–
Selbstorganisierende Proteine zur Herstellung funktionaler Beschichtungen
D I S S E R T A T I O N
Zur Erlangung des akademischen Grades
Doctor rerum naturalium (Dr. rer. nat.)
vorgelegt
der Fakultät Mathematik und Naturwissenschaften der Technischen Universität Dresden
von
Dipl. biol. Jan Tobias Günther
geboren am 09.03.1979 in Wurzen Eingereicht am 25.6.2014
Diese Dissertation wurde in der Zeit von 02/2008 bis 05/2012 im Institut für Ressourcenöko- logie und von 06/2012 bis 01/2014 im Helmholtz-Institut für Ressourcentechnologie
am Helmholtz-Zentrum Dresden-Rossendorf angefertigt
1. Gutachter: Prof. Dr. Alexander Eychmüller 2. Gutachter: Prof. Dr. Michael Göttfert
An dieser Stelle möchte ich all jenen danken, die zum Gelingen dieser Arbeit beigetragen und mich während dieser Zeit begleitet haben.
Herrn Dr. Johannes Raff möchte ich danken für die Betreuung meiner Arbeit und seine aktive Unter- stützung, in allen Bereichen meiner Forschungstätigkeit. Danke – für die vielen guten Gespräche, die immer wieder Stoff für neue Ideen lieferten.
Frau Dr. Katrin Pollmann danke ich ebenfalls für die Betreuung meiner Arbeit und die fachliche Un- terstützung in Form von zahlreichen Diskussionen.
Ich danke beiden, dass ich im Rahmen des Projekts NanoFoto die Möglichkeit erhielt, meine Disserta- tion zu diesem spannenden Thema anzufertigen. Vielen Dank auch für die Anschaffung des Raster- kraftmikroskops.
Für die Begutachtung der Arbeit danke ich Herrn Prof. Alexander Eychmüller, der mir in seiner Vorle- sung die Welt der Nanopartikel näher brachte und über die Jahre viel Geduld mit mir hatte. Herrn Prof. Michael Göttfert danke ich ebenfalls für die Begutachtung der Arbeit.
Ein besonderes Dankeschön geht an meine Kollegen der ehemaligen Nachwuchsforschergruppe Na- noBio, welche zwar mittlerweile verteilt auf das Institut für Ressourcenökologie des HZDR und das Helmholtz-Institut Freiberg für Ressourcentechnologie aber dennoch Hand in Hand an spannenden und aktuellen Forschungsthemen arbeiten. Speziell danke ich Frau Dr. Sabine Kutschke, Frau Caroline Bobeth, Frau Monika Dudek und Herrn Falk Lehmann für die unterbrechungsfreie Bereitstellung von S-Layer-Proteinen. Frau Dr. Ulrike Weinert danke ich für zahlreiche fruchtbare Diskussionen und Un- terstützung in hektischen Momenten. Herrn Matthias Suhr danke ich für die perfekte Zusammenar- beit bei dem Thema Zellimmobilisierung. Frau Dr. Franziska Lederer danke ich ebenfalls für viele gute Diskussionen und die tolle Zusammenarbeit bei verschiedenen Themen.
Für die Durchführung von Lichtstreuexperimenten danke ich Herrn Stephan Weiß. Für Analysen mit- tels Elektronenmikroskopie und Röntgenphotoelektronenspektroskopie bedanke ich mich bei Frau Elfie Christalle, Herrn Dr. Arndt Mücklich sowie Herrn Dr. Hellfried Reuther.
Ganz herzlich danke ich auch der Projektgruppe BioBASE, allen voran Herrn Dr. Jürgen Hofinger, wel- cher durch unermüdliche Beratung und Unterstützung maßgeblich für den Erfolg und das Voran- kommen der zukünftigen Firma Biconex verantwortlich war und ist.
Meine Familie und meine Freunde haben immer hinter mir gestanden. Dafür möchte ich ihnen an dieser Stelle auch meinen Dank aussprechen. Danke für die moralische und tatkräftige Unterstüt- zung.
Diese Arbeit wurde finanziert über die Projekte NanoFoto, HEF BioBASE und EFT BioBASE. Die Förde- rung dieser Projekte erfolgte vom BMBF und BMWi. Vielen Dank für die Bereitstellung der For- schungsgelder.
I
Inhaltsverzeichnis
1 Einleitung ... 1
1.1 Morphologie der S-Layer ... 1
1.2 Lokalisation und Verankerung in der Zellhülle ... 3
1.3 Selbstassemblierung und Isolierung ... 4
1.4 Funktionen von S-Layern in der Zelle ... 5
1.5 Einsatzmöglichkeiten von S-Layer-Proteinen in der Nanotechnologie ... 7
1.6 Ursprung der verwendeten Proteine ... 10
2 Zielstellung ... 11
3 Funktionsweise von S-Layer-Proteinen ... 12
3.1 Ergebnisse ... 12
3.1.1 Abbildung nativer S-Layer auf lebenden Organismen ... 12
3.1.2 S-Layer-Rekristallisation auf technischen Oberflächen ... 18
3.2 Diskussion ... 52
3.2.1 Abbildung nativer S-Layer auf lebenden Organismen ... 52
3.2.2 S-Layer-Rekristallisation auf technischen Oberflächen ... 55
4 Anwendungsbeispiele mit S-Layer-Proteinen ... 60
4.1 Anwendungsbeispiel (I) – Beschichtung von Nickelmikrosieben ... 62
4.2 Anwendungsbeispiel (II) – Pt-Nanopartikelbeschichtungen ... 66
4.3 Anwendungsbeispiel (III) – Pd-Nanopartikelbeschichtung für CNT-Synthese ... 72
4.4 Anwendungsbeispiel (IV) – Polyelektrolythohlkugeln ... 76
4.5 Anwendungsbeispiel (V) – Veredelung von technischen Textilien ... 82
4.6 Anwendungsbeispiel (VI) – Metallbeschichtungen auf Kunststoffen ... 85
4.6.1 Vorbeschichtung von IXEF ... 87
II
4.6.2 Metallisierung und Prüfung der Haftfestigkeit ... 91
5 Zusammenfassung und Ausblick ... 94
6 Material und Methoden ... 98
6.1 Chemikalienliste ... 98
6.2 Geräteliste ... 99
6.3 Proteinbiochemische Methoden ... 101
6.3.1 SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) ... 101
6.3.2 Proteinquantifizierung nach Lowry ... 102
6.3.3 Isolierung der bakteriellen Hüllproteine ... 103
6.4 Oberflächenreinigung und Oberflächenmodifizierung ... 105
6.4.1 Reinigung ... 105
6.4.2 Aminosilanisierung ... 106
6.4.3 Polyelektrolytbeschichtung ... 107
6.4.4 Beschichtung mit S-Layer-Proteinen ... 108
6.4.5 S-Layer-basierte Nanopartikel-Synthese ... 110
6.5 Methoden für die Charakterisierung von Oberflächeneffekten und Oberflächen ... 111
6.5.1 Rasterelektronenmikroskopie und XPS ... 111
6.5.2 Transmissionselektronenmikroskopie ... 111
6.5.3 Rasterkraftmikroskopie (AFM) ... 112
6.5.4 QCM-D ... 115
6.6 Dynamische Lichtstreuung und Molmassenbestimmung ... 119
6.7 Polyelektrolythohlkugelsynthese ... 120
6.8 Kunststoffgalvanik ... 122
Verzeichnisse ... 126
Abkürzungsverzeichnis ... 126
Abbildungsverzeichnis ... 127
III Tabellenverzeichnis ... 130 Literatur ... 132 Publikationen ... 138 Anhang ... A Herstellung von Monomerlösungen – Molmassenbestimmung ... A Aufnahmen der S-Layer am lebenden Bakterium ... C Ermittlung der Höhe einer S-Layerschicht ... D Einfluss der Proteinkonzentration auf die SLP-Rekristallisation ...E Anwendungsbeispiel (II) – XPS-Übersichtsspektren ... F Anwendungsbeispiel (IV) – Formelherleitung ... G Anwendungsbeispiel (VI) – weitere QCM-D-Ergebnisse ... G
1
1 Einleitung
Ein häufiges Merkmal von Vertretern aller phylogenetischen Gruppen von Prokaryonten ist das Auftreten von Hüllproteinen. Bakterielle surface layer Proteine (SLP) bilden bei vielen Bakterien und nahezu allen Archaeen einen Teil meist die äußere Lage der Zellhülle. Diese mit Poren durchsetzte Schicht umschließt die gesamte Zelle und wird als „surface layer“ bzw.
S-Layer bezeichnet1. Während Nutzen und Funktion der Proteinschicht bei verschiedenen Organismen sehr unterschiedlich sein können, ist die Selbstassemblierung eine gemeinsame Eigenschaft der Proteine2. Erstmals beschrieben wurden S-Layer 1952 von A. L. Houwink und J. B. Le Poole bei der Betrachtung von Zellwand-Fragmenten des Bakterienstamms Spirillum serpens. Die erstmalige Beschreibung bei Archaeen erfolgte 1956 ebenfalls durch Houwink bei dem Stamm Halobacterium salinarum3. Als gemeinsamer Zellwandbestandteil bei eini- gen grampositiven Bakterien wurden die regelmäßigen Proteingitter erst Jahre später identi- fiziert4.
1.1 Morphologie der S-Layer
S-Layer sind parakristalline Proteinmembranen, die in der Regel aus identischen Unterein- heiten gebildet werden. Auf Grund der mangelnden Fernordnung der Untereinheiten spricht man von Parakristallinität. In der Regel handelt es sich um einschichtige Strukturen, wobei auch mehrlagige Mehrkomponenten-S-Layer nachgewiesen wurden, etwa bei Lampropedia hyalina5. Eine Klassifizierung der S-Layer erfolgt nach Gesichtspunkten der Symmetrie. Abbil- dung 1 zeigt diese Unterteilung der S-Layer, bei denen es sich um Homopolymere mit regel- mäßig angeordneten Untereinheiten handelt. Aus der Symmetrie ergibt sich auch die Anzahl der Untereinheiten je Einheitszelle. In der Natur wurden bisher schräge Symmetrien mit ein oder zwei Untereinheiten, hexagonale Symmetrien mit drei oder sechs Untereinheiten sowie quadratische Symmetrien mit vier Untereinheiten gefunden.
2 Abbildung 1: Schematische Darstellung der möglichen Gitter-Symmetrien nach Sleytr et. al. 4
S-Layer können in folgenden Symmetrien kristallisieren: schräg (p1 und p2), trigonal (p3), tetragonal (p4) oder hexagonal (p6)
Da die S-Layer in der Regel aus identischen Untereinheiten gebildet werden, weisen die Pro- teinschichten Poren einheitlicher Form und Größe auf. Bei vielen S-Layern können zwei oder mehr Porenklassen unterschieden werden, welche im Bereich von etwa 2-8 nm liegen6-11.
Abbildung von S-Layern auf Organismen
Der Beginn der Abbildung von S-Layer-Proteinen mittels mikroskopischer Methoden war zugleich ihre Entdeckung. In den Anfängen der Forschung um diese strukturierten Proteinla- gen kamen vor allem die Transmissions- und Rasterelektronenmikroskopie mit komplexen Anforderungen an die Probenvorbereitung zum Einsatz. Letztere in Kombination mit der aufwendigen Gefrierbruchtechnik8. Mit der Entwicklung der Rasterkraftmikroskopie im Jahre 198612 entstand eine Technik, die es erstmals ermöglichte biologische Proben unter physio- logischen Bedingungen zu untersuchen und zu manipulieren13-16. Es folgten intensive Unter- suchungen der verschiedensten Organismen und ihrer S-Layer17-20. Die Rasterkraftmikrosko- pie erfordert die Fixierung der Probe auf einem glatten Probenträger. Die Untersuchung der S-Layer erfolgte daher häufig in vitro nach der Isolierung vom Bakterium. Eine größere Her- ausforderung ist dagegen die Abbildung auf den Organismen, da diese sich auf Grund ihrer Größe schlechter auf dem Substrat immobilisieren lassen. Es gibt eine Reihe von Ansätzen
3 mit diversen Vor- und Nachteilen. Das Antrocknen ganzer Zellen ist eine eher ungünstige Methode, da der Wasserverlust zu Defekten wie Deformierungen und Zellwandbrüchen an den empfindlichen Proben kommen kann21, 22. Selbst Kritisch-Punkt-Trocknung kann bei fei- nen Strukturen Probleme verursachen23. Darüber hinaus führt Trocknung sehr oft zur Inakti- vierung der Organismen24-26 was ungünstig für in vivo Beobachtungen ist. Besser geeignete Methoden wie die Adsorption der Zellen an modifizierte Substrate sind häufig nicht für alle Organismen gleichermaßen geeignet27. Eine sehr effektive Methode ist die Immobilisierung von Zellen in Filtermembranen28, 29. Sie eignet sich jedoch nur für kugelförmige Zellen und nicht für die filamentösen Bacillus und Lysinibacillus Stämme.
1.2 Lokalisation und Verankerung in der Zellhülle
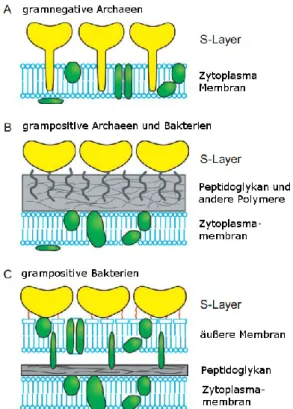
Die Zellwände grampositiver Bakterien enthalten in der Regel eine Vielzahl von Polysaccha- riden, welche zum Großteil kovalent an das Peptidoglykan-Netzwerk (PG) gebunden ist. Un- terteilt werden diese Polymere in Teichonsäuren, Teichuronsäuren und andere neutrale oder saure Polymere, die zu keiner der beiden Gruppen gehören. Diese Polymere werden in der Literatur als sekundäre Zellwand-Polymere (SCWPs) bezeichnet. SCWPs sind in der Ver- gangenheit als Bindeglieder zwischen dem PG-Netzwerk und den S-Layern identifiziert wor- den. Die S-Layer sind nicht kovalent mit dem darunter liegenden SCWPs verbunden. Diese Verankerung vermitteln Motive im Protein, die als Zellwand-Bindedomäne (cell wall binding domain, CWBD) bezeichnet werden. Derartige Motive in der Primästruktur der Proteine, die so genannten SLH-Domänen (surface layer homology domain), wurden erstmals bei SLP von Bacillus sp. und Clostridium sp. identifiziert30. SLH-Domänen finden sich aber nicht nur bei SLPs sondern auch bei anderen extrazellulären Proteinen von grampositiven Bakterien und bei Membranproteinen der äußeren Membran bei gramnegativen Bakterien30-32. Diese SLH- Domänen der SLPs von Bacillus sp. und Clostridium sp. binden spezifisch jedoch nicht kova- lent an das Peptidoglycan33 oder an Peptidoglycan assoziierte SCWP34-36 . Andere SLP, bei- spielsweise von Vertretern der Gattung Lactobacillus, beinhalten keine SLH-Domäne, tragen jedoch andere CWBDs für die Verankerung in Zellwandstrukturen, wobei eine Bindung an Teichonsäuren vermutet wird37-39.
4 Die Verankerung der SLPs bei Archaeen erfolgt durch säulenartige Fortsätze direkt in der Cytoplasmamembran, welche häufig aus Tetraetherlipiden bestehen und so bis zu 70 nm von der Cytoplasmamembran entfernt sein10, 40-42 können.
Abbildung 2: Schematische Darstellung des Zellwandaufbaus von Prokaryonten nach Sleytr et. al.4
1.3 Selbstassemblierung und Isolierung
Die Proteine haben die Fähigkeit zur Selbstassemblierung, was zu zweidimensionalen porö- sen Proteinanordnungen führt. Dieser Vorgang ist ein entropiegetriebener Prozess. Die Selbstorganisation in vivo bzw. der Einbau neuer Monomere während des Zellwachstums erfolgt im Bereich der Wachstumszonen der darunter liegenden Schichten der Zellwand. Die Einlagerung kann bei grampositiven und -negativen Bakterien an definierten Domänen erfol- gen oder ist statistisch verteilt43-45. Störungen der Gitterstruktur, wie sie an Zellpolen zu fin- den sind, ergeben sich aus rein geometrischen Erfordernissen. Diese Bereiche wurden als bevorzugte Einbaustellen vermutet und bestätigt46-48.
Methoden zur Isolierung der Proteine unterscheiden sich je nach Aufbau der Zellwand und der Verankerung der Proteine in den unterliegenden Schichten. Es hat sich gezeigt, dass die Stabilität der Untereinheiten zueinander größer ist als zur unterliegenden Zellwandkompo-
5 nente. Proteine, deren Gitterstruktur nicht wie bei vielen Archaeen durch kovalente Bindun- gen stabilisiert ist, können in aller Regel durch hohe Konzentrationen chaotroper Salze wie Guanidin-Hydrochlorid oder Harnstoff desintegriert werden. Gereinigte desintegrierte Pro- teinmonomere können durch Entfernen der chaotropen Substanzen wieder hochgeordnete Strukturen bilden. Die Gitterkonstanten entsprechen dabei meist denen der in vivo vorkom- menden S-Layer, wobei Unterschiede der Gitterkonstanten zwischen in vivo und in vitro as- semblierten S-Layern dokumentiert sind49-52. Die in vitro Rekristallisation kann zur Bildung von planen, zylindrischen oder sphärischen Mikrostrukturen führen8.
1.4 Funktionen von S-Layern in der Zelle
Eine Funktion, die allen S-Layern gemein ist, konnte bisher nicht identifiziert werden. Tat- sächlich sind die Funktionen der S-Layer in verschiedenen Organismen sehr unterschiedlich.
Insbesondere bei Archaeen hat die Proteinstruktur die Funktion der Formgebung und Stabili- sierung. SLP bilden eine Proteinhülle, die ähnlich einem Exoskelett für eine erhöhte Stabilität der Zellhülle verantwortlich ist3. In diesem Zusammenhang wurde auch eine erhöhte Resis- tenz gegen osmotischen Druck in hypertonen Umgebungen diskutiert53. Auf Grund der ein- heitlichen Porengröße der Proteinhülle kann diese auch als Filterschicht für Makromoleküle dienen. Die zahlreichen Aminosäurereste der Proteine können die Zelle durch Adsorption von Schwermetallen vor deren schädlicher Wirkung schützen.
Bei vielen Archaeen bildet die Proteinhülle mit ihren langen säulenartigen Fortsätzen in Kombination mit der äußeren porösen Deckschicht einen Periplasma-ähnlichen Reaktions- raum. Die einheitliche Ausschlussgrenze für Partikel hat auch Einfluss auf die Verteilung von extrazellulären Enzymen, welche aus dem Organismus ausgeschleust werden. Die regelmä- ßige Proteinmatrix kann für den Organismus als Trägerstruktur zur Bindung von Exoenzymen dienen54.
S-Layer können aber auch eine gewichtige Rolle bei der Erkennung und Adhäsion an Oberflä- chen spielen. Im Falle einiger S-Layer wurde ihre Beteiligung bei der Adhäsion und Invasion von Wirtszellen im Beispiel von Bacillus anthracis55, 56 oder Bacteroides forsythus57 beschrie- ben. Besonders diese Beteiligung der S-Layer an der Pathogenität der Organismen verdeut-
6 licht auch die Rolle der Proteinhülle als Virulenzfaktor. Die Virulenz von Hüllproteinschichten ist jedoch nicht auf Zellerkennung und -bindung beschränkt. In ihrer Schutzfunktion für das Bakterium können S-Layer Phagozytose verhindern und somit zu Resistenzen der Organis- men gegen das lytische System des Wirtes führen54, 58, 59
. Die Veränderungen der Hüllpro- teinschicht durch Rekombination wurden sowohl bei pathogenen als auch nicht pathogenen Prokayonten beobachtet und dienen vermutlich in beiden Fällen zur Anpassung an veränder- te Umweltbedingungen60-62. Bei pathogenen Organismen bedeutet das, trotz der Abwehrre- aktionen im Wirt zu überleben61. Bei nichtpathogenen Organismen wurde dieser Vorgang zum Beispiel bei Erhöhung des Sauerstoffgehalts im Medium beobachtet. Dabei wird die energieaufwendige Stressantwort durch Bildung kleinerer SLP und damit einhergehender Energieeinsparung unterstützt62.
Prokaryoten, deren zelluläre Mechanismen zur Selbsterhaltung und Vermehrung nur durch ihre Zellwand von ihrer mitunter feindlichen Umgebung getrennt sind, benötigen Strategien und Mechanismen sich vor Umwelteinflüssen zu schützen. S-Layer erfüllen in einigen Orga- nismen auch die Aufgabe einer Schutzschicht. Es wurde bei den S-Layer-tragenden Bakteri- enstämmen der Gattungen Aquaspirillum serpens und A. sinuosum sowie Caulobac- ter crescentus eine Resistenz gegenüber den Stämmen der Bacterivoren Bdellovib- rio bacteriovorus festgestellt, die bei ihren S-Layer-freien Pendants nicht vorhanden war 63. Im Falle des SLP von Lactobacillus acidophilus ATCC4356 wurden anhand von Sequenzver- gleichen mit den SLP-Genen von L. helveticus und L. crispatus zwei Domänen identifiziert. In weiteren Untersuchungen wurden die Funktionen der Domänen beleuchtet. Während eine Domäne die Verankerung in der Zellhülle des Bakteriums sicherstellt, ist die andere Domäne für die Zusammenlagerung der Monomere verantwortlich. Darüber hinaus zeigte letztere eine deutliche proteolytische Stabilität, die auf eine entsprechende Schutzfunktion für das Bakterium hinweist37.
Die Matrix der Hüllproteine kann an der Bildung von anorganischen Nanostrukturen und im Weiteren an einer Mineralbildung beteiligt sein. Cyanobakterienstamm Synechococcus strain GL24 bildet beispielsweise Coelestin und Strontianit auf seiner Zelloberfläche, welche mit
7 einem hexagonalen S-Layer bedeckt ist64, 65. Darüber hinaus gilt eine Beteiligung der S-Layer an der Bildung von Manganknollen auf dem Meeresboden als sehr wahrscheinlich66.
1.5 Einsatzmöglichkeiten von S-Layer-Proteinen in der Nanotechnologie Während viele Ansätze bei der Herstellung von Sub-Mikrometer-Strukturen dem „top down“-Ansatz folgen, geht der Trend zu Selbstorganisationsprozessen für „bottom up“- Strukturierung von Oberflächen. Der „top-down“-Ansatz stellt einen Weg dar, bei dem mak- roskopische Substrate mit Strukturen im Nanometerbereich versehen werden. Diese Verfah- ren haben ihre Ursprünge in der Mikrosystemtechnik. Ein „bottom up“-Prozess nutzt bei der Sub-Mikrometer-Strukturierung chemisch-physikalische Eigenschaften von Atomen und Mo- lekülen, um durch Selbstorganisation dieser Bausteine Nanostrukturen zu erzeugen.
SLPs stellen auf Grund ihrer Eigenschaft, hochgeordnete Proteinschichten mit Gitterkon- stanten im ein- bis zweistelligen Nanometerbereich auszubilden, einen interessanten Ansatz für „bottom up“-Prozesse in der Nanotechnologie dar. Da die Selbstorganisation eine intrin- sische Fähigkeit der Proteine ist, werden keine komplizierten Technologien benötigt, um diese regelmäßigen Proteinstrukturen auf Oberflächen herzustellen. In den vergangenen Jahren entstanden zahlreiche Ansätze zur Anwendung dieser Eigenschaften in technischen Prozessen und zur Herstellung neuartiger Biokomposit-Materialien. Bei diesen Ansätzen stand wiederum häufig die Natur Pate indem die Funktionen der S-Layer auf Anwendungen umgelegt wurden.
S-Layer als Filter
Während S-Layer für die Zelle wie ein Filter wirken können, ist eine Anwendung als Filterma- terial in technischen Prozessen in zweierlei Hinsicht denkbar. Durch die einheitliche Poren- verteilung und den definierten Porendurchmesser könnten S-Layer zur Filterung wässriger Medien verwendet werden67-70. S-Layer als Mikro- oder Nanosiebe auf geeigneten Support- Materialien wurde als Anwendungsidee bereits patentiert71.
Ein anderer Ansatz macht sich herausragende adsorptive Eigenschaften der Proteinschichten zunutze um Metallionen selektiv aus einer vorbeiströmenden wässrigen Phase zu entfernen.
8 Dabei kommt die Eigenschaft der Proteine zum Tragen, die in der Natur die Zelle vor dem Eindringen von toxischen Schwermetallen schützen, Spurenelemente jedoch passieren las- sen72-74.
S-Layer als Matrix zur Anordnung von funktionellen Molekülen
Die Nutzung der Selbstorganisation zur geordneten Anbindung von funktionellen Molekülen ist ein weiteres Anwendungsfeld der SLPs. Auch hier sind mehrere Vorgehensweisen denk- bar. Zum einen bieten Proteine eine Vielzahl von Aminosäureresten, die unter zu Hilfenahme von geeigneten Quervernetzern für die Anbindung von funktionellen Molekülen genutzt werden können. Dabei muss man weiterhin unterscheiden, ob die Assemblierung vor oder nach der Bindung erfolgt. Wird die Kopplung vor der Gitterbildung durchgeführt, kann es zu sterischen Behinderungen und somit zu Störungen bei der Selbstorganisation kommen. Die- se Methode wurde bereits für die Herstellung von Förster-Resonanz-Energie-Transfer- basierten Biosensoren diskutiert und erprobt75. Zum anderen ist es möglich, mit genetischen Methoden Fusionsproteine zu erzeugen, welche zur Selbstorganisation fähig sind und weite- re funktionelle Polypeptide oder Proteine tragen. Auch hier gibt es bereits entsprechende Arbeiten76-79. Beispielsweise wurde ein Fusionsprotein, bestehend aus dem S-layer Protein SbsB von Geobacillus stearothermophilus PV72und Streptavidin, hergestellt, welche erfolg- reich in Suspension kristallisiert wurden76, 77.
SLP zur Herstellung von Vakzinen
Erfolgt eine Immobilisierung von Antigenen auf den S-Layern, werden diese als Vakzine inte- ressant. Studien legen einen unterstützenden Effekt der S-Layer als Adjuvanz nahe. In diesen Arbeiten wurden Birkenpollenallergene mit S-Layer-Proteinen fusioniert78-80. Die Eigenschaft der S-Layer als Virulenzfaktoren in einigen Organismen macht die Proteinschichten ohne Zusätze bereits zum potenziellen Impfstoff. Mit Erfolg wurde dies schon im Falle von Aero- monas salmonicida und A. hydrophilia gezeigt, welche als Fischpathogene in der Lachszucht Probleme verursachen können81.
9 SLP zur Stabilisierung funktioneller Lipidmembranen
Viele biologische Prozesse laufen an Kompartimentgrenzen ab. Diese Grenzen sind meist durch Membranen definiert, welche mit funktionellen Molekülen durchsetzt sind. Spezifi- sche Erkennungs- und Transportprozesse laufen über derartige funktionelle Membranen, was diese Membranen für bioanalytische und biomimetische Anwendungen interessant macht. Durch die geringe mechanische Stabilität künstlich hergestellter Membransysteme waren deren Einsatzgebiete stark begrenzt 82, 83. S-Layer wurden in vivo häufig als Stützstruk- tur auf Membranen identifiziert und stellen eine interessante Möglichkeit dar, die Stabilität auch bei künstlichen Membransystemen zu verbessern. Eine Verbesserung der Stabilität durch S-Layer wurde in Verbindung mit Phospholipiddoppellagen und Tetraehterlipidmono- lagen nachgewiesen84.
SLP als Matrix bei Biomineralisation und Herstellung von Nanopartikeln
Es ist bekannt, dass die Matrix der Hüllproteine auf Prokaryoten an der Bildung von anorga- nischen Nanostrukturen beteiligt sein kann. So wurde gezeigt, dass sich die Proteinschichten zur Herstellung regelmäßiger Silikatstrukturen eignen, die morphologisch dem Proteinnetz- werk entsprachen85. Aber auch Nanopartikel können mit den Proteinstrukturen gebildet werden.
Als Nanopartikel bezeichnet man üblicherweise Verbände von wenigen bis einigen tausend Atomen. Nanopartikel zeigen häufig neue elektrische, optische oder chemische Eigenschaf- ten, die im Bulkmaterial nicht zu finden sind. Durch das extrem verschobene Verhältnis von Oberfläche zu Volumen hin zur Oberfläche kommen bei Nanopartikeln Oberflächeneigen- schaften wie Oberflächenladung und Oberflächenkräfte wesentlich stärker zum Tragen. Bei der Katalyse mit Übergangsmetallen steigt die Aktivität oft mit dem Zerteilungsgrad des Ka- talysators. Problematisch ist jedoch häufig die Tendenz eines solchen kolloidalen Systems zur Aggregation zu größeren Partikeln. Die Adsorption von Stabilisatoren hat sich als effektive Gegenmaßnahme etabliert86. Mögliche Stabilisatoren sind hierbei Lösungsmittel, Polymere und Dendrimere, aber auch niedermolekulare Verbindungen wie Thiole, Detergenzien, or- ganische Säuren und ionische Flüssigkeiten87-92. S-Layer-Proteine bieten hier einen beson- ders interessanten Ansatz. Durch die räumliche Ordnung der Proteine mit definiertem Git-
10 teraufbau und damit auch definierten Poren bieten sie die Möglichkeit, Nanopartikel zu se- parieren. Darüber hinaus können die Proteinschichten auch gleichzeitig für die Synthese ge- nutzt werden. Proteine binden Metallionen, insbesondere Schwermetalle, an den zahlrei- chen Aminosäureresten. Was bei einigen Bakterien als Schutz vor Schwermetallen funktio- niert, kann in technischen Prozessen zur Synthese von Nanopartikeln und Nanopartikelarrays genutzt werden. Die Eignung als Matrix wurde in vitro bereits nachgewiesen. Dazu wurden gereinigte S-Layer in Suspension mit Metallionen inkubiert. In weiteren Schritten erfolgt die Reduktion der Metallionen, was zur Ausbildung von Nanopartikeln führt. Die Herstellung der Partikel kann beispielsweise durch Reduktion mit Natriumazid in Suspension als auch mittels Elektronenstrahl im Transmissionselektronenmikroskop93, 94 erfolgen. Mit Hilfe von S-Layern erzeugte Nanopartikel wurden auch bereits für mögliche Anwendungen in Betracht gezogen.
Ein Beispiel ist die Oxidation von Kohlenstoffmonoxid. Dabei erfolgte der Reduktionsschritt bei der Herstellung der Nanopartikel, mit Dimethylaminoboran (DMAB)95. Goldnanopartikel konnten unter Zuhilfenahme der S-Layer-Proteine mittels Reduktion mit Wasserstoff erzeugt werden96.
1.6 Ursprung der verwendeten Proteine
Der Ursprung der in dieser Arbeit verwendeten Bakterienstämme und SLP ist die Halde Ha- berland in der Nähe von Johanngeorgenstadt. Bei den 1997 isolierten Bakterien handelt es sich um die Stämme Lysinibacillus sphaericus JG-A12, Lysinibacillus sphaericus JG-B53 und Viridibacillus arvi JG-B58, welche dort in der schwermetallkontaminierten Abraumhalde des Uranerzbergbaus gefunden wurden73, 97. Die Proteine der genannten Organismen sind SlfB für L. sphaericus JG-A12 und Slp1 für L. sphaericus JG-B53.Im Falle von JG-B58 ist nicht klar welcher S-Layer die Zelle bedeckt, da mehrere aktive S-Layergene im Genom gefunden wur- den.
11
2 Zielstellung
Um das Potenzial dieser speziellen S-Layer-Proteine für den Einsatz in technischen Anwen- dungen zu untersuchen, wurde ein zweigeteilter Ansatz gewählt.
(I) Die S-Layer sollten auf der Oberfläche der Bakterien beobachtet werden. Um daraus Er- kenntnisse über ihre natürliche Morphologie zu gewinnen. Die Bakterien sollten dazu lebend auf geeigneten Oberflächen immobilisiert und mittels Rasterkraftmikroskopie hochaufgelöst abgebildet werden. Für die Immobilisierung sollte eine geeignete Methode ausgewählt oder entwickelt werden. Nach der Untersuchung der Proteine am lebenden Organismus, sollte Rekristallisation der gereinigten Proteine auf technischen Oberflächen erfolgen. Ziel der Un- tersuchungen war, eine universelle aber einfache Methode zu entwickeln, die es erlaubt S- Layer-Proteine als selbstorganisierende Beschichtung für die Funktionalisierung von Oberflä- chen zu nutzen. Die Entwicklung der Methode sollte sehr anwendungsnah erfolgen und eine spätere Hochskalierung erlauben. Dabei sollte gleichzeitig ein tieferes Verständnis für die- Funktionsweise der Proteine geschaffen werden.
(II) Die Erkenntnisse des ersten Teils sollten im zweiten Teil angewendet werden, um die Eignung der S-Layer-Proteine für ausgewählte Fragestellungen zu demonstrieren. Damit soll- te geprüft werden, ob sich S-Layer-Proteine zur Herstellung von Beschichtungen auf unter- schiedlichsten technischen Werkstoffen eignen. Zur Verwendung kommen sollten die S- Layer-Proteine der Lysinibacillus Stämme L. sphaericus JG-A12 und L. sphaericus JG-B53 so- wie Viridibacillus arvi JG-B58. Die Anwendungsnähe sollte durch die Zusammenarbeit mit Partnern aus Forschung und Industrie erreicht werden. Es sollten Möglichkeiten zum Einsatz der S-Layer-Technologie und gegebenenfalls die weitere Funktionalisierung der Beschichtung in konkreten Anwendungsbeispielen untersucht und beurteilt werden.
12
3 Funktionsweise von S-Layer-Proteinen
3.1 Ergebnisse
3.1.1 Abbildung nativer S-Layer auf lebenden Organismen
Biotemplate natürlichen Ursprungs sollten zum besseren Verständnis von Form und Funkti- on in ihrer natürlichen Umgebung beobachtbar sein. Im Falle der S-Layer-Proteine bedeutet das, eine Abbildung der Proteinschichten auf den Ursprungsbakterien. Die Untersuchungen an getrockneten Bakterien sind dabei nicht zielführend, da strukturelle Desintegration der Oberfläche durch Wasserentzug erfolgen würde und die native Struktur der bakteriellen Zellhülle im Regelfall zerstört wird. Mittels AFM ist eine Beobachtung der lebenden Orga- nismen im wässrigen Medium möglich. Dazu müssen die Bakterien auf einem flachen Sub- strat immobilisiert werden. Verschiedene Methoden zur Immobilisierung wurden bereits in der Einleitung vorgestellt. Methoden, welche einen Trocknungsschritt beinhalteten wurden verworfen und eine Methode mit gelatinebeschichteten Substraten wurde erfolglos getes- tet. Daher wurde eine neue Methode in dieser Arbeit entwickelt, welche universell für Mik- roorganismen einsetzbar ist. Die Methode basiert auf einer mehrlagigen Polyelektrolytbe- schichtung auf glatten, für AFM-Untersuchungen geeigneten Substraten und der anschlie- ßenden Adsorption der Zellen an dieser Beschichtung, durch Zentrifugieren.
3.1.1.1 Vorbereitung und Immobilisierung
Die Substrate wurden mittels Rotationsbeschichtung mit fünfzehn Lagen Polyelektrolyt be- schichtet. Beginnend mit PEI wurden immer im Wechsel PSS und PEI bei 6.500 U/min aufge- schleudert. Jeweils ein Tropfen (etwa 20 µl) der Polyelektrolytlösungen mit 3 g/l PEI oder PSS wurden mit einer Tropfpipette auf den rotierenden Wafer (5 mm x 5 mm) getropft. Zwischen den Schritten wurde der Wafer jeweils mit etwa 5 Tropfen Reinstwasser gespült. Die einzel- nen Schritte dauerten nur wenige Sekunden, bis keine sichtbare Flüssigkeit mehr auf dem Substrat war.
13 In Abbildung 3 sind die weiteren Schritte der Probenpräparation schematisch dargestellt. Für den Zentrifugationsschritt wurden handelsübliche Reaktionsgefäße (1,5 ml) mit Heißleim gefüllt. Um einen schrägen Boden zur Auflage des Wafers herzustellen, wurde 1 g Heißleim in ein Reaktionsgefäß eingewogen (Abbildung 3 - 1.), anschließend in einem Heizblock bei 90°C verflüssigt (Abbildung 3 - 2.) und danach für zwei Minuten mit 10.000*g zentrifugiert, damit beim Erstarren des Heißklebers ein schräger Boden entsteht (Abbildung 3 - 3.). Die modifizierten Wafer wurden in den vorbereiteten Reaktionsgefäßen platziert. Nach Zugabe der Bakteriensuspension (Abbildung 3 - 4.) wurden die Reaktionsgefäße mit 15.000*g bei 4°C eine Stunde zentrifugiert. Bei diesem Schritt erfolgt die Sedimentierung der Mikroorganis- men auf der Waferoberfläche Abbildung 3 - 5.). Abschließend wird der Wafer mit Wasser aus einer Spritzflasche gespült, um lose Bakterien zu entfernen.
Abbildung 3: Schema zur Herstellung und Benutzung von Reaktionsgefäßen mit geneigtem Boden.
1. Einwaage von 1 g Heißkleber; 2. Schmelzen des Klebers; 3. Zentrifugation der verflüssigten Klebmasse und Erstarrung; 4. Platzieren des Wafers mit Bakteriensuspension im Reaktionsgefäß; 5. Sedimentieren der Mik- roorganismen auf dem beschichteten Wafer durch Zentrifugieren.
3.1.1.2 Kontrolle der Bakteriendichte
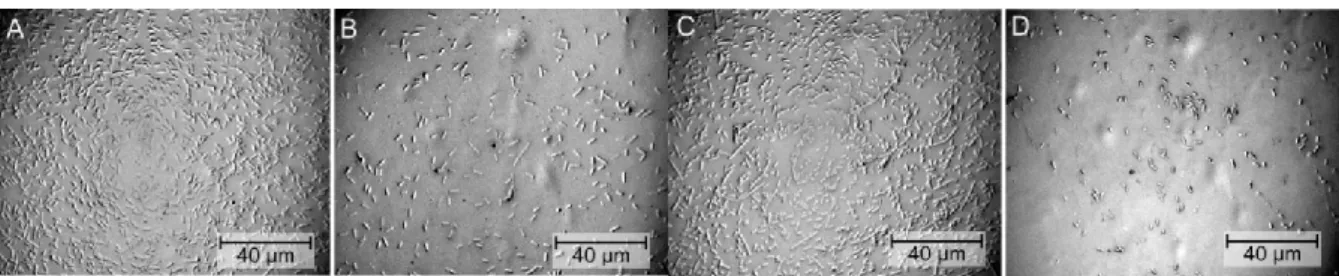
Ein Großteil der Bakterien in Suspension wird auf dem Wafer sedimentiert. Die Bakterien- dichte auf dem Wafer sollte daher kontrolliert werden. Im Falle einer zu hohen Zelldichte auf der Oberfläche kann es zu Deformationen der übereinanderliegenden Zellen kommen. Im Spülschritt wird loses Zellmaterial abgespült, und zurück bleibt die untere deformierte Bak- terienlage. Eine schnelle Kontrolle der Zelldichte kann bei SiO2-Substraten mit einem Auf- lichtmikroskop erfolgen. Lichtmikroskopische Aufnahmen von Präparaten, hergestellt mit unterschiedlich dichten Bakteriensuspensionen, zeigt Abbildung 4. Bei Bakterien war eine optische Dichte bei 600 nm (OD600) von 0,5 - 1 in der Regel ausreichend, um gut separierte aber nicht zu weit verteilte Zellen auf der Oberfläche zu erhalten.
14 Abbildung 4: Lichtmikroskopische Kontrolle der Mikroorganismendichte auf dem Wafer.
(A) Viridibacillus arvi JG-B58, OD600=0.1; (B) Lysinibacillus sphaericus JG-B53, OD600=0.05; (C) E. coli OD600=0.1;
(D) E. coli OD600=0.05
3.1.1.3 Aufnahmen der S-Layer am lebenden Bakterium
Die Bakterien wurden im AFM in einer Durchflusszelle abgebildet, wobei phosphatgepufferte Saline (PBS) als Medium eingesetzt wurde. Die Aufnahmen entstanden mit einem BL- AC40TS-C2 (Biolever mini) Cantilever von Olympus im AC-Mode.
Abbildung 5: AFM-Aufnahmen (Amplitudenbilder) von immobilisierten Bakterien und deren Oberflächen.
Links: (A) Übersichtsscan von Lysiniacillus sp. JG-B53; (B) Detailaufnahme der Oberfläche von Lysinibacillus sp. JG-B53 mit gut sichtbarem S-Layer; (C) Übersichtsscan von Viridibacillus arvi JG-B58; (D) Detailaufnahme der Oberfläche von Viridibacillus arvi JG-B58 mit gut sichtbarem S-Layer; E: Nahaufnahme einer Bakterienzel- le (Viridibacillus arvi JG-B58) mit sichtbaren Disklinationen im S-Layer-Gitter.
Abbildung 5 zeigt die Aufnahmen zweier Stämme. In (A) und (C) sind Übersichtsscans der immobilisierten Bakterien dargestellt, die zur Orientierung dienten. Die Bilder (B) und (D) zeigen hochaufgelöste Aufnahmen der bakteriellen Zelloberfläche des jeweiligen Bakteri- ums. Deutlich zu erkennen ist das Proteingitter des S-Layers. In (E) ist ein Ende einer stäb- chenförmigen Bakterienzelle abgebildet. Dabei ist die komplette Umhüllung des Bakteriums
15 mit dem S-Layer zu sehen. Der S-Layer wird in seiner Kristallinität durch Disklinationen in den Wachstumsbereichen unterbrochen, was insbesondere an den Polen der Bakterien der Fall ist. Disklinationen im S-Layer-Gitter ergeben sich aus der Geometrie der Pole sowie der Stre- ckung der Zelle durch Wachstum und sind in der Literatur mehrfach beschrieben47, 48 und konnten im Falle der Bacillus Stämme ebenfalls beobachtet werden. Die restliche Zelle ist von einem S-Layer mit nahezu einheitlicher Gitterorientierung umhüllt.
Bei dem Bakterium Bacillus sp. JG-B58 ist es mit der hier entwickelten Methode gelungen, Veränderungen in der S-Layer-Umhüllung über einen längeren Zeitraum zu beobachten. Die lebenden Organismen wurden über Nacht bei Raumtemperatur in der Durchflusszelle des Rasterkraftmikroskops belassen. Wie in Abbildung 6 zu sehen ist, verändert sich der S-Layer der Zelle stark. Während vitale Zellen einen sehr gleichmäßigen geschlossenen S-Layer auf- weisen, zeigen alternde Zellen unter Sauerstoff- und Nährstoffmangel (Medium PBS) viele kleine Kristallite auf der Oberfläche. Bereits diese Veränderung ist sehr interessant. Jedoch zeigt sich bei genauerer Analyse ein weiterer S-Layer unter der äußeren Schicht. Beide S- Layer weisen eine tetragonale Symmetrie auf, wie in Abbildung 6 erkennbar ist. Eine mögli- che Ursache der Veränderung könnte die Expression eines anderen SLP unter Stress sein, ausgelöst durch oben genannte Mängel. Eine Überexpression von SLP, ausgelöst durch die allgemeine Stressantwort im Bakterium, ist ebenfalls denkbar.
Abbildung 6: AFM-Amplitudenbilder einer gealterten Zelle mit zweilagigem S-Layer.
AFM-Amplitudenbilder vom Ausschnitt einer Zelle (A) und einem Detailbild (B), auf dem klar im mittleren Bereich der untere S-Layer mit tetragonaler Symmetrie zu erkennen ist.
1.0
0.8
0.6
0.4
0.2
0.0
µm
1.0 0.8 0.6 0.4 0.2 0.0
µm
-3 -2 -1 0 1 2 nm
600
500
400
300
200
nm
600 500 400 300 200
nm
-1.0 -0.5 0.0 0.5 nm
16 3.1.1.4 Weitere Möglichkeiten der Methode
Mit Hilfe der entwickelten Methode wurden weitere Untersuchungen an Mikroorganismen durchgeführt, um Möglichkeiten und Grenzen zu beurteilen. Abgebildet wurden dafür Mik- roorganismen sowohl lebend als auch fixiert mit Glutardialdehyd. Mit der Fixierung können die Mikroorganismen in verschiedenen Wachstumsstadien morphologisch untersucht wer- den, wie dies am Beispiel von Pichia pastoris erfolgt ist. Abbildung 7 zeigt in (A) eine Mutter- zelle mit zwei Tochterzellen. Die Knospungsstelle wurde in einer Detailaufnahme (B - D) ab- gebildet.
Abbildung 7: AFM-Aufnahmen einer Knospung bei Pichia pastoris.
(A) Höhenbild von Pichia pastoris, (B-D) Detailaufnahme: Höhenbild (B), Amplitudenbild (C), Phasenbild (D)
Eine weitere Möglichkeit ist lebende Bakterien über einen längeren Zeitraum zu beobachten und beispielsweise Zellteilungen zu beobachten, wie dies im Fall von E. coli erfolgt ist. In Ab- bildung 8 sind drei Bilder von immobilisierten E. coli Zellen, die im Abstand von 40 Minuten aufgenommen wurden. Deutlich zu erkennen sind beginnende Einschnürungen, die im zeitli- chen Verlauf der Aufnahmen fortschreiten und letztendlich in einer Zellteilung enden.
17 Abbildung 8: AFM-Aufnahmen (Amplitudenbilder) von in Teilung befindlichen lebenden E. coli Zellen.
Die Aufnahmen der drei Bilder erfolgten im Abstand von jeweils 40 min.
18 3.1.2 S-Layer-Rekristallisation auf technischen Oberflächen
Sollen S-Layer als biofunktionale Beschichtung auf technischen Oberflächen zu Einsatz kom- men, erfordert dies ein zuverlässiges und vor allem universelles Beschichtungsverfahren.
Bekannt sind die Fähigkeit der S-Layer zur Selbstassemblierung und die Eigenschaft sich be- vorzugt an Phasengrenzen anzuordnen. Unter Ausnutzung dieser Eigenschaften wurde in dieser Arbeit die Beschichtung von Oberflächen mit S-Layer-Proteinen durch Rekristallisation untersucht. Dies erfolgte mit verschiedenen Methoden: Während der zeitliche Verlauf sehr gut an der Quarzmikrowaage beobachtbar ist, kann die Ausbildung des Proteingitters nur mittels AFM eindeutig nachgewiesen werden. AFM-Messungen decken nur sehr kleine Be- reiche ab, während mittels QCM-D der größte Teil der Sensorfläche erfasst wird. AFM- Messungen erlauben somit direkte punktuelle Schichtdickenmessungen. Durch die wirken- den Kräfte und damit geringfügige Kompression der weichen Proteinschicht während des Scanvorgangs kann der erhaltene Wert der Schichtdicke etwas geringer ausfallen. Die QCM-D liefert hingegen nur mittlere Schichtdicken. Der S-Layer weist eine feste Schichtdicke auf. Der Bedeckungsgrad ist bei Beginn der Messung Null und steigt während des Rekristalli- sationsprozesses an. Damit geben mittlere Schichtdicken bei geordneter Rekristallisation einen Hinweis auf den Bedeckungsgrad der Oberfläche mit Proteinen. Einflüsse, die einer so strikten Betrachtung entgegenlaufen, sind ungeordnete Adsorption der Proteine und die Adsorption von Verunreinigungen, was punktuell zu anderen Schichtdicken führen kann, aber nur gemittelt über die Sensorfläche erfasst wird. Die Modellierung der Schichtdicke erfolgte jeweils nach Sauerbrey und nach Voigt. Es wurden sowohl verschiedene Substrate als auch Beschichtungs- und Aktivierungsmethoden untersucht.
19 3.1.2.1 Herstellung von Monomer-Lösungen
Die zur Rekristallisation benötigten Monomere wurden mit verschiedenen Methoden herge- stellt und stabilisiert. Stabilisierung bedeutet hier, die Monomere an einer Polymerisierung zu hindern. Die Desintegration der Proteingitter erfolgt schonend und reversibel mit cha- otropen Substanzen wie Harnstoff oder Guanidin-Hydrochlorid, indem intramolekulare hyd- rophobe Wechselwirkungen durch hohe Salzkonzentrationen gestört werden. Darin gleichen sich alle Methoden. Die weitere Vorgehensweise ist abhängig von den Anforderungen, die an die Proteinlösung gestellt werden. Damit es nicht in der hergestellten Lösung zur Rekris- tallisation kommt, müssen die zur Selbstorganisation benötigten zweiwertige Ca2+-Ionen entfernt werden. Die Entfernung kann mit Hilfe von Komplexbildnern erfolgen. Bei den ein- gesetzten Proteinen ist diese Methode nur bedingt geeignet, da die Entfernung von Kom- plexbildnern wie EDTA und EGTA aus der Proteinlösung problematisch ist*. Im Laufe der Ar- beiten wurde untersucht, ob die Entfernung der chaotropen Salze notwendig ist, um eine Rekristallisation auf Oberflächen zu erreichen. Zum Einsatz kamen hochkonzentrierte Mo- nomerlösungen mit Konzentrationen von 30 g/l Protein. Bei einer Proteinendkonzentration von 0,1 g/l wird die eingesetzte Lösung 300fach verdünnt. Die Konzentration der chaotropen Salze sinkt somit von 6 Mol/l auf 20 mMol/l. Diese geringe Konzentration hat auf Adsorption und Kristallisation auf den angebotenen Oberflächen keinen messbaren Einfluss mehr. Mo- nomerlösungen mit 5 g/l konnten ebenfalls problemlos eingesetzt werden, obwohl die Ver- dünnung der Lösung nur noch Faktor 50 beträgt und damit die Konzentration von Guanidin- hydrochlorid noch 120 mM beträgt. Gleiches gilt für Monomerlösungen welche mit 8 M Harnstoff hergestellt werden. Eine Konzentration von 160 mM Harnstoff nach der Verdün- nung wirkte sich nicht nachteilig auf die Versuche aus.
Die vollständige Desintegration der Proteine durch Guanidinhydrochlorid wurde am Beispiel von Slp1 (Lysinibacillus sphaericus JG-B53) mittels statischer Lichtstreuung (SLS) überprüft.
Mit diesen Methoden wurde eine Molmassenbestimmung der Proteine in den Monomerlö-
* persönliche Mitteilung Bo Li, HZDR: In spektroskopischen Untersuchungen fanden sich störende Signale von EDTA in SLP-Monomerlösungen, welche zur Entfernung von Calcium aus den Proteinen mit EDTA versetzt wur- den.
20 sungen durchgeführt. Die Ergebnisse der SLS-Messungen sind Tabelle 1 zu entnehmen. Wei- terführende Daten zu Messung und Berechnung befinden sich in Anhang A - B.
Tabelle 1: Molmassenbestimmung anhand von SLS-Messungen in Monomerlösungen von Slp1. Ergebnisse aus Debye-Plot mit Zetasizer (Rückwärtsstreuung bei 173°)
Probe Molmasse (kDa) Fehler (%)
1 128 21,9
2 169 39,6
3 114 21,8
Die ermittelten Werte für die molare Masse der vermessenen Proteinlösungen Slp1 sind den berechneten und mittels SDS-PAGE ermittelten Proteingrößen ähnlich (SDS PAGE: 150 kDa;
berechnet: 116 kDa)98. Daher kann von einer vollständigen Desintegration der Proteine in 6M Guanidinhydrochlorid ausgegangen werden.
3.1.2.2 Reinigung von Substratoberflächen für Beschichtung und Analysen
Für die Untersuchung von Oberflächenbeschichtungen und -prozessen mittels AFM mussten diese gereinigt werden, um den Einfluss von Kontaminationen zu minimieren. Für diese Zwe- cke wurden verschiedene Reinigungsmethoden getestet und deren Eignung für jedes Mate- rial bewertet. Bei diesen Versuchen erwiesen sich für säure- und laugenfeste Materialien herkömmliche Methoden wie Peroxomonoschwefelsäure (Piranha-Lösung) und die in der Wafer-Reinigung bewährte RCA-Reinigung als sehr gut geeignet. Vorteile der RCA-Reinigung sind neben dem sehr guten Reinigungsergebnis die einfache Durchführung, der geringe Chemikalienverbrauch und damit eine geringe Abfallmenge. Die RCA-Reinigung wurde in der Folge für die Reinigung von Siliziumwafern und Saphirwafern eingesetzt. Kunststoffoberflä- chen wurden mit der kommerziellen Reinigungslösung Hellmanex III® (Hellma GmbH & Co.
KG, Müllheim) gereinigt. Ein Beispiel für eine RCA-gereinigte Oberfläche ist auf Seite 34 in Abbildung 18 gezeigt.
21 3.1.2.3 Rekristallisation auf unbehandelten Oberflächen
Die Rekristallisation auf Siliziumdioxid ist für SLP von Bacillus sphaericus CCM 2177 in der Literatur beschrieben. Bei den hier verwendeten Proteinen zeigte sich in AFM-Versuchen, dass die Rekristallisation an gereinigten Siliziumdioxidoberflächen langsam und mit geringem Bedeckungsgrad erfolgt. Der Rekristallisationsprozess wurde mittels AFM in Durchflussver- suchen untersucht. Nach 18,5 Stunden betrug der Bedeckungsgrad zwischen 50 und 60 %.
Eine AFM-Aufnahme der Oberfläche wird in Abbildung 9 gezeigt. Deutlich sichtbar ist ein dendritisches Wachstum der Proteinaggregate auf der Oberfläche. Nach 20 Minuten hat die Rekristallisation bereits begonnen, und ein Teil der Oberfläche ist bedeckt. Der Anfang der Rekristallisation ist daher relativ schnell, der Verlauf verlangsamt sich aber zunehmend.
Abbildung 9: AFM- Höhenbilder von S-Layern auf einer Siliziumdioxidoberfläche.
Fortschritt der Rekristallisation nach 20 min (A) und 3 h (B) mit SlfB; Keine Veränderung der Bedeckung nach 21 h (C)
10
8
6
4
2
0
µm
10 8
6 4
2 0
µm
-4 -2 0 2 4 nm 5
4
3
2
1
0
µm
5 4 3 2 1 0
µm
-4 -2 0 2 4 nm
5
4
3
2
1
µm
5 4 3 2 1
µm
-4 -2 0 2 4 nm
22 QCM-D Versuche zum Adsorptionsverhalten der S-Layer zeigen ein ähnliches Bild. Der Ver- lauf eines vergleichbaren Versuchs in der Durchflusszelle der QCM-D ist in Abbildung 10 dar- gestellt.
Abbildung 10: QCM-D-Graph einer Rekristallisation von SlfB auf einem SiO2-Sensor.
Das Substrat wurde ohne jegliche Modifizierung eingesetzt. Equilibrieren des Sensors erfolgte mit RK8- Puffer. Zugabe der Proteine (0,1 g/l in RK8-Puffer) erfolgte bei Zeitmarke 6:00. Der Spülvorgang mit RK8- Puffer erfolgte bei Zeitmarke 1:05:00. Daten modelliert nach Kelvin-Voigt (rot) und berechnet nach Sauer- brey (Graustufen, Berechnung auf Grundlage der Obertöne 3 – 13 im Diagramm S3 – S13)
Die QCM-D-Versuche wurden immer etwa eine Stunde nach Zugabe der Proteinlösung be- endet. Bis zu diesem Zeitpunkt hat sich auf dem Sensor bereits eine Proteinschicht gebildet.
Die Berechnungen auf Grundlage der Messdaten ergaben die Massezunahme auf der Sen- soroberfläche. Diese wurde in eine mittlere Schichtdicke umgerechnet und deren Verlauf als Diagramm dargestellt. Die Darstellung der Schichtdicke anstelle der Masse erfolgt, um den Schichtbildungsprozess zu veranschaulichen. Dabei wurde eine Proteindichte von 1410 kg/m³ angenommen. Die Ermittlung der Proteindichte erfolgte rechnerisch nach einer For- mel aus der Fachliteratur99. Bei einer mittels AFM bestimmten S-Layer-Höhe von etwa 10 nm, wie sie in der Literatur angegeben17 und später auch für den hier verwendeten S-Layer im Folgenden bestätigt wird (Seite 36), kann man von einer mittleren Schichtdicke direkt auf den ungefähren Bedeckungsgrad schließen. Damit ergeben die QCM-D-Messungen einen Bedeckungsgrad von ca. 50%. Die Messung berücksichtigt mehrere Obertöne, die mittels Sauerbrey-Gleichung in eine Schichtdicke umgerechnet werden. Des Weiteren wird aus mehreren Obertönen unter Berücksichtigung der entsprechenden Dissipationen die Schicht- dicke nach dem Modell von Kelvin und Voigt modelliert. Beide Berechnungsgrundlagen un- terschieden sich nicht signifikant, daher kann der Proteinfilm in erster Näherung als starr
23 betrachtet werden. Generell ist bei Versuchen mit diesen Proteinen aufgrund der geringen Schichtdicken die Berechnung nach Sauerbrey gut geeignet.
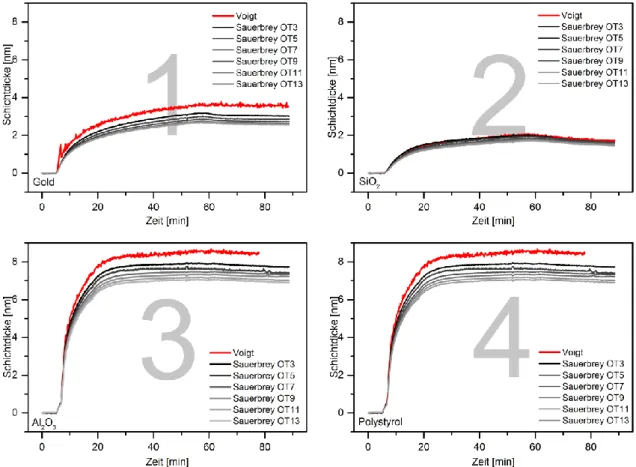
Es wurden parallel Sorptionsversuche mit verschiedenen Substraten durchgeführt. Die Er- gebnisse dazu sind in Abbildung 11 für SlfB und Abbildung 12 für Slp1 dargestellt. Zusätzlich zu SiO2-Oberflächen erfolgten Adsorptionsversuche an Gold, Al2O3 und Polystyrol.
Abbildung 11: QCM-D Rekristallisationsversuche mit SlfB auf unmodifizierten Substraten.
Sensor 1: Au-Oberfläche, Sensor 2: Edelstahl-Oberfläche, Sensor 3: Al2O3-Oberfläche, Sensor 4: Polystyrol- Oberfläche. Equilibrieren der Sensoren erfolgte mit RK8-Puffer. Zugabe der Proteine (0,1 g/l in RK8-Puffer) erfolgte bei Zeitmarke 1:00. Der Spülvorgang mit RK8-Puffer erfolgte bei Zeitmarke 52:00.
In der Anfangsphase erfolgt ein schnelles Schichtwachstum, was nach wenigen Minuten deutlich langsamer wird. Dieser Prozess ist abhängig von dem gewählten Substrat. QCM-D- Messungen mit SlfB und Slp1 belegen, dass über den betrachteten Zeitraum keine geschlos- sene Proteinschicht auf den unmodifizierten Sensoroberflächen adsorbiert. Die Zugabe der SLP erfolgt wenige Minuten nach Beginn der Messung, deutlich zu erkennen an der Fre- quenzabnahme sowie der Zunahme des Dissipationsfaktors. Nach etwa 50 Minuten wurde
24 die Rekristallisation beendet, indem das System mit Puffer gespült wurde. Die verwendeten Substrate waren reine Gold-, SiO2- (Abbildung 10), Al2O3- sowie Polystyrol- und Edelstahl- oberflächen. In Abbildung 12 sind die QCM-D-Graphen für Messungen mit Slp1 abgebildet.
Die Proteine adsorbieren nur langsam an den Gold- und Siliziumdioxidoberflächen und bil- den nur dünne Schichten, während auf Aluminiumoxid- und Polystyroloberflächen die ad- sorbierte Proteinmenge bzw. Schichtdicke deutlich höher ist.
Abbildung 12: QCM-D-Rekristallisationsversuchen mit Slp1 auf unmodifizierten Substraten.
Sensor 1: Au-Oberfläche, Sensor 2: SiO2-Oberfläche, Sensor 3: Al2O3-Oberfläche, Sensor 4: Polystyrol- Oberfläche. Equilibrieren der Sensoren erfolgte mit RK8-Puffer. Zugabe der Proteine (0,1 g/l in RK8-Puffer) erfolgte bei Zeitmarke 1:35. Der Spülvorgang mit RK8-Puffer erfolgte bei Zeitmarke 52:20.
Die Oberflächenladung von Aluminiumoxid ist bei dem eingestellten pH-Wert positiv. Auf- grund der negativen Nettoladung der Proteine kommt es auf diesen Oberflächen zu einer verstärkten Adsorption. Daher stehen diese Ergebnisse im Einklang mit den Erwartungen. Im Vergleich der beiden Proteine fällt auf, dass die adsorbierte Schicht an Goldoberflächen im Vergleich zum Slp1 größer ist. Die Werte für Al2O3 hingegen zeigen eine verringerte Schicht- dicke für SlfB im Vergleich zu Slp1. Auch das SlfB zeigt gute Affinität zu Polystyroloberflä-
25 chen. Die Proteinadsorption, welche mittels QCM gemessen wird, erlaubt keine Aussage über die Gitterbildung der SLP. Das belegen AFM-Aufnahmen von proteinbelegten Sa- phirwafern, wie in Abbildung 13 gezeigt ist. Die Proteinschicht weist nicht die typischen Git- terstrukturen, sondern kleinere Aggregate auf.
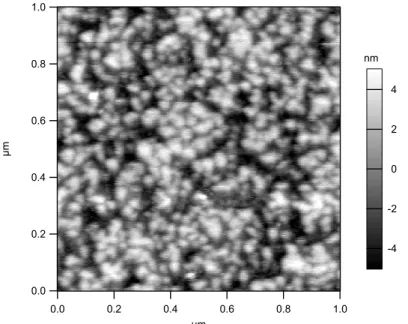
Abbildung 13: AFM Höhenbild einer mit SlfB belegten Al2O3-Oberfläche (Saphirwafer).
Ein Saphirwafer wurde mit 5 g/l A12 SlfB inkubiert (0,5 mM Tris, 10 mM CaCl2, pH 9,0).
In Abbildung 14 sind AFM-Höhenbilder gezeigt, welche sehr detailliert die tetragonale Struk- tur der S-Layer zeigen, aber auch deutliche Unterschiede aufweisen. Die linke Aufnahme stammt von dem Rekristallisationsexperiment von SlfB auf unmodifiziertem Siliziumdioxid.
Das rechte Bild entstand nach Adsorbtion einer SlfB Polymersuspension auf einem Silizium- dioxidwafer. Deutlich zu erkennen ist der Unterschied zwischen beiden Aufnahmen, insbe- sondere ein deutlicher Unterschied in der Oberflächenrauigkeit. Die Rauigkeiten sind in Ta- belle 2 aufgelistet. Die Höhe der Proteinschichten unterschied sich ebenfalls. Während die rekristallisierten Proteinschichten eine Höhe von ca. 10 nm aufwiesen, waren im Fall der adsorbierten Proteinsuspension nur vereinzelte Kristallite auf der Oberfläche zu finden, wel- che jedoch eine Höhe von ca. 19 nm aufwiesen, was auf eine S-Layer-Doppelschicht deutet.
Im Falle des S-Layer-Proteins SbpA von Lysinibacillus sphaericus CCM2177 wurde Ähnliches beobachtet. Es handelt sich hierbei um die zwei Seiten des S-Layers, welche unterschiedli- che Eigenschaften und Morphologie aufweisen17. Während es sich bei der rekristallisierten Schicht um eine Monolage handelt, sind die 19 nm dicken Schichten Doppellagen, welche
1.0
0.8
0.6
0.4
0.2
0.0
µm
1.0 0.8
0.6 0.4
0.2 0.0
µm
-4 -2 0 2 4 nm
26 möglicherweise durch Kollabieren von in Suspension gebildeten röhrenförmigen Proteinag- gregaten entstanden sind.
Abbildung 14: AFM Höhenbilder von den zwei unterschiedlichen S-Layer-Seiten.
SlfB auf Siliziumdioxid rekristallisiert (A) und in Suspension rekristallisiertes SlfB auf Siliziumdioxid adsor- biert (B).
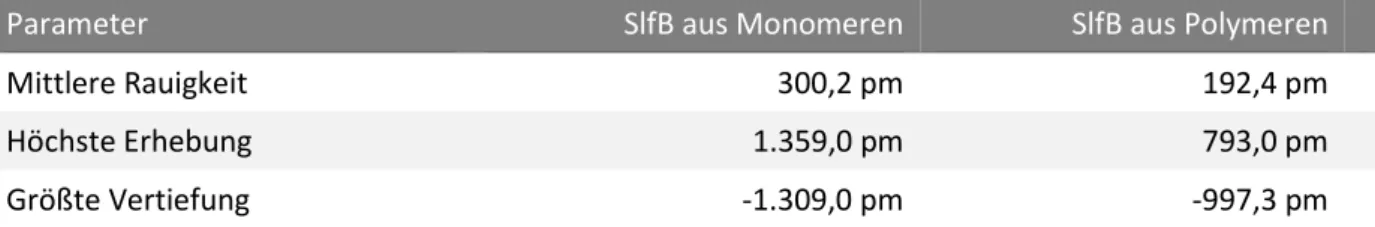
Tabelle 2: Unterschiede der Rauigkeit der zwei Seiten eines kristallisierten SlfB-S-Layers.
Parameter SlfB aus Monomeren SlfB aus Polymeren
Mittlere Rauigkeit 300,2 pm 192,4 pm
Höchste Erhebung 1.359,0 pm 793,0 pm
Größte Vertiefung -1.309,0 pm -997,3 pm
Die Beschichtung von gereinigtem Siliziumdioxid mit S-Layern war ohne weitere Vorberei- tung der Oberfläche nicht zufriedenstellend. Daher wurden verschiedene Methoden zur Modifizierung der Oberflächen getestet. Zwei Methoden kamen nach Vorversuchen in die engere Auswahl und werden im Folgenden diskutiert.
140
120 100 80
60 40
20 0
nm
140 120 100 80 60 40 20 0
nm
-600 -400 -200 0 200 400 600 pm
140
120 100
80
60 40
20
0
nm
140 120 100 80 60 40 20 0
nm
-600 -400 -200 0 200 400 600 pm
27 3.1.2.4 Modifizierung von Oberflächen
Für die Beschichtung von Oberflächen kann deren Modifizierung und Aktivierung ein wichti- ger Schritt sein. Daher wurden verschiedene Modifikationen durchgeführt und die erhalte- nen Oberflächen untersucht. Die Modifikationen unterscheiden sich in kovalente Bindungen von Aminogruppen und nicht kovalente Beschichtungen mit Polymeren, ebenfalls zur Bereit- stellung von funktionellen Gruppen wie Amino-, Carboxyl- und Sulfonsäureresten. Für eine nachfolgende Beschichtung mit Proteinen ist eine Oberfläche mit möglichst vielen positiven Ladungen am günstigsten. Da auch in der Natur weit verbreitet, sollten hier Aminogruppen zum Einsatz kommen.
3.1.2.4.1 Silanisierung
Die Silanisierung erfolgte mit Aminopropyltrietoxysilan (APTES) und wurde mit drei verschie- denen Methoden durchgeführt. Die Methoden wurden von bekannten Methoden abgeleitet und sind im Kapitel „Material und Methoden“ ab Seite 106 beschrieben.
Die erzeugten Oberflächen wurden mittels AFM und XPS untersucht. Kriterien hierbei waren Oberflächenmorphologie (AFM) und Stickstoffgehalt der Oberfläche (XPS). Ziel war es, die Oberflächenrauigkeit nicht zu erhöhen jedoch eine möglichst gute Aminofunktionalisierung zu erreichen. Die beschichteten Oberflächen wurden mittels AFM auf ihre Topographie un- tersucht und die Rauigkeiten berechnet. Bei der Silanisierung kann es zur Bildung von Polysi- loxanpartikeln kommen, was hier nicht erwünscht ist. Das Ausmaß der Partikelbildung auf der Oberfläche korreliert mit ansteigender Oberflächenrauigkeit. Die Messung erfolgte tro- cken an Luft. Der Vergleich der Proben erfolgte des Weiteren mit XPS zur Elementanalyse der Oberfläche. In diesen Vergleich wurden die Beschichtungen mit Polyelektrolyten mit einbezogen. Die Auswertung erfolgt ab Seite 35. Die Analyse der Adsorption durch QCM-D Messungen war aufgrund der schlechten Chemikalienbeständigkeit der Dichtungen in den Messzellen nicht möglich.
Methode I ist die direkte Applikation von APTES auf die Siliziumdioxidoberfläche.
Methode II ist eine Inkubation der Oberfläche mit 1% APTES in Xylol.
Methode III ist die Inkubation der Oberfläche in gasförmigem APTES.
28 AFM-Analysen der silanisierten Oberflächen
Abbildung 15: AFM-Höhenbilder von silanisierten Siliziumwafer-Oberflächen.
Dargestellt sind jeweils ein 1 µm x 1 µm Bereich (A, C, E) und ein 10 µm x 10 µm Bereich. (B, D, F). Obere Reihe: Methode I (A, B), Mittlere Reihe: Methode 2 (C, D), Untere Reihe: Methode III (E, F)
29 Bei den AFM-Analysen wurden Areale von 1 µm x 1 µm (Detailbild) und 10 µm x 10 µm (Übersichtsbild) gescannt. Die AFM-Höhenbilder sind in Abbildung 15 gegenübergestellt. Die Abbildung zeigt die Ergebnisse der drei Methoden im Vergleich.
Die nach Methode 1 modifizierte Oberfläche weißt im Übersichtsbild kleinere Verunreini- gungen auf. Vermutlich handelt es sich hierbei um Polysiloxanpartikel, welche bei der Me- thode entstehen. Im Detailbild von Oberflächenbereichen ist eine sehr ebenmäßige Oberflä- che zu sehen wie sie mit der Methode angestrebt wird.
Methode 2 zeigt im Vergleich zu Methode 1 deutlichere Tendenzen zur Bildung von Polysilo- xanpartikeln auf der Oberfläche. Diese erhöhte Rauigkeit ist für nachfolgende Beschichtun- gen hinsichtlich der späteren Analytik sehr ungünstig. Die Oberflächen zeigen sowohl im Übersichtsbild als auch im Detailbild deutlich mehr Partikel auf der Oberfläche als die mit Methode 1 präparierten Oberflächen.
Methode 3 hat nicht das Potenzial eine ideal silanisierte Oberfläche zu produzieren. Hier sind ebenfalls im Übersichtsbild größere Partikel zu beobachten. Im Detailbild, welches in einem partikelfreien Bereich aufgenommen wurde, zeigt sich eine Oberflächenstruktur, welche bis auf wenige Störungen mit jener aus Methode 1 vergleichbar ist. Visuell erscheint somit Me- thode 1 die beste Methode zu sein, gefolgt von Methode 3 und Methode 2 an dritter Stelle.
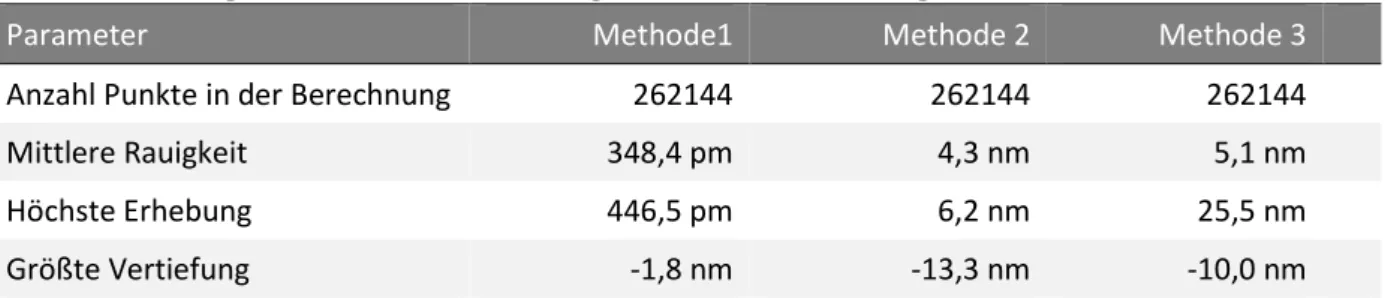
Tabelle 3: Messergebnisse der Oberflächenrauigkeit nach der Silanisierung mit APTES
Parameter Methode1 Methode 2 Methode 3
Anzahl Punkte in der Berechnung 262144 262144 262144
Mittlere Rauigkeit 348,4 pm 4,3 nm 5,1 nm
Höchste Erhebung 446,5 pm 6,2 nm 25,5 nm
Größte Vertiefung -1,8 nm -13,3 nm -10,0 nm
Die Berechnung der Rauigkeit anhand der Detailbilder, dargestellt in Tabelle 3, zeigt jedoch, dass Methode 2 eine geringere Rauigkeit aufweist. Ursache hierfür sind wenige größere Par- tikel auf einer nach Methode 3 modifizierten Oberfläche.