Heinrich-Heine-Universität Düsseldorf
Bachelorarbeit
Auf der Suche nach Kreuzungspunkten zwischen niedrig liegenden elektronischen Singulettzuständen
des trans- Butadiens
Maximilian Vogtland
Angefertigt am
Institut für Theoretische Chemie und Computerchemie
Zeitraum: 07.2012 – 09.2012
1. Gutachter: Prof. Dr. Christel Marian 2. Gutachter: Jun.-Prof. Dr. Jörg Tatchen
VII
Eidesstattliche Erklärung
Ich versichere an Eides Statt durch meine eigenhändige Unterschrift, dass ich die vorliegende Arbeit selbstständig, ohne fremde Hilfe angefertigt habe. Alle aus fremden Quellen wortgetreu oder sinngemäß übernommenen Gedanken sind als solche kenntlich gemacht. Ich versichere außerdem, dass ich keine anderen als die angegebenen Quellen und Hilfsmittel verwendet habe. Diese Versicherung bezieht sich auch auf alle in der Arbeit enthaltenen Zeichnungen, Skizzen, bildlichen Darstellungen und dergleichen. Diese Arbeit wurde bisher in gleicher oder ähnlicher Fassung keiner anderen Prüfungsbehörde vorgelegt und auch nicht veröffentlicht.
________________ __________________
Ort, Datum Maximilian Vogtland
VIII
IX
Inhaltsverzeichnis 1. Einleitung
2. Bisherige Forschungsergebnisse 3. Theorie
3.1. Photochemische Reaktionen 3.2. Potentialhyperflächen
3.2.1. Born-Oppenheimer-Näherung 3.2.2. Stationäre Punkte
3.2.2.1. Optimierungsmethoden
3.2.2.1.1. Quasi-Newton Methode 3.2.3. Konische Durchschneidung
3.3. Dichtefunktionalthoerie (DFT) 3.3.1. Funktionale
3.4. Konfigurationswechelwirkung (CI) 3.4.1. MRCI
3.5. DFT/MRCI 4. Auswertung
4.1. Gleichgewichtsgeometrien des Grund-, 1Bu und 2Ag Zustandes 4.2. Potentialschnittflächen der Grundzustandsgeometrie
4.3. Potentialschnittflächen entlang der Torsion für die 2Ag Geometrie 4.4. Torsion um nur eine Doppelbindung
4.5. Minima des S1 und S2 Zustandes
4.6. Pyramidalisierung und Kreuzung mit dem Grundzustand 4.7. Interpolation
5. Zusammenfassung 6. Quellenverzeichnis 7. Anhang
8. Orbitalbilder
X
Abkürzungen und Verweise
[x] Verweis auf das Quellenverzeichnis [A x] Verweis auf den Anhang
[O x] Verweis auf die Orbitalbilder
D, 4 Doppelanregung mit vier offenen Schalen
1
1. Einleitung
Das s-trans-1,3- Butadien ist das kürzeste konjugierte System. Es wird daher gerne zur Untersuchung der elektronischen Struktur von π-Systems sowohl in der experimentellen, als auch in der theoretischen Forschung verwendet. Die Besonderheit des s-trans- Butadiens besteht in der Tatsache, dass obwohl ein elektronischer Übergang in den hellen 1Bu Zustand stattfindet, es zu keiner Fluoreszenz kommt [1-2]. Desweiteren konnte in der Umgebung des 1Bu Zustands ein optisch dunkler Ag Zustand detektiert werden. Die genaue Lage dieses 2Ag Zustandes zum 1Bu Zustand ist jedoch nach wie vor umstritten. Theoretische Untersuchungen ergaben, dass es infolge einer Änderung der Geometrie zu einer Kreuzung zwischen diesen Zuständen kommt. Desweiteren berichten Fuß et al. [3] und auch Levine und Martínez [4] über eine mögliche Kreuzung des 2Ag mit dem Grundzustand. Diese Kreuzung würde die fehlende Fluoreszenz erklären.
Die vorliegende Arbeit soll daran anknüpfen und das s-trans-Butadien genauer mit der DFT/MRCI Methode untersuchen, besonders im Hinblick auf das Verhalten des Grundzustandes und der ersten beiden angeregten Zustände. Das Ziel dieser Arbeit ist, die Lage der ersten angeregten Singulettzustände zu bestimmen, sowie die oben genannten Kreuzungspunkte zu lokalisieren, um eine Erklärung für Verlauf und Mechanismus des strahlungslosen Überganges in den Grundzustand zu finden. Hierfür werden Schnitte der Potentialhyperfläche entlang ausgewählter interner Koordinaten erstellt, sowie die Strukturminima der ersten beiden angeregten Singulettzustände berechnet.
Von den Ergebnissen erhofft man sich auch, Anhaltspunkte für das Verhalten größerer konjugierter Systeme zu gewinnen, sowie die Anwendbarkeit der DFT/MRCI Methode für diese Systeme festzustellen.
2
2 Bisherige Forschungsergebnisse
Erste Ergebnisse über Lage und Kreuzung des 2Ag und 1Bu Zustandes konnten Chadwick et al. [5] liefern, indem sie das Raman- Spektrum des mit UV- Licht angeregten s-trans- Butadiens gemessen haben. Sie konnten feststellen, dass es infolge der symmetrischen C-C-C Bindungswinkeländerung offenbar zu einer Kopplung zwischen dem 2Ag und dem 1Bu Zustand kommt. Aus ihren Ergebnissen schlossen sie weiterhin, dass der 2Ag Zustand unterhalb des 1Bu Zustandes liegt, während Elektronenenergieverlustspekten (EELS), sowie Multiphotonen- Ionisation (MPI) die umgekehrte Reihenfolge ergaben [6-7]. Lediglich die EELS Messungen von Palmer und Walker [8] stimmen mit den Ergebnissen der Raman Messung überein. Theoretische Berechnungen ergeben in Abhängigkeit von Methode, Basissatz, Level der Elektronenkorrelation [9-12] sowohl die eine, als auch die andere Möglichkeit. Sie stimmen jedoch darin überein, dass beide Zustände energetisch nah beieinander liegen.
Die genaue Lage ist somit nachwievor sowohl aus experimenteller, als auch aus theoretischer Sicht ungeklärt.
Das Interesse an den beiden Zuständen beruht desweiteren auf ihrem Zusammenspiel während des photophysikalischen Prozesses. So ist wie eingangs erwähnt nach einer elektronischen Anregung des s-trans- Butadiens keine Fluoreszenz beim Übergang in den Grundzustand festzustellen [1-2]. Das Absorptionsspektrum zeigt jedoch eine intensive Bande mit einem Maximum bei 208 nm, die der Anregung in den optisch hellen 1Bu Zustand zugeschrieben wird [13-14]. Die Absorption aus dem Grund- in den 2Ag Zustand ist hingegen symmetrie-verboten. Die Breite der 1Bu Absorptionsbande von etwa 1000 cm-1 deutet auf eine kurze Lebensdauer des 1Bu Zustandes hin [14-15].
Dies deckt sich mit den Beobachtungen von Chadwick et al. und den Dynamikuntersuchungen von Levine und Martínez [4] in denen sie unter Relaxation der Doppelbindungen eine direkte Kreuzung zwischen 1Bu und 2Ag Zustand etwa 40-50 fs nach der Anregung feststellen konnten. Die darauffolgende Kreuzung des 2Ag
Zustandes mit dem Grundzustand erklärt womöglich die fehlende Fluoreszenz[3-4, 16].
Sie konnten zudem feststellen, dass je nach Geometrie und Ladungsverteilung im S1
Zustand die Kreuzung mit dem Grundzustand zu verschiedenen Photoprodukten führen kann (Abb. 1).
3
Abb. 1 [4]: Schema der möglichen Kreuzungsmechanismen
Um den Verlauf der Kreuzung zu erklären und die Kreuzungspunkte zu lokalisieren, nutzten Ostojić und Domcke [17] Potentialschnittflächen der internen Koordinaten, welche einzelnen Schwingungsmoden entsprechen. Aufbauend auf den Ergebnissen von Ostojić et al. wurde die folgende Arbeit erstellt.
4
3. Theorie
3.1 Photochemische Reaktionen [18]
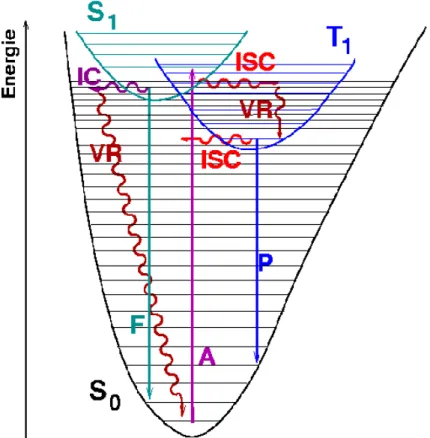
Bei einer photochemischen Reaktion kommt es zu einer Anregung eines Moleküls in einen elektronisch- und schwingungsangeregten Zustand durch die Absorption eines Photons. Der Rückweg aus dem angeregten Zustand in den Grundzustand kann anschließend auf verschiedene Wege erfolgen (Abb. 2).
www.uni-duesseldorf.de/Jahrbuch/2003/Marian/Grafik/Marian03.gif
Abb. 2: Jablonski-Schema
S0: Singulettgrundzustand, S1: Erste angeregte Singulettzustand, T1: Erste angeregte Triplettzustand, IC: internal conversion, ISC: intersystem crossing, VR:
Vibrationsrelaxation, F: Fluoreszenz, P: Phosporeszenz, A: Absorption
Mittels Fluoreszenz kann die aufgenommene Energie als Strahlung aus dem S1 Zustand emittiert werden. Die Übertragung der Energie von Singulett zu einem nahe gelegenem Triplett Zustand oder umgekehrt findet durch Umkehr des Elektronenspins statt (intersystem crossing). Eine Übertragung auf andere Moleküle ist ebenfalls durch
5
Kollision möglich. Dieser Prozess wird als quenching bezeichnet. Die Emission aus dem T1 Zustand wird als Phosphoreszenz bezeichnet. Sowohl Fluroreszenz, als auch Phosphoreszenz finden nur aus den jeweils niedrigsten angeregten Zuständen (S1 und T1) statt. Einen strahlungslosen Prozess zwischen Zuständen gleicher Multiplizität stellt die interne Konversion dar (internal conversion). Bei ihr wird Energie in Molekülschwingungen anstatt in elektromagnetische Strahlung umgesetzt. Aus den schwingungsangeregten Zuständen kann anschließend die Relaxation in den niedrigsten Schwingungszustand stattfinden. Die Geschwindigkeit der Mechanismen unterscheidet sich dabei sehr. Während die Phosphoreszenz bis zu mehreren Sekunden dauern kann, finden die strahlungslosen Übergänge im Picosekundenbereich statt.
3.2 Potentialhyperflächen [19-20]
Die Potentialhyperfläche eines Moleküls beschreibt alle möglichen Reaktionswege des betrachteten Systems und liefert somit wertvolle Informationen. Sie ist eine Funktion der Kern- Koordinaten und der Energie des Moleküls. Die Dimension der Potentialhyperfläche ergibt sich aus den Freiheitsgraden des Moleküls mit N Atomen zu 3N plus dessen Energie. Die direkte analytische Berechnung von Potentialhyperflächen ist oft zu aufwendig, stattdessen werden häufig nur sogenannte Schnitte entlang einzelner wichtiger Koordinaten durchgeführt und an diese Punkte eine analytische Funktion angepasst.
Um die Einzelpunktrechnungen für konstante Kernkoordinaten zu erleichtern, werden zuvor Elektronen- und Kernposition abhängige Terme des Hamiltonoperators separiert.
ℋ = − 1
2 ∇ −
1
2 ∇−
| − | −
|− |
+ 1
−
+
−
= + + ! + ! + ! (2.1) Allgemeiner Hamiltonoperator
3.2.1 Born-Oppenheimer-Näherung [19-21]
6
Die Born-Oppenheimer-Näherung ermöglicht diese Separierung. Sie geht von der Annahme aus, dass sich die Elektronen " aufgrund des Massenunterschiedes um ein vielfaches schneller bewegen als die Kerne # und sich daher instantan der Kernbewegung anpassen. Näherungsweise lässt sich sagen, dass die Kernkoordinaten aus Sicht der Elektronen konstant sind und diese Elektronen sich im elektrischen Feld der fixierten Kerne bewegen. Folglich lässt sich die elektronische Energie (Eigenwerte zu , ! , ! ) als Funktion der Elektronenkoordinaten und der konstanten Kernkoordinaten % ansehen. Sie wird im elektronischen Hamiltonoperator ℋ &
zusammengefasst.
ℋ &'(); (% )+ = + ! + ! (2.2)
Zur Beschreibung des allgemeinen Hamiltonoperators ℋ werden die kinetische Energie der Kerne (Eigenwert zu ) und deren Coulombwechselwirkung (Eigenwert zu !), welche den nuklearen Hamiltonoperator ℋ bilden, sowie der elektronische Hamiltonoperator summiert.
ℋ'( )+ = + ! (2.3)
ℋ = ℋ+ ℋ & (2.4)
Bei Betrachtung fixierter Kernkoordinaten wird die kinetische Energie der Kerne (Rotation, Translation und Vibration) vernachlässigt, sodass sich die Gesamtenergie des Moleküls ,-.- aus der Coulombwechselwirkung der Kerne und der elektronischen Energie zusammensetzt.
,-.-= , &+ ∑ ∑ %001 2
3%1
(2.5)
Diese Energie wird bei den Potentialhyperflächen verwendet. In der Nähe von Kreuzungen elektronischer Zustände gilt die Born-Oppenheimer-Näherung infolge der Kopplung von Kern- und Elektronenbewegung jedoch nicht.
3.2.2 Stationäre Punkte [19-20]
Minima und Sattelpunkte der Potentialhyperfläche nehmen einen besonderen Stellenwert ein und werden auch stationäre Punkte genannt. Ein Minimum entspricht einer Gleichgewichtsgeometrie des Moleküls, während ein Sattelpunkt erster Ordnung einem Übergangszustand zwischen zwei Minima entspricht. Voraussetzung für einen stationären Punkt ist, dass der Gradient der Potentialfläche Null wird. Charakterisieren lassen sich die stationären Punkte über die Hessematrix, die der 2. Ableitung in
7
Richtung aller Koordinaten entspricht. Bei einem Minimum sind alle Eigenwerte der Hessematrix positiv. Bei einem Sattelpunkt n-ter Ordnung sind n-Eigenwerte negativ, während alle anderen positiv sind. Die n negativen Eigenwerte führen zu genau n imaginären Eigenfrequenzen.
Ob es sich um eine lokale oder globale Extremstelle handelt, kann nicht unterschieden werden. Zudem ist es meist nur möglich die zur Ausgangsgeometrie nächstgelegene Extremstelle anzulaufen.
3.2.2.1 Optimierungsmethoden [20, 22]
Um die Gleichgewichtsgeometrie eines Moleküls 45 6 zu erreichen, wird die Ausgangsgeometrie 457 um den Schrittvektor 85 verändert.
45 6 = 457 +85 (2.6)
Dies geschieht über mehrere Zwischenschritte 9, da kein analytischer, sondern nur ein iterativ genähert Schrittvektor bestimmt werden kann.
45:= 45 +85 (2.7)
45 6 ≈ 457+ ∑ 85 (2.8)
Der Schrittvektor jedes Iterationsschrittes 85 setzt sich aus dem Richtungsvektor <5 und der Schrittlänge = zusammen.
85 = <5=
Diese Werte können über verschiedene Methoden ermittelt werden. Im Folgenden soll die Quasi-Newton Methode erläutert werden, welche zur Optimierung der angeregten Zustände benutzt wurde.
3.2.2.1.1 Quasi-Newton Methode [20, 22-23]
Beim Newton-Raphson Verfahren wird der Schrittvektor 85 aus dem Produkt der inversen analytischen Hessematrix >3 und dem analytischen Gradienten ?5 berechnet, sodass sich für einen Iterationsschritt
45:= 45 − >3?5 (2.9)
ergibt. Da die Berechnung der analytischen Hessematrix in jedem Schritt sehr rechenintensiv ist, wird sie im Zuge der Quasi-Newton Methode iterativ genähert. Die
8
inverse iterative Hessematrix >:3 des Punktes 45:wird hierbei aus der inversen Hessematrix >3, dem Schrittvektor 8 und den Gradienten ?5 und ?5: ermittelt, wobei verschiedene Ansätze entwickelt wurden. In dieser Arbeit wird der Ansatz nach Broyden, Fletcher, Goldfarb und Shanno (BFGS) verwendet.
Für den ersten Schritt der Quasi-Newton Methode muss eine Starthessematrix H0 vorgegeben werden, deren Wahl maßgeblichen Einfluss auf die Effizienz der Methode nimmt.
Mit dem Programm Gong werden die Gradienten mit einem parallelisierten Algorithmus numerisch berechnet. Die Berechnung verläuft dabei wahlweise über Zwei- Punkte- bzw. in Minimums Nähe über Drei-Punkte- Gradientenverfahren.
3.2.3 Konische Durchschneidungen [23-26]
Als konische Durchschneidungen werden die Bereiche bezeichnet, bei denen zwei elektronische Zustände energetisch entartet vorliegen. Aufgrund dieser Entartung kommt es zu einer starken Kopplung zwischen Elektronen- und Kernbewegung, sodass diese nicht mehr getrennt betrachtet werden können. Dies hat zur Folge, dass die Born- Oppenheimer-Näherung nicht länger gelten kann. Diese Art der Kopplung wird als nicht-adiabatische Kopplung bezeichnet.
Bei dem Durchlaufen einer konischen Durchschneidung wird die elektronische Energie so komplett in Schwingungsenergie umgesetzt, was als interne Konversion bezeichnet wird. Es erfolgt somit ein ultraschneller strahlungsloser Übergang zwischen den beiden elektronischen Zuständen.
3.3 Dichtefunktionaltheorie (DFT) [20, 27]
Bei der DFT Methode wird zur Beschreibung der Energie die Elektronendichte A'+, die als Funktion von der Position r der Elektronen abhängt, anstatt der Schrödingergleichung genutzt. Kohn und Sham haben die Energie in wechselwirkende bzw. nicht-klassische (ΔC, ΔD ) und nicht-wechselwirkende bzw. klassische Terme (C, D , D ) aufgeteilt.
,EA'+F = CEA'+F + D EA'+F + D EA'+F + ΔCEA'+F + ΔD EA'+F (2.10)
9
Mit C als kinetische Energie nicht-wechselwirkender Elektronen, D als Coulombpotential zwischen Elektronen und Kernen, bzw. bei D zwischen Elektronen, ΔC als kinetische Wechselwirkungsenergie und ΔD als Korrektur aller nicht- klassischen Energien des Coulombpotentials. ΔD und ΔC werden meist als Austauschkorrelationsenergie ,GH zusammengefasst. Da für die Dichte der nicht- wechselwirkenden Terme gilt,
A = ∑ IJ |JK (2.11)
ergibt sich die Orbitalschreibweise mit N für die Anzahl der Elektronen ,EA'+F = LMJN−1
2 ∇NJO − PJQ
|− |
RH&
QJST
+ UJV1
2 W A'X+
| − X| <XVJY
+ ,GHEA'+F (2.12)
Bei dem folgenden Kohn-Sham self-consistent field (SCF) Formalismus werden die Kohn-Sham Orbitale J ähnlich wie beim Hartree-Fock-Formalismus über die Kohn- Sham-Gleichung ermittelt.
ℎ[\J = ]J (2.13)
Dabei ist der Kohn-Sham-Einteilchenoperator ℎ[\ definiert als
ℎ[\ = −∇− ∑RH& |03^^|+ _|`a3bcb|<X+ DGH (2.14) mit DGH =ded`fg
Über diesen Formalismus können die Terme C, D , D exakt berechnet werden. Die Austauschkorrelationsenergie Exc muss hingegen über Funktionale bestimmt werden.
3.3.1 Funktionale [20, 27]
Die Funktionale beschreiben die Austauschkorrelationsenergie aus Sicht des uniformen Elektronengases, wobei es drei verschiedene Arten von Funktionalen gibt. Zum einen die lokale Dichte Näherung (LDA) bzw. lokale Spin-Dichte Näherung (LSDA) unter Einbeziehung des Elektronenspins, zum anderen die generalisierte Gradienten Näherung (GGA), welche eine Änderung der Dichte mit einbezieht. Die dritte Art stellen Hybridfunktionale (ACM) dar, bei denen die Austauschkorrelationsenergie zu einem
10
Teil aus der DFT Austauschkorrelationsenergie,GHhij, zum anderen Teil aus dem Hartree-Fock-Austauschterm ,Gki besteht.
In dieser Arbeit wurden die Hybridfunktionale B3-LYP [28] für die Optimierung des Grundzustandes und BH-LYP [29] für die DFT/MRCI Rechnungen benutzt, welche sich wie folgt zusammensetzen:
,GHlmnop = q7',Gki− ,Gn\hr+ + qG∆,Gltt+ qH',Hnop− ,Hn\hr+ (2.15) mit q7 = 0,2 , qG= 0,72 und qH = 0,81
,GHlknop = q7',Gki− ,Gltt+ + qH,Hnop (2.16) mit q7 = 0,5
B88 ist ein semi-empirisches GGA Funktional nach Becke, LYP ein nicht-empirisches Funktional nach Lee, Yang und Parr. Ec entspricht der Korrelationsenergie, Ex der Austauschenergie.
3.4 Konfigurationswechselwirkung (CI) [20-21, 27]
Bei der CI Methode werden zur Beschreibung des Moleküls mehrere Slaterdeterminaten mit verschiedenen Konfigurationen linear kombiniert. Bei den Slaterdeterminanten handelt es sich zum einen um die Slaterdeterminante des Grundzustandes yki, wie sie bei der HF-Methode verwendet wird, und zum anderen um die Slaterdeterminanten angeregter Zustände y. Ihr Einfluss wird durch den Koeffizienten q gewichtet.
Ψ-.- = q7yki+ qy
{.
.HH.
+ q}y}
{.
~}
.HH.
~
+ q}-y}-
{.
~}~- .HH.
~~
+ ⋯ (2.17)
Durch die Linearkombination werden sowohl der Grundzustand als auch die angeregten Zustände besser beschrieben. Da die Doppelanregung und Einfachanregungen den größten Einfluss haben, werden meist nur diese mitberechnet.
3.4.1 Multi-Referenz-Konfigurationswechselwirkung (MRCI) [21, 27]
Im Gegensatz zur CI Methode, bei der die Hartree-Fock-Wellenfunktion als Referenz und Ausgang für die Anregungen benutzt wird, setzt sich der Referenzzustand y7 bei der MRCI Methode aus Wellenfunktionen verschiedener Konfigurationen
11
(Configurated state functions (CSF)), also aus mehreren Referenzen, zusammen, welche mit dem Faktor q skaliert werden.
y7 = q+ q+ ⋯ (2.18)
Die CSFs werden über den sogenannten Referenzraum festgelegt, welcher mit dem Complete Active Space (CAS) vergleichbar ist. Neben der Elektronen- und Orbitalanzahl, welche den Referenzraum bilden sollen, wird sätzlich noch der Wert für die maximal n-fache Anregung (Einfach-, Zweifach-, Dreifachanregung,…) mitgegeben.
Abb. 3 [27]: Begrenzungen der an der Anregung beteiligten Orbitale
Wenn wir beispielsweise den complete active space aus Abb. 3 als Referenzraum wählen und maximal Doppelanregungen erlauben, setzt sich die multi- Referenz aus allen möglichen Anordnungen der vier Elektronen in die zwei höchstbesetzten (HOMO- 1, HOMO) und niedrigsten unbesetzten Orbitale (LUMO, LUMO+1) zusammen, die durch Einfach- und Doppelanregungen erhalten werden.
Bei der DFT/MRCI Methode kann der genaue Referenzraum anstatt durch den User durch eine erste Testrechnung ermittelt. Dies garantiert, dass alle wichtigen
12
Konfigurationen, die zu den zu untersuchenden Zuständen beitragen, berücksichtigt werden.
3.5 DFT/MRCI Methode [21, 30-31]
Bei der DFT/MRCI Methode werden die Hartree-Fock Orbitale durch die Kohn-Sham- Orbitale ersetzt und diese für den Aufbau des DFT/MRCI Hamiltonoperators >hij genutzt, da durch die DFT Werte die dynamische Korrelation berücksichtigt wird und so durch den MRCI Ansatz nur noch die statische Korrelation mit einfließen muss. Dies hat zur Folge, dass deutlich weniger CSFs benötigt werden um das System zu beschreiben, aber auch dass die Doppelnennung der dynamischen Elektronenkorrelation durch den MRCI Ansatz verhindert werden muss.
Abb. 4 [27]: Struktur einer CI Matrix mit blockweiser Anordnung der Determinanten der einzelnen Anregungsarten und deren Wechselwirkung
Die DFT/MRCI-Matrix wird wie in Abb. 4 nach Art der Anregung aufgeteilt und durch CSFs aufgebaut, welche durch einen Besetzungszahlvektor für den Raumanteil und einen Spinkopplungsanteil charakterisiert sind. Aufgrund der Charakterisierung werden die Matrixelemente wie folgt eingeteilt
• CSF mit gleichem Raum- und Spinkopplungsanteil (Diagonalelemente) (Ia)
13
• CSF mit gleichem Raum- , aber unterschiedlichem Spinkopplungsanteil (Außerdiagonalelement) (Ib)
• Ein-Elektronen Differenz im Raumanteil (Außerdiagonalelemente) (IIa)
• Zwei-Elektronen Differenz im Raumanteil (Außerdiagonalelemente) (IIb)
Die Diagonalelemente des DFT/MRCI Hamiltonoperators >hijergeben sich aus der exakt bestimmten Hartree-Fock-Rechnung, welche DFT spezifisch korrigiert wird.
>hij− ,hij
= > − ,ki − HHki− HH[\
fg H
+ ki− [\
fg
+ 1
" GH 'qq|+ − E#7F
fg H fg
'q|q+
(2.19)
Die Gleichung besteht aus zwei Teilen. Im ersten Teil werden die Hartree-Fock- Orbitale durch die Kohn-Sham-Orbitale ersetzt. Der zweite Teil besteht aus skalierten Zwei-Elektronen-Beiträgen zur Einfachanregungsenergie.
ki und [\ sind Matrixelemente des Hartree-Fock- bzw. Kohn-Sham Fock-Operators.
Der Index c bezieht sich auf einen Erzeugungsoperator, während sich a auf einen Vernichtungsoperator bezieht. " GH steht für das Anregungslevel (z.B.
Dreifachanregung). Der Faktor der Zweielektronenintegrale ist ein angepasster Parameter, dessen Wert Tab. 1 entnommen werden kann. Er entspricht ungefähr dem Wert = 1 − 8ki vom Faktor 8ki des gewählten Funktionals (für BH-LYP 8ki = 0,5). Der Faktor E#7F der Korrektur für das Austauschintegral hängt von der Multiplizität und der Anzahl der offenen Schalen #7 der CSF ab. Für Singulettzustände gilt
E#7F = pE0F + N7 (2.20)
für Triplettzustände
E#7F = N7m (2.21)
Wobei folgende Werte für die Koeffizienten ermittelt wurden.
Tab. 1 [31]: Optimierte Parameter der DFT/MRCI Näherung mit BH-LYP Funktional
14
Für die Außerdiagonalelemente (IIa, IIb) werden die exakten Werte über die Energiedifferenz der Diagonalelemente der CSFs und X gewichtet und gehen so in den DFT/MRCI Hamiltonoperator >hij mit ein.
>hij XX = > XX32eb (2.22)
Durch diese Skalierung wird zum Großteil die Doppelgewichtung der dynamischen Elektronenkorrelation separiert, während die statische Elektronenkorrelation auf Grund der geringen Energiedifferenz ihrer Zustände mit einbezogen wird.
Für die nicht-Diagonalelemente des Typs Ia werden die exakten Werte genommen, ohne Skalierung.
4 Ergebnisse und Diskussion
Im folgenden Kapitel werden Potentialschnittflächen für die Geometrien des Grund- und 2Ag Zustandes diskutiert, die Lage und Kreuzungen des 1Bu und 2Ag, sowie des Grundzustandes lokalisieren und erklären sollen.
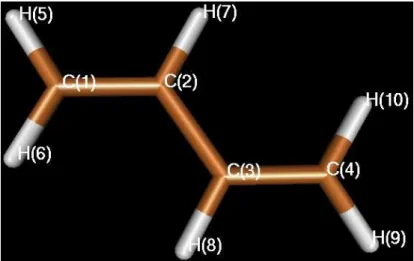
Alle Rechnungen wurden mit einem TZVP Basissatz [32] durchgeführt. Für alle Tabellen und Schaubilder gilt die Nummerierung der Atome aus Abb. 5, desweiteren sind alle Energien in eV relativ zur DFT/MRCI Energie des Grundzustandes in seiner Gleichgewichtsgeometrie angegeben [A1].
15
Abb. 5: Nummerierung der Atome des s-trans-Butadiens
4.1 Gleichgewichtsgeometrien des Grund-, 1Bu und 2Ag Zustandes
Für eine erste Betrachtung wurden die Geometrien des Grund- (1Ag), 1Bu und 2Ag
Zustandes optimiert und für jede Geometrie die Anregungsenergien berechnet, wobei die Geometrieoptimierung des Grundzustandes mit dem B3-LYP Funktional [28] und die der angeregten Zustände mit dem BH-LYP [29] Funktional durchgeführt wurden.
Die Optimierung erfolgt mit einer Gradientenkonvergenznorm von 0,003 Hartree für den S1 und 0,001 Hartree für den S2 Zustand.
Es muss jedoch beachtet werden, dass die Geometrien derangeregten Zustände nur in C1
Symmetrie optimiert werden können. Dies hat zur Folge, dass die planaren Geometrien nicht genau der C2h Symmetrie entsprechen. Die berechneten Strukturen sind auch nicht exakt planar, bei der 2Ag Struktur z. B. ergeben sich leichte Abweichungen von ca. 0.5- 2 Grad. (für die genauen Koordinaten siehe [A2-4]).
16
Abb. 6 [A1]: Energien der des Grund
Für die Grundzustandsgeometrie
(Abb. 6). Dies entspricht der Reihenfolge, festgestellt wurde, während in
Reihenfolge beobachtet wurde
Zustand und 2Ag Zustand ist in Übereinstimmung mit sehr gering (ca. 0,2 eV). Bei der 1B
Reihenfolge der angeregten Zustände feststellen, 2A
stärkste Änderung der Energien und der Struktur ist für die 2A verzeichnen. Auch hier ist 2A
Zugleich ist der S0 Zustand angehoben und 2A Abstand zwischen S1 und S0.
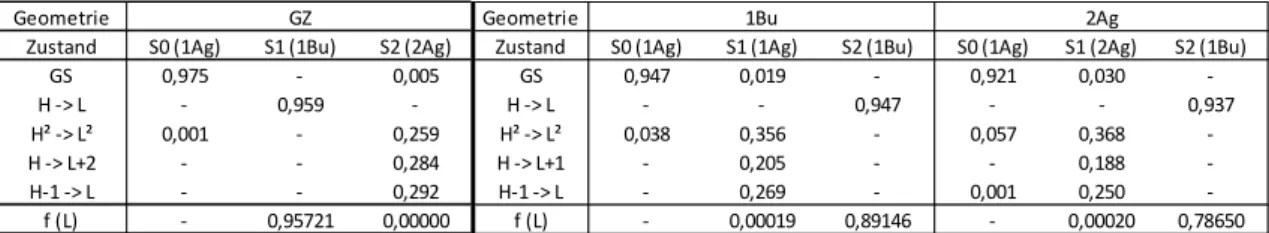
Unterscheiden lassen sich die Zust Oszillatorstärke f (L). Der optisch helle Anregung charakterisiert (Tab. 2)
Grundzustandsgeometrie zu etwa gleichen Anteilen aus HOMO LUMO+2 und HOMO²
1Bu und 2Ag Zustände tauschen die Orbitale LUMO+1 und LUMO+2 die Position (siehe Orbitalbilder im Anhang),
Strukturen mit HOMO LUMO+1 bezeichnet
Zustand charakteristisch. Die beiden Einzelanregungen
Doppelanregung [34] und finden zwischen Orbitalen gleicher Symmetrie st : Energien der des Grund-, 1Bu und 2Ag Zustandes für deren
Gleichgewichtsgeometrien
Grundzustandsgeometrie befindet sich der 1Bu Zustand unter dem ies entspricht der Reihenfolge, die auch in Ethen und s-trans
, während in größeren konjugierten Systemen die umgekehrte Reihenfolge beobachtet wurde [33-36]. Der Energieunterschied zwischen
ist in Übereinstimmung mit bisherigen Forschung
Bei der 1Bu Geometrie lässt sich eine Umkehr in der Reihenfolge der angeregten Zustände feststellen, 2Ag ist hier der stabilere Zustand
Energien und der Struktur ist für die 2Ag Geometrie zu verzeichnen. Auch hier ist 2Ag stabiler und deutlich niedriger in der Energie als 1B
Zustand angehoben und 2Ag abgesenkt, somit verringert sich der
terscheiden lassen sich die Zustände anhand der Art der Anregung und der optisch helle 1Bu Zustand ist durch eine HOMO
charakterisiert (Tab. 2). Der optisch dunkle 2Ag Zustand setzt sich
zu etwa gleichen Anteilen aus HOMO-1 LUMO, HOMO LUMO² Anregungen zusammen. In den Geometrien der tauschen die Orbitale LUMO+1 und LUMO+2 die Position (siehe Orbitalbilder im Anhang), darum ist die entsprechende Anregung in diesen
LUMO+1 bezeichnet. Die Doppelanregung ist für den . Die beiden Einzelanregungen dienen zur Stabi
] und finden zwischen Orbitalen gleicher Symmetrie st Zustandes für deren
Zustand unter dem 2Ag Zustand trans-Hexatrien n die umgekehrte Der Energieunterschied zwischen dem 1Bu
bisherigen Forschungergebnissen Geometrie lässt sich eine Umkehr in der ist hier der stabilere Zustand. Die Geometrie zu stabiler und deutlich niedriger in der Energie als 1Bu. abgesenkt, somit verringert sich der
Anregung und der Zustand ist durch eine HOMO LUMO setzt sich bei der LUMO, HOMO LUMO² Anregungen zusammen. In den Geometrien der tauschen die Orbitale LUMO+1 und LUMO+2 die Position hende Anregung in diesen
ist für den Ag
dienen zur Stabilisierung der ] und finden zwischen Orbitalen gleicher Symmetrie statt: HOMO-
17
1 und LUMO haben Au Symmetrie, HOMO und LUMO+2 (LUMO+1) entsprechend Bg Symmetrie. In allen oben genannten Fällen handelt es sich um → ∗ - Anregungen.
Zu bemerken ist, dass für solche stabilisierenden Effekte normalerweise die HOMO LUMO+1 anstatt der HOMO LUMO+2 Anregung typisch ist, wie es für die Grundzustandsgeometrie der Fall ist.
Tab. 2: Anteil der einzelner Konfigurationen des Grund-, 1Bu und 2Ag Zustandes und deren Oszillatorstärke f (L) jeweils für deren optimierte Geomtrien
In der 1Bu und 2Ag Struktur tauschen die Orbitale LUMO+1 und LUMO+2 die Position. Bei der HL+1 und HL+2 Anregung sind also die gleichen Orbitale beteiligt (siehe Text)
Durch die Geometrieänderung vom Grundzustand über 1Bu zur 2Ag Struktur kommt es zu einer leichten Stabilisierung der Doppelanregung, sowohl im 2Ag, als auch im Grundzustand. Der Anteil der Einzelanregungen in dieser Reihe nimmt hingegen ab.
Ebenfalls kommt es zu einer Änderung der Oszillatorstärke. Während sie für den hellen 1Bu Übergang deutlich abnimmt, wird sie für den 2Ag Übergang leicht von Null verschieden.
Erklären lassen sich diese Beobachtungen mit einer Änderung der Bindungsalternanz, die für den 2Ag Zustand am deutlichsten ausfällt (Abb. 7). Bei diesem kommt es zu einer kompletten Umkehr von Einfach- und Doppelbindungen, während sich für den 1Bu Zustand ein stark konjugiertes System bildet.
Geometrie Geometrie
Zustand S0 (1Ag) S1 (1Bu) S2 (2Ag) Zustand S0 (1Ag) S1 (1Ag) S2 (1Bu) S0 (1Ag) S1 (2Ag) S2 (1Bu)
GS 0,975 - 0,005 GS 0,947 0,019 - 0,921 0,030 -
H -> L - 0,959 - H -> L - - 0,947 - - 0,937
H² -> L² 0,001 - 0,259 H² -> L² 0,038 0,356 - 0,057 0,368 -
H -> L+2 - - 0,284 H -> L+1 - 0,205 - - 0,188 -
H-1 -> L - - 0,292 H-1 -> L - 0,269 - 0,001 0,250 -
f (L) - 0,95721 0,00000 f (L) - 0,00019 0,89146 - 0,00020 0,78650
GZ 1Bu 2Ag
18
Abb. 7 [A2-4]: C-C Bindun
Dieses Verhalten ist auf die zurückzuführen. Diese hat Doppelbindungen zur Folge und
Einfachbindungen. Da im Falle des 2A Doppelanregung beteilig ist
Verlängerung der Doppelbindungen als im 1B
Bindungen des 2Ag Zustandes sind auf die beiden Einzelanregungen (H HL+1) zurückzuführen, da die K
C-C Bindungen gehen [O3, 5,
Tab. 3 [A2-4]: Bindungslängen ( des planar optimierten Grund
Koordinate C1C2 C2C3 C4H9 C4H10
C3H8 H5H6C1 C1C2C3 H8C3C4 C3C4H10
C Bindungslängen der optimierten Geometrien
→ ∗- Anregungen aus dem HOMO [O2] ins LUMO [O3]
eine Verringerung der Elektronendichte an den und führt zu einer Zunahme von Elektronendichte
Einfachbindungen. Da im Falle des 2Ag Zustandes die HOMO²
nregung beteilig ist, kommt es für diesen Zustand zu einer stärkeren Verlängerung der Doppelbindungen als im 1Bu Zustand. Die insgesamt längeren C
Zustandes sind auf die beiden Einzelanregungen (H ) zurückzuführen, da die Knotenebenen des LUMOs und LUMO+1 C Bindungen gehen [O3, 5, ].
Bindungslängen (Å) und Bindungswinkel (°) des planar optimierten Grund-, 1Bu und 2Ag Zustandes
Koordinate GZ 1Bu 2Ag
1,341 1,417 1,488
1,458 1,381 1,404
1,086 1,074 1,070
1,088 1,077 1,072
1,090 1,080 1,076
116,6 118,3 119,1
124,3 124,9 123,3
119,4 117,2 117,1
121,5 121,2 120,5
gslängen der optimierten Geometrien
HOMO [O2] ins LUMO [O3]
Verringerung der Elektronendichte an den nendichte an den LUMO² , kommt es für diesen Zustand zu einer stärkeren Die insgesamt längeren C-C Zustandes sind auf die beiden Einzelanregungen (H-1L und notenebenen des LUMOs und LUMO+1 durch die
19
Die C-H Bindungen werden in beiden Fällen gleichmäßig kürzer, was zu einer Weitung der H-C-H Winkel führt. Die C-C-C Winkel werden hingegen nur für den 1Bu Zustand weiter, während sie für den 2Ag Zustand schmaler werden.
4.2 Potentialschnittflächen der Grundzustandsgeometrie
Um die Kreuzung der 1Bu und 2Ag Zustände zu lokalisieren und die verantwortlichen Koordinatenänderungen zu detektieren, wurden ausgehend von der Grundzustandsgeometrie die Potentialschnittflächen für die in Abb. 8 dargestellten Koordinaten erstellt. Diese Potentialschnittflächen wurden bereits von B. Ostojić [17]
mittels CASPT2 berechnet und sollen als Referenz herangezogen werden. Im Folgenden sollen jedoch nur die Potentialschnittflächen diskutiert werden, die entweder zu einer Kreuzung führen oder sich von den Ergebnissen von B. Ostojić signifikant unterscheiden. Die Restlichen Daten finden sich im Anhang [A5].
symmetrisch
anti-symmetrisch
C1C2 + C3C4
C2C3
C1C2C3 sym. zu C2C3C4
C1C2C3 antisym. zu C2C3C4 [A5]
H5C1C2C3 konrot. zu H9C4C3C2
H5C1C2C3 disrot. zu H9C4C3C2
Abb. 8: Koordinatenänderungen der untersuchten Potentialschnittflächen
20
Abb. 9 [A6]: Potentialschnittflächen für die simultane Bindungslängenänderun Doppelbindung
Abb. 9 zeigt die Potentialflächen für die Änderung entlang der Doppelbindungskoordinaten.
führt bereits zu einer Kreuzung des 1B
Zustandes liegt dabei bei längeren Bindungslängen als das des 1B stimmt mit den Beobachtungen, die für die
überein.
: Potentialschnittflächen für die simultane Bindungslängenänderun Doppelbindungen C1C2 und C3C4
zeigt die Potentialflächen für die Änderung entlang der Eine Verlängerung der Doppelbindungen um
ührt bereits zu einer Kreuzung des 1Bu und 2Ag Zustandes. Das Minimum des 2A Zustandes liegt dabei bei längeren Bindungslängen als das des 1Bu Zustandes. Dies
Beobachtungen, die für die optimierten Geometrien gemacht wurden, : Potentialschnittflächen für die simultane Bindungslängenänderung der
zeigt die Potentialflächen für die Änderung entlang der Eine Verlängerung der Doppelbindungen um nur 0,05 Å Das Minimum des 2Ag
Zustandes. Dies gemacht wurden,
Abb. 10 [A7]: Potentialschnittflächen für die Bindungslängenänderung
Oben: Ergebnis von B. Ostoji
Die Veränderung der C
zeigt jedoch, dass eine Verkürzung für den 1B
bevorzugt wird und der Änderung der Bildungsalternanz entspricht.
B. Ostojić zeigen sich jedoch Abweichungen.
den angeregten Zuständen
während bei B. Ostojić der entgegengesetzte Trend zu beobachten ist.
: Potentialschnittflächen für die Bindungslängenänderung der Einfachbindung C2C3
Oben: Ergebnis von B. Ostojić [6] (man beachte hierbei das größere Intervall)
Die Veränderung der C-C Einfachbindung (Abb. 10) führt zwar zu keiner Kreuzung, eigt jedoch, dass eine Verkürzung für den 1Bu und 2Ag Zustand i
bevorzugt wird und der Änderung der Bildungsalternanz entspricht. Beim Vergleich mit zeigen sich jedoch Abweichungen. So wird die Energiedifferenz zwischen gten Zuständen von einer kurzen hin zu einer langen Bindung geringer,
der entgegengesetzte Trend zu beobachten ist.
21
: Potentialschnittflächen für die Bindungslängenänderung
beachte hierbei das größere Intervall)
führt zwar zu keiner Kreuzung, Zustand im gleichen Maße Beim Vergleich mit wird die Energiedifferenz zwischen von einer kurzen hin zu einer langen Bindung geringer, der entgegengesetzte Trend zu beobachten ist.
22
Abb. 11 [A8]: Potentialschnittflächen für die symmetrische Bindungswinkeländerung der C1C2C3 und C2C3C4 Bindun
Die symmetrische Änderung der C
von Chadwick et al. [5] beobachteten Kreuzung
Kreuzung jedoch mit einer Energiezunahme verbunden ist, ist sie für die Suche nach dem Kreuzungspunkt mit dem Grundzustand uninteressant.
Abb. 12 [A9]: Potentialschnittflächen für die disrotatorische Torsion der H5C1C2C3 und H9C4C3C2 Torsionswinkel
Aus Darstellungszwecken wurde der Ausgangwert von 180° gleich Null gesetzt : Potentialschnittflächen für die symmetrische Bindungswinkeländerung
der C1C2C3 und C2C3C4 Bindungswinkel
Die symmetrische Änderung der C-C-C Bindungswinkel (Abb. 11) führt zu der bereits beobachteten Kreuzung des 1Bu und 2Ag Zustandes.
Kreuzung jedoch mit einer Energiezunahme verbunden ist, ist sie für die Suche nach em Kreuzungspunkt mit dem Grundzustand uninteressant.
: Potentialschnittflächen für die disrotatorische Torsion der H5C1C2C3 und H9C4C3C2 Torsionswinkel
Aus Darstellungszwecken wurde der Ausgangwert von 180° gleich Null gesetzt : Potentialschnittflächen für die symmetrische Bindungswinkeländerung
führt zu der bereits Zustandes. Da die Kreuzung jedoch mit einer Energiezunahme verbunden ist, ist sie für die Suche nach
: Potentialschnittflächen für die disrotatorische Torsion
Aus Darstellungszwecken wurde der Ausgangwert von 180° gleich Null gesetzt
Abb. 13 [A10]: Potentialschnittflächen für die konrotatorische Torsion der H5C1C2C3 und H9C4C3C2 Torsionswinkel
Aus Darstellungszwecken wurde der Ausgangwert von 180° gleich Null gesetzt
Sowohl für die disrotatorische
13) kommt es zu einer Kreuzung der angeregten Zustände Potentialschnittflächen nicht wesentlich
dass sich die planare Struktur (0°) an optimierten planaren Geometrien des 1B
einem Sattelpunkt. Desweiteren kommt es durch die Torsion zu einer deutlichen Energieerhöhung des Grundzustandes, ebenso wie zu einer signifikanten Verri
der Energiedifferenz zwischen
für die Torsion ist wie die Änderung der Bindungsalternanz auf die Verringerung der Elektronendichte an den Doppelbindungen zurückzuführen.
Doppelbindung einem Bindungsbruch vergleichbar ist Grundzustandes deutlich stärker an
Auf Grund dieser Beobachtung Kreuzung zwischen Grund zwischen 1Bu und 2A
zurückzuführen. Dies scheint plausibel, da Geometrie geringer ist und zudem eine
als eine Torsion. Zusätzlich befindet sich das s
: Potentialschnittflächen für die konrotatorische Torsion der H5C1C2C3 und H9C4C3C2 Torsionswinkel
Aus Darstellungszwecken wurde der Ausgangwert von 180° gleich Null gesetzt
disrotatorische (Abb. 12), als auch für die konrotatorische kommt es zu einer Kreuzung der angeregten Zustände, wobei
Potentialschnittflächen nicht wesentlich unterscheiden. Besonders hervorzuheben ist, sich die planare Struktur (0°) an einem Maximum der Energie b
Geometrien des 1Bu und 2Ag Zustandes befinden sich somit auf einem Sattelpunkt. Desweiteren kommt es durch die Torsion zu einer deutlichen Energieerhöhung des Grundzustandes, ebenso wie zu einer signifikanten Verri
Energiedifferenz zwischen dem Grund- und 2Ag Zustand. Der energetische Verlauf für die Torsion ist wie die Änderung der Bindungsalternanz auf die Verringerung der Elektronendichte an den Doppelbindungen zurückzuführen. Da die Drehung um eine einem Bindungsbruch vergleichbar ist, steigt die Energie des Grundzustandes deutlich stärker an als die der angeregten Zustände.
Auf Grund dieser Beobachtungen wird daher vermutet, dass die Torsion zur erwarteten Kreuzung zwischen Grund- und 2Ag Zustand führt. Die vorhergehende Kreuzung und 2Ag Zustand ist jedoch auf die Bindungslängenänderung zurückzuführen. Dies scheint plausibel, da zum einen die benötigte Änderung der Geometrie geringer ist und zudem eine Relaxation der Bindungslängen schne
Zusätzlich befindet sich das s-trans- Butadien bezüglich der Torsion
23
: Potentialschnittflächen für die konrotatorische Torsion
Aus Darstellungszwecken wurde der Ausgangwert von 180° gleich Null gesetzt
, als auch für die konrotatorische Torsion (Abb.
wobei sich die beiden Besonders hervorzuheben ist, befindet. Die bereits Zustandes befinden sich somit auf einem Sattelpunkt. Desweiteren kommt es durch die Torsion zu einer deutlichen Energieerhöhung des Grundzustandes, ebenso wie zu einer signifikanten Verringerung Der energetische Verlauf für die Torsion ist wie die Änderung der Bindungsalternanz auf die Verringerung der die Drehung um eine , steigt die Energie des
, dass die Torsion zur erwarteten Die vorhergehende Kreuzung Zustand ist jedoch auf die Bindungslängenänderung zum einen die benötigte Änderung der ndungslängen schneller abläuft Butadien bezüglich der Torsion
24
auf einem relativ flachen Maximum, sodass die benötigte Torsion von 30° bis 40° nur langsam eintreten wird.
4.3 Potentialschnittflächen
Weiterhin werden die disrotatorische (Abb.
ausgehend von der planar optimierten Torsion von +-90° untersucht.
Abb. 14 [A11]: Potentialschnittfl
auf einem relativ flachen Maximum, sodass die benötigte Torsion von 30° bis 40° nur
Potentialschnittflächen entlang der Torsion für die 2Ag Geometrie
disrotatorische (Abb. 15) und konrotatorische (Abb.
planar optimierten Geometrie des 2Ag Zustandes untersucht.
: Potentialschnittfläche der konrotatorischen Torsion für die 2A Geometrie
auf einem relativ flachen Maximum, sodass die benötigte Torsion von 30° bis 40° nur
(Abb. 14) Torsion Zustandes bis zu einer
für die 2Ag
Abb. 15 [A12]: Potentialschnittfläche der disrotatorischen Torsion
Wie erwartet liegt der 2A Fällen so weit unter dem 1B
Zustände mehr kommt. Beim Vergleich fällt auf, dass die Energieänderungen deutlich kleiner ausfällt (
zusammenhängt. Bei 60°
Grundzustand in allen Modellen (Abb. 12
zwischen S1 und S0, und damit auch die Lage ein
Torsion nicht maßgeblich durch die Bindungslänge beeinflusst. Der Energieabstand von S1 und S2 zum Grundzustand wird hingegen fast nur durch die Änderung der Grundzustandsenergie bestimmt.
Erst bei einer Torsion größer Zustandes und infolge dessen
dem Grundzustand. Besonders bei der konrotatorischen Torsion setzt dieser Energieabfall abrupt ein
höheren Zustände (5A oder 6A) zu erklären, wurden die
zwischen 70° und 90° Torsion genauer betrach
: Potentialschnittfläche der disrotatorischen Torsion Geometrie
Wie erwartet liegt der 2Ag (2A) Zustand durch die Bindungslängenänderung
em 1Bu (3A) Zustand, dass es zu keiner Kreuzung der beiden Beim Vergleich des Bereiches +- 60° mit Abb. 13 bzw. Abb. 12 fällt auf, dass die Energieänderungen während der Torsion für den Grundzustand deutlich kleiner ausfällt (ca. 1 eV), was wiederum mit der Bindungslängenänderung
60° beträgt die Energiedifferenz zwischen dem 2A
in allen Modellen (Abb. 12-15) etwa 2,7 eV, d. h. der Energieunterschied , und damit auch die Lage einer möglichen Durchdringung, wird bei Torsion nicht maßgeblich durch die Bindungslänge beeinflusst. Der Energieabstand von zum Grundzustand wird hingegen fast nur durch die Änderung der Grundzustandsenergie bestimmt.
Erst bei einer Torsion größer 70° kommt es zu einem starken Energieabfall des 2A Zustandes und infolge dessen im Bereich von 90° fast zu der erwarteten Kreuzung
Besonders bei der konrotatorischen Torsion setzt dieser Energieabfall abrupt ein, während es bei der disrot. Torsion scheint, als würde einer der höheren Zustände (5A oder 6A) diabatisch nach unten verschoben. Um dieses Verhalten zu erklären, wurden die vertikalen Anregungen im Bereich des steilen Energieabfalls zwischen 70° und 90° Torsion genauer betrachtet. Auffallend ist, dass
25
: Potentialschnittfläche der disrotatorischen Torsion für die 2Ag
rch die Bindungslängenänderung in beiden (3A) Zustand, dass es zu keiner Kreuzung der beiden mit Abb. 13 bzw. Abb. 12 für den Grundzustand , was wiederum mit der Bindungslängenänderung zwischen dem 2Ag und dem 15) etwa 2,7 eV, d. h. der Energieunterschied er möglichen Durchdringung, wird bei Torsion nicht maßgeblich durch die Bindungslänge beeinflusst. Der Energieabstand von zum Grundzustand wird hingegen fast nur durch die Änderung der
70° kommt es zu einem starken Energieabfall des 2Ag erwarteten Kreuzung mit Besonders bei der konrotatorischen Torsion setzt dieser isrot. Torsion scheint, als würde einer der . Um dieses Verhalten des steilen Energieabfalls dass der 2Ag Zustand
26
durch eine Doppelanregung mit vier offenen Schalen (D, 4; HOMO-1, HOMO LUMO, LUMO+1) dominiert wird und in Richtung 90° deren Anteil stark zunimmt (Tab. 4)
Tab. 4: Anteil der dominanten Doppelanregung mit vier offenen Schalen der disrotatorischen und konrotatorischen Torsion
Der Energieabfall scheint also direkt mit dem Anteil der Doppelanregungen zusammen zu hängen. Dies lässt sich mit der Berechnung des DFT/MRCI Hamiltonoperators über die Formeln (2.19) und (2.20) zur Berechnung der Diagonalterme und (2.22) zur Berechnung der Außerdiagonalterme erklären. In Formel (2.20) wird der Faktor E#7F für die Korrektur des Austauschintegrals in Abhängigkeit der Anzahl der offenen Schalen N0 berechnet. Im Fall von vier offenen Schalen ergibt sich somit:
E#7F = pE0F + N7 = 0,595 + 4 ∗ 0,106 = 1,019
Dies hat zur Folge, dass bei Berechnung der Formel (2.19) die Diagonalterme kleiner werden und somit die Außerdiagonalterme an Einfluss gewinnen. Zusätzlich gibt es für die D, 4 Anregung zwei mögliche Singulettkonfigurationen bei denen dieser Effekt auftritt. In Formel (2.22) werden die Außerdiagonalterme über den Energieunterschied der Diagonalelemente skaliert. Da beide Singulettkonfigurationen sich jedoch nur in ihrem Spin und nicht in ihrem Raumanteil unterscheiden (Typ Ia, vgl. Einleitung), kommt es bei ihnen zu keiner Skalierung, sondern zu einer vollen Gewichtung in den Außerdiagonaltermen.
>hij XX = > XX
Diese Effekte, speziell der letztere, haben zur Folge, dass der Einfluss der Elektronenkorrelation, welche durch die Außerdiagonalelemente beschrieben wird, so groß wird, dass die berechneten Energien zu klein und somit als Artefakte betrachtet
Torsion (°) konrot. disrot.
70 0,54 0,32
75 0,71 0,65
80 0,85 0,83
85 0,93 0,92
90 0,95 0,95
Anteil D,4
werden müssen. D, 4
gleichem Raumanteil bilden können, Methode.
4.4 Torsion um nur eine Doppelbindung
Bisher wurde die Torsion
Doppelbindungen durchgeführt. Im nächsten Schritt wurden die Torsionen Doppelbindungen unabhängig voneinander
Gleichgewichtsgeometrie des Grundzustandes Potentialfläche des S1 Zustand
Abb. 16 [A13]
ausgehend vo
Die Diagonalen entsprechen den bisher betrachteten Torsionen plötzlicher Energieabfall in den Ecken gut zu erkenn
der Sattelpunkt (hier als Maximum) Geometrie kommt es bei
Energieabfall ohne jegliche Barriere und S3 Zustand [A13].
D, 4 und höhere Anregungen, die mehrere Konfigurationen mit Raumanteil bilden können, sind somit ein generelles Problem der DFT/MRCI
Torsion um nur eine Doppelbindung
her wurde die Torsion gemäß der Schwingungen simultan für beide Doppelbindungen durchgeführt. Im nächsten Schritt wurden die Torsionen Doppelbindungen unabhängig voneinander betrachtet ausgehend von der Gleichgewichtsgeometrie des Grundzustandes. Abb. 16 zeigt eine 3D Darstellung der
Zustands.
[A13]: S1 Zustand bei Torsionen der Doppelbindungen ausgehend von der Grundzustandsgeometrie
Die Diagonalen entsprechen den bisher betrachteten Torsionen (Abb. 12
licher Energieabfall in den Ecken gut zu erkennen ist. Ebenfalls gut zu erkennen ist (hier als Maximum) für die planare Geometrie. Ausgehend von dieser Geometrie kommt es bei einer Torsion um nur eine Doppelbindung
ohne jegliche Barriere bis hin zu 90° Torsion, dies gilt auch für den S
27
mehrere Konfigurationen mit sind somit ein generelles Problem der DFT/MRCI
simultan für beide Doppelbindungen durchgeführt. Im nächsten Schritt wurden die Torsionen der ausgehend von der 16 zeigt eine 3D Darstellung der
Zustand bei Torsionen der Doppelbindungen
(Abb. 12-13), deren ist. Ebenfalls gut zu erkennen ist für die planare Geometrie. Ausgehend von dieser Torsion um nur eine Doppelbindung zu einem stetigen bis hin zu 90° Torsion, dies gilt auch für den S2
28
Abb. 17 [A14 ausgehend vo
Bei der Betrachtung der Potentialschnittfläche für ebenfalls ein stetiger Energieabfall des S
Zustandes (4A) zu erkennen.
um nur eine Doppelbindung keine Kreuzung festges
der Torsion um beide Doppelbindungen noch zu einer Kreuzung des 1B Zustandes gekommen war. Der 1B
2Ag Zustand. Für den S3 Zustand
durch die Torsion zu mehreren Kreuzungen zwischen den höher angeregten Zuständen kommt und diese sehr nah beieinander liegen
der höher angeregten Zustände ist, der von der Torsion profitiert.
Die Änderung der Energie des Grundzustandes der Torsion um beide Doppelbindungen
Zustände größer ist, sodass bei 90° nur noch 2 eV Grund
Neben der Energieänderung sind noch weitere Effekte während der Torsion zu beobachten.
1. Ab einer Torsion von 60 S2 Zustände. Während sich charakteristischen Anregungen
[A14]: Torsion um die C3-C4 Doppelbindung ausgehend von der Grundzustandsgeometrie
Bei der Betrachtung der Potentialschnittfläche für die Torsion einer Doppelbindung Energieabfall des S1 (2A), S2 (3A) und eines höher angeregten
Für den S1 und S2 Zustand fällt auf, dass bei der Torsion um nur eine Doppelbindung keine Kreuzung festgestellt werden kann, während es bei der Torsion um beide Doppelbindungen noch zu einer Kreuzung des 1B
Zustandes gekommen war. Der 1Bu Zustand bleibt somit energetisch stets unter dem Zustand ist eine eindeutige Zuordnung nicht möglich
durch die Torsion zu mehreren Kreuzungen zwischen den höher angeregten Zuständen und diese sehr nah beieinander liegen. Auffallend ist jedoch, dass er der Einzige der höher angeregten Zustände ist, der von der Torsion profitiert.
Energie des Grundzustandes ist vergleichbar mit der Änderung bei der Torsion um beide Doppelbindungen, während der Energiegewinn für die angeregten , sodass bei 90° nur noch 2 eV Grund- und S1 Zustand trennen.
nergieänderung sind noch weitere Effekte während der Torsion zu
Ab einer Torsion von 60° ändert sich zunehmend der Charakter de . Während sich die S1 und S2 Zustände aus den für 1B eristischen Anregungen zusammensetzen, besteht der
die Torsion einer Doppelbindung ist (3A) und eines höher angeregten Zustand fällt auf, dass bei der Torsion tellt werden kann, während es bei der Torsion um beide Doppelbindungen noch zu einer Kreuzung des 1Bu und 2Ag
Zustand bleibt somit energetisch stets unter dem ht möglich, da es durch die Torsion zu mehreren Kreuzungen zwischen den höher angeregten Zuständen Auffallend ist jedoch, dass er der Einzige
ist vergleichbar mit der Änderung bei der Energiegewinn für die angeregten Zustand trennen.
nergieänderung sind noch weitere Effekte während der Torsion zu
der S0, S1 und aus den für 1Bu und 2Ag
der S0 Zustand
29
letztendlich zu etwa gleichen Teilen aus der Grundzustandskonfiguration und der HOMO² LUMO² Anregung (Tab. 5).
Tab. 5: Anteil der Anregungen an S0, S1 und S2 Zustand abhängig von der Torsion
2. S1 und S2 Zustand bilden zudem ab 50° einen starken Dipolcharakter aus, was als sudden polarization effect [37] bezeichnet wird (Tab. 5). Die Dipolmomente für den S1 und S2 Zustand zeigen dabei in entgegengesetzte Richtungen [38]
(Abb. 18), sodass für den S1 Zustand die gedrehte Methylengruppe negativ ist, während sie für den S2 Zustand positiv geladen ist. Laut den Dynamiksimulationen von Levine und Martínez [4], sowie den Ergebnissen von Malrieu und Trinquier [39] ist die Ladung dabei nicht über das Allylsystem delokalisiert, sondern auf die C-Atome der verdrehten Doppelbindung beschränkt. Dieser sudden polarization effect wird sowohl bei Ethen, als auch bei größeren konjugierten Systemen beobachtet [40-41].
Tab. 6 Dipolmomente des S0, S1 und S2 Zustand abhängig von der Torsion
GZ GWG
Torsion (°) GS H -> L H² -> L² GS H -> L H² -> L² GS H -> L H² -> L²
60 0,921 - 0,056 0,006 0,820 0,048 0,031 0,065 0,354
70 0,862 0,001 0,105 0,027 0,739 0,119 0,076 0,148 0,487
80 0,720 0,003 0,228 0,139 0,497 0,271 0,110 0,400 0,379
90 0,435 0,016 0,482 0,379 0,309 0,202 0,093 0,572 0,167
S0 S1 S2
GZ GWG
Torsion (°) S0 S1 S2
50 0,00 0,71 0,34
60 0,00 1,46 0,74
70 0,00 3,07 2,52
80 0,00 4,87 5,13
90 0,27 4,79 4,84
Dipolmoment (debye)
30
Abb. 18: Dipolmoment bei 90° Torsion der
(Dipolmoment zeigt von der negativen zur positiven Ladung)
Der Ursprung dieser Effekte
zurückzuführen. In der planaren Geometrie hat LUMO Au Symmetrie besitzt. Die Torsion führt Dadurch wird jegliche Linear
weiterhin eine Angleichung der Gestalt und Energie de zur Folge (Abb. 19, Tab. 7)
Abb. 19: HOMO (links) und LUMO und
Zur besseren Darstellung des LUMO
ei 90° Torsion der optimierten S0 Geometrie für den Zustand
(Dipolmoment zeigt von der negativen zur positiven Ladung)
Der Ursprung dieser Effekte ist auf einen Symmetrieverlust von C
n der planaren Geometrie hat das HOMO Bg Symmetrie, während das Symmetrie besitzt. Die Torsion führt zur Aufhebung der Orbitalysmmetrie.
inearkombination dieser Orbitale ermöglich [40,
der Gestalt und Energie der HOMO und LUMO Orbitale
: HOMO (links) und LUMO für die optimierte S0 Geometrie und bei 90° Torsion derselben
Zur besseren Darstellung des LUMOs bei 90° Torsion wurde das Molekül um 180° in der Vertikalen gedreht
Geometrie für den S1 und S2
(Dipolmoment zeigt von der negativen zur positiven Ladung)
auf einen Symmetrieverlust von C2h zu C1
, während das zur Aufhebung der Orbitalysmmetrie.
, 42] Dies hat r HOMO und LUMO Orbitale
Geometrie (oben)
ertikalen gedreht
31
Tab. 7 Orbitalenergien
4.5 Minima des S1 und S2 Zustandes
Ausgehend von der Struktur bei 90° wurden die Geometrien des S1 und S2 Zustandes mit einer Konvergenznorm von 0,003 Hartree (S1) bzw. 0,005 Hartree (S2) nochmals frei optimiert [A15].
Abb. 20 [15]: Overlay der S1 (blau) und S2 (rot) Geometrien
Die beiden Geometrien unterscheiden sich nur geringfügig voneinander, dies lässt sich anhand der Überlagerung der Strukturen in Abb. 20 erkennen lässt. Die C2-C3 Diederwinkel und die Bindungslängen zeigen geringe Abweichungen [A15].
HOMO (eV) LUMO (eV)
GZ GWG -7,62 0,21
90° Torsion -5,50 -2,00
![Abb. 3 [27]: Begrenzungen der an der Anregung beteiligten Orbitale](https://thumb-eu.123doks.com/thumbv2/1library_info/4531469.1596278/17.892.325.625.397.836/abb-begrenzungen-anregung-beteiligten-orbitale.webp)
![Abb. 4 [27]: Struktur einer CI Matrix mit blockweiser Anordnung der Determinanten der einzelnen Anregungsarten und deren Wechselwirkung](https://thumb-eu.123doks.com/thumbv2/1library_info/4531469.1596278/18.892.240.617.450.862/struktur-matrix-blockweiser-anordnung-determinanten-einzelnen-anregungsarten-wechselwirkung.webp)
![Abb. 6 [A1]: Energien der des Grund](https://thumb-eu.123doks.com/thumbv2/1library_info/4531469.1596278/22.892.182.675.101.395/abb-a-energien-der-des-grund.webp)
![Abb. 7 [A2-4]: C-C Bindun](https://thumb-eu.123doks.com/thumbv2/1library_info/4531469.1596278/24.892.151.699.106.435/abb-a-c-c-bindun.webp)
![Abb. 9 [A6]: Potentialschnittflächen für die simultane Bindungslängenänderun Doppelbindung](https://thumb-eu.123doks.com/thumbv2/1library_info/4531469.1596278/26.892.152.700.106.427/abb-a-potentialschnittflächen-simultane-bindungslängenänderun-doppelbindung.webp)
![Abb. 10 [A7]: Potentialschnittflächen für die Bindungslängenänderung](https://thumb-eu.123doks.com/thumbv2/1library_info/4531469.1596278/27.892.206.741.104.731/abb-a-potentialschnittflächen-für-die-bindungslängenänderung.webp)
![Abb. 11 [A8]: Potentialschnittflächen für die symmetrische Bindungswinkeländerung der C1C2C3 und C2C3C4 Bindun](https://thumb-eu.123doks.com/thumbv2/1library_info/4531469.1596278/28.892.155.701.104.428/abb-a-potentialschnittflächen-symmetrische-bindungswinkeländerung-c-c-bindun.webp)
