:
Kernspin- Anteil). Wie die Elektronenwellenfunktion ΨEkann Ψ durch ihr Sym- metrieverhalten gekennzeichnet werden (vgl. [2], S.290):
Molek¨ulwellenfunktionen werden mit Ψ− bzw. Ψ+ gekennzeichnet, je nach- dem ob die Symmetrieoperation
”Inversion aller Koordinaten“ das Vorzeichen der Wellenfunktion ¨andert oder nicht.
Wellenfunktionen homonuklearer Molek¨ule besitzen eine weitere Symmetrieei- genschaft: Sie werden mit Ψa(antisymmetrisch) bzw. Ψs(symmetrisch) gekenn- zeichnet, je nachdem ob die Symmetrieooperation
”Austausch der Kernkoordi- naten“ das Vorzeichen der Wellenfunktion ¨andert oder nicht.
Betrachtet man die Rotationsniveaus beim ¨Ubergang X → B so sieht man,
dass die Auswahlregeln (61) und (62) nichts Neues gegen¨uber (59) bringen.
Wohl aber schr¨ankt (60) (59) dahingehend ein, dass nur Rotations¨uberg¨ange mit ∆J =±1 m¨oglich sind.
Bezeichnet man die Rotationsquantenzahlen J00 des Grundzustands mitJ, erh¨alt man mit dieser Auswahlregel folgende zwei Linienserien (Zweige):
∆J = +1 :σ=σ0+Fv0(J+ 1)−Fv00(J) =:R(J)R-Zweig (63)
∆J=−1 :σ=σ0+Fv0(J−1)−Fv00(J) =:P(J)P-Zweig (64) (vgl. Abb.7). Mit Gleichung (58) erh¨alt man daraus unter Vernachl¨assigung der Dehnungsterme Dv:
R(J) =σ0+Bv0(J+ 1)(J+ 2)−Bv00J(J+ 1)
=σ0+ 2Bv0 + (3Bv0 −B00v)J+ (Bv0 −B00v)J2 (65)
P(J) =σ0+Bv0(J−1)J −Bv00J(J−1)
=σ0−(Bv0 +Bv00)J+ (Bv0 −Bv00)J2 (66) Mitm :=J + 1 f¨ur den R- Zweig und m:= −J f¨ur den P- Zweig k¨onnen beide Formeln durch eine einzige dargestellt werden:
σ=σ0+ (B0v+Bv00)m+ (Bv0 −Bv00)m2 (67) F¨urBv0 6=Bv00liegen die Wellenzahlenσ=σ(m) auf einer Parabel.
Abbildung 7:Fortrat-Parabel der Rotationslinien des Schwingungs¨uber- gangsv00= 0→v0= 36
Quelle: [1], S. 28a
P(J) =−0,058J−0,016J2
R(J) = 0,042 + 0,026J−0,016J2 (68) Formeln und Bild folgen aus (66),(65),(47),(48), sowie den Rotationskon- stanten aus [10] und [11].
Dies hat zur Folge, dass die Wellenzahlenσentweder zu großen oder zu klei- nen Wellenzahlen hin f¨ur ein gewissesm=mseinen Maximalwert (Scheitelwert) annehmen.
Man erh¨altms aus dmdσ = 0 zu:
ms=− B0v+Bv00
2(Bv0 −Bv00) (69)
Setzt manmsin Gleichung (67) ein, so erh¨alt man f¨ur den Abstand zwischen der ”Nulllinie“ σ0 (sie entspricht dem - verbotenen, vgl. (59) - rotationslosen Ubergang zwischen den beiden betrachteten Schwingungsniveaus, d.h.¨ J00 = J0= 0 und dem Scheitel
σs−σ0=−(B0v+Bv00)2
4(Bv0 −B00v) (70)
Man kann unterscheiden:
σs> σ0, d.h. der Scheitel liegt auf der kurzwelligen (blauen) Seite vonσ0, wennB0v< Bv00
σs< σ0, d.h. der Scheitel liegt auf der langwelligen (roten) Seite vonσ0, wennB0v> Bv00
Inwieweit kann die vorliegende spektroskopische Apparatur die Rotationsstruktur aufl¨osen?
Die Aufl¨osung des verwendeten Spektrometers liegt bei ca. 0,4 nm. Kann man damit zwei benachbarte (∆m = 1) Rotationslinien aufl¨osen? Ihr Abstand be- tr¨agt:
∆σ:=σ(m)−σ(m−1) = 2Bv00+ 2m(B0v−Bv00) (71) Setzt man hier die aus der Literatur bekannten (vgl. [10],[11]) Werte der Rotati- onskonstanten der beiden Jodzust¨ande ein (B00e = 0,037 cm−1,B0e= 0,029 cm−1, α0e= 0,00016 cm−1), so erh¨alt man etwa f¨urm≈30: ∆σ≤0,6 cm−1. Dies ent- spricht bei einer mittleren Wellenl¨ange von 570 nm einer Wellenl¨angendifferenz von ∆λ≈0,015 nm. Die Aufl¨osung des Spektrometers reicht also bei Weitem nicht aus, um einzelne Rotationslinien aufzul¨osen.
Dies hat zur Folge, dass die Vielzahl an Rotations¨uberg¨angen eines bestimmten Schwingungs¨ubergangs im Spektrum nur als breite Absorptions
”banden“ sicht- bar wird, die auf einer Seite durchσs, den
”Bandenkopf“, scharf begrenzt wird.
Da f¨ur die beiden betrachteten Jodzust¨ande Bv0 < Bv00 ist, istσs > σ0, d.h. es
ist zu erwarten, dass die Absorptionsbanden des Jod- Molek¨uls nach der blauen Seite hin scharf begrenzt und
”rotschattiert “sind.
Der Abstand zwischen dem Bandenkopf und der Nulllinie betr¨agt mit der Formel (70)σs−σ0≤0,13 cm−1, d.h.σsundσ0liegen innerhalb der Meßungenauigkeit der Apparatur.
Tr¨agt man die Fortrat-Parabel auf und betrachtet die Verteilung der Pro- jektionen auf die σ-Achse, so sieht man, dass unter der Voraussetzung, dass die Intensit¨aten der Rotations¨uberg¨ange einer Schwingungsbande von gleicher Gr¨oßenordnung sind, in der Gegend des Bandenkopfes und der Nulllinie beson- ders viel Licht absorbiert wird. N¨aheres dazu im AbschnittBesetzungsverteilung auf die Rotationsniveaus.
Die Schwingungsstruktur des ¨Ubergangs
Zur Untersuchung der Schwingungsstruktur h¨alt man beide betrachteten Zust¨ande rotationslos, d.h.J0 =J00= 0 , alsoFv00(J00)−Fv0(J0) = 0. Mit Gleichungen (57) und (35) erh¨alt man dann:
σ=Te0+G0(v0)−G00(v00)
=Te0+ωe0(v0+1
2)−ωe0x0e(v0+1
2)2+ω0ey0e(v0+1 2)3+. . .
−(ωe00(v00+1
2)−ωe00x00e(v00+1
2)2+ωe00y00e(v00+1
2)3+. . .)
(72)
Es gibt keine strengen Auswahlregeln f¨ur die Schwingungsquantenzahlv, es kann im Prinzip jedes Schwingungsniveau des unteren mit jedem Schwingungs- niveau des oberen Zustands kombinieren.
Wenn die Schwingungsniveaus relativ zumv= 0-Niveau jedes Zustands gemes- sen werden, erh¨alt man aus den Gleichungen (35) und (36):
σ=σ0+ω00v0−ω00x00v02+ω00y00v03+· · ·−(ω000v00−ω000x000v002+ω000y000v003+. . .) (73) Dabei gibt:
σ00=Te0+G0(0)−G00(0)
=Te0+1 2ωe0 −1
4ωe0x0e+1
8ωe0y0e+. . .
−(1 2ω00e−1
4ω00ex00e+1
8ωe00ye00+. . .)
(74)
die Wellenzahl des ¨Ubergangsv0= 0→v00= 0 an.
Zur Analyse der Schwingungsstruktur ist es n¨utzlich, Banden in
”Progressionen“
zusammen zu fassen: Zu einer Progression geh¨oren ¨Uberg¨ange, die von einem bestimmten Schwingungsniveau eines Zustands (z.B. v00 = 0) zu aufeinander-
folgenden Niveaus des anderen Zustands (z.B.v0 =. . . ,6,7,8, . . .) f¨uhren. Eine
”v00-Progression“ besteht aus Banden, deren Wellenzahlen mit wachsendem v0 zunehmen. Sie endet in einem Kontinuum, wo der obere elektronische Zustand dissoziiert.
Um die Energie zu berechnen, bei der das Jod-Molek¨ul dissoziiert, findet man durch Betrachtung von Abb.8:
Ediss=σ00−G00(0) +D0e (75)
Abbildung 8:Veranschaulichung aller Energien f¨ur den Grundzustand und den ersten angeregten Zustand
2.3 Zur Interpretation der beobachteten Intensit¨ aten der Absorptionsbanden
Nachdem die Zahl der m¨oglichen ¨Uberg¨ange durch die Auswahlregeln einge- schr¨ankt wurde, bleibt nun noch zu untersuchen, wovon die St¨arke der Absorp- tion einer bestimmten (festen) einfallenden Lichtintensit¨at der Wellenzahlσmn
auf einem erlaubten ¨UbergangEm→En abh¨angt.
Man muss dazu laut Abschnitt 2.1 die zahlenm¨aßige Verteilung der Jod- Mo- lek¨ule auf die m¨oglichen unteren Niveaus und die Gr¨oße der ¨Ubergangsmomente wissen.
2.3.1 Wie sind die Jod- Molek¨ule auf die Schwingungs- und Rotati- onsniveaus des Grundzustands verteilt?
Jeder Energiezustand eines zweiatomigen Molek¨uls sei identifiziert durch die QuantenzahlenJ undv, sowie eine Zahl , die den elektronischen Zustand an- gibt. Dann befinden sich nach dem Maxwell- Boltzmann- Verteilungsgesetz von N vorliegenden Molek¨ulen, die sich bei einer Temperatur T in thermischem Gleichgewicht befinden, ein Anteil von:
NvJ =N gvJ
exp(−E(,v,J)kT ) Qi
(76) Molek¨ulen in einem bestimmten Energiezustand E(, v, J). Dabei ist
Qi= X
,v,J
gvJexp(−E(, v, J)
kT ) (77)
die”innere Verteilungsfunktion“. Die statistischen GewichtegvJ geben die Entartung der Niveaus (, v, J) an.
Man nimmt nun an, dass die gesamte innere Energie eines Molek¨uls separat aufgespalten werden kann in
E(, v, J) =Ee() +Ev(v) +Er(J) (78) Dies impliziert, dass das Molek¨ul separate elektronische, Schwingungs- und Ro- tationsfreiheitsgrade besitzt, die unabh¨angig angeregt werden k¨onnen.
(Dass diese Annahme nur N¨aherungscharakter hat, sieht man z.B. daran, dass die Rotationskonstanten Bv = Be−αe(v+ 12) + . . . sehr wohl von v abh¨angen und weiter die Konstanten Be, αe, . . . vom elektronischen Zustand abh¨angen). Unter dieser Annahme kann man zeigen (vgl. [6], S. 216 ff), dass sichNvJ schreiben l¨asst als
NvJ =N(g
exp(−EekT())
Qe
)
| {z }
f
(gv
exp(−EkTv(v)) Qv
)
| {z }
fv
(gJ
exp(−EkTr(J)) Qr
)
| {z }
fJ
(79)
Dabei gebenQe, Qv undQJ die Verteilungsfunktionen f¨ur verschiedene Ar- ten von Freiheitsgraden an:
Qe=X
gexp(−Ee() kT ), Qv =X
v
gvexp(−Ev(v) kT ), Qr=X
J
gJexp(−Er(J) kT )
(80)
g, gv, gJ geben die Entartungen des betrachteten elektronischen, Schwingungs-
und Rotationszustands an.
Die Faktorenf, fv undfJ sind die Bruchteile von Molek¨ulen im elektroni- schen Zustand, im Schwingungszustandv und im RotationszustandJ bei der TempearaturT.
Diese Faktoren und deshalb auch die relative Verteilung der Molek¨ule in jedem Freiheitsgrad sind (in der betrachteten N¨aherung) unabh¨angig voneinan- der.
Wie sieht nun die Verteilung beim Jod- Molek¨ul aus?
1. Bei 300 K betr¨agt kT = 0,2585 eV (208,5 cm−1).
Der erste angeregte elektronische Zustand des Jodmolek¨uls A3Π1u liegt ca. 12000 cm−1uber dem Grundzustand, so dass sich¨ fE≈10−26bei 300 K ergibt. Es befinden sich also so gut wie alle Molek¨ule im elektronischen Grundzustand.
2. Interessant ist beim Jod- Molek¨ul die Besetzungsverteilung auf die Schwin- gungsniveaus des Grundzustands:
F¨ur die Schwingungen eines zweiatomigen Molek¨uls istgv= 1.
Da die (unteren) Schwingungsniveaus durch die FormelG00(v) =ω00e(v+
1
2)−ω00ex00e(v+12)2gut beschrieben werden und nach [10]ω00e = 214,5 cm−1 undω00ex00e = 0,61cm−1 betragen, ergibt sich:
Qv=X
v
exp(−Ev(v) kT ) =X
v
exp(−G00(v)hc
kT )≈0,9357 (81) und f¨ur die Besetzungsverteilung auf die Schwingungsniveaus des Grund- zustands das Bild.
Infolge der großen Masse des Jod- Molek¨uls ist das Schwingungsquantω00e so klein, dass bei Zimmertemperatur auch die Schwingungsniveausv00 = 1,2, . . . merklich besetzt sind.
3. Besetzungsverteilung auf die Rotationsniveaus:
Der GewichtsfaktorgJl¨asst sich als Produkt schreiben:gJ=gn·(2J+ 1).
2J+ 1 gibt die Entartung des J-ten Rotationsniveaus an, denn der Dre- himpulsvektorJ~ mit |J~|=p
J(J+ 1)~ kann (2J+ 1) verschiedene Ori- entierungen im Raum annehmen (vgl. [6], S.179). Um den Gewichtsfaktor gn zu ber¨ucksichtigen, muss man bei der Gesamtmolek¨ulwellenfunktion noch einen Kernanteil Ψn als Faktor ber¨ucksichtigen. Ψn wird aus den jeweils (2I + 1)- fach entarteten Wellenfunktionen der einzelnen Kerne konstruiert (I: Kernspin). Mit wachsendem J ist Ψn abwechselnd sym- metrisch und antisymmetrisch bzgl. Kernaustausch. Man kann nun zei- gen, dass bei homonuklearen Molek¨ulen symmetrische Wellenfunktionen Ψsn auf gsn = (2I + 1)(I + 1) verschiedene Weisen aus den Einzelkern- wellenfunktionen konstruiert werden k¨onnen, w¨ahrend antisymmetrische Wellenfunktionen Ψan aufgna = (2I+ 1)I verschiedene Weisen.
Abbildung 9: Thermische Besetzungsverteilung der Schwingungsnive- aus des Jodgrundzustands beiT = 300K.
Diese Variation ingn ¨andert die Besetzung aufeinander folgender Rotati- onsniveaus im Verh¨altnis
gsn
gan =I+ 1
I (82)
Da der KernspinI beim Jod- AtomI= 52 bet¨agt, lautet diese Verh¨altnis beim Jod- Molek¨ul 7:5.
gnbleibt aber im Weiteren unber¨ucksichtigt, da der Einfluss dieser Varia- tion l¨angst nicht aufgel¨ost werden kann und andererseits die Grobstruktur einer Schwingungsbande nicht beeinflusst wird.
fJ lautet dann:
fJ = (2J+ 1)exp(−F(J)hckT ) Qr
mitQr= X∞ J=0
(2J+ 1) exp(−F(J)hc kT )
(83)
Qrkann man f¨urT = 300 K f¨ur das Jod- Molek¨ul (Be= 0,037 cm−1, vgl.
[10]) zu 2,82·103 approximieren (vgl. [6], S.187).
F¨ur die unteren Schwingungszust¨ande des Grundzustands ergibt sich mit diesemQr, F(J) = BeJ(J + 1)(v klein!) und T = 300 K als Besetzungs- verteilung der Rotationsniveaus.
![Abbildung 3: Veranschaulichung der Approximation des realen Poten- Poten-tials durch das des harmonischen Oszillator und der Morse-Funktion [2]](https://thumb-eu.123doks.com/thumbv2/1library_info/5087763.1654105/15.892.302.592.191.497/abbildung-veranschaulichung-approximation-realen-poten-harmonischen-oszillator-funktion.webp)