Erlangung der Doktorwurde
Naturwissenschaftlich - Mathematischen Gesamtfakultatder Ruprecht - Karls - Universitatder
Heidelberg
vorgelegt von
Diplom-Physiker Guido Mieen aus Paderborn
Tag der mundlichen Prufung: 17.11.2000
Simulation der Koksverbrennung unter Verwendung
detaillierter Reaktionsmechanismen
Gutachter: Prof. Dr. Dr. h. c. Jurgen Warnatz Prof. Dr. Bernhard Schramm
Inhaltsverzeichnis
1 Einleitung 3
1.1 Eigenschaften von Kohle . . . 3
1.2 Kohleverbrennung . . . 3
1.3 Zielsetzung der Arbeit . . . 5
2 Grundlagen 7
2.1 Gasphase . . . 72.1.1 Erhaltungsgleichungen . . . 7
2.1.2 Zustandsgleichung . . . 8
2.1.3 Transportprozesse . . . 8
2.1.4 Thermochemie . . . 10
2.1.5 Reaktionskinetik . . . 12
2.2 Oberache . . . 13
2.2.1 Erhaltungsgleichungen an der Phasengrenze . . . 13
2.2.2 Thermochemie . . . 15
2.2.3 Reaktionskinetik . . . 15
2.3 Simulation und Numerik . . . 19
2.3.1 Staupunktstromung . . . 19
2.3.2 Ortsdiskretisierung und Losungsverfahren . . . 21
2.3.3 Sensitivitats{ und Reaktionsuanalyse . . . 21
2.3.4 Struktur des Programmpaketes . . . 21
3 Koksmodell 23
3.1 Graphit als Koksmodell . . . 233.2 Eigenschaften von Graphit . . . 23
3.3 Oberachenkomplexe . . . 24
3.3.1 Ubersicht . . . 24
3.3.2 Eigenschaften . . . 27
4 Heterogene Bildung von Kohlenmonoxid und -dioxid 29
4.1 Bildung von Kohlenmonoxid . . . 294.1.1 Reaktionsmechanismus . . . 29
4.1.2 Simulationsergebnisse . . . 32
4.1.3 Reaktionsu- und Sensitivitatsanalyse . . . 36
4.2 Bildung von Kohlendioxid . . . 40
4.2.1 Reaktionsmechanismus . . . 40
4.2.2 Simulationsergebnisse . . . 41
4.2.3 Reaktionsu- und Sensitivitatsanalyse . . . 43
5 Heterogene Bildung von Stickstooxiden 46
5.1 Oberachenkomplexe . . . 46 5.2 Reaktionsmechanismus . . . 47 5.3 Simulationsergebnisse . . . 49
6 Zusammenfassung und Ausblick 54
Literatur 57
1 Einleitung
1.1 Eigenschaften von Kohle
Kohle stellt seit Jahrtausenden eine wichtige Energiequelle fur die Menschheit dar. Das Ausgangsmaterial der Kohle besteht aus panzlichen Uberresten, die durch biochemische und geochemische Prozesse im Laufe von Jahrmillionen trans- formiert werden. Dieser Transformationsproze wird als Inkohlung bezeichnet.
Das Ausma der Inkohlung bestimmt den Grad, zu dem das Panzenmaterial die reine Graphitstruktur erreicht hat [1].
Kohle ist ein Gemisch aus den verschiedensten Bestandteilen, deren genaue chemische Struktur noch nicht vollstandig geklart ist. Es wird davon ausgegan- gen, da Kohle aus Gerusten von carbozyklischen und heterozyklischen Ringen besteht, die grotenteils aromatischen Charakter haben [2{6]. Die Ringe sind teil- weise durch aliphatische Ketten verbunden [1]. Die Hauptbestandteile von Kohle variieren je nach Kohleart wie folgt [7]:
Kohlensto 65-95%
Wassersto 2-7%
Sauersto25%
Sticksto 1-2%
Schwefel 10%
Asche (mineralische Bestandteile) 5-15%
Wasser 2-20%
Der Kohlenstoanteil ist um so hoher, je alter die Kohle ist. Braunkohle enthalt 65 bis 75%, Steinkohle 75 bis 90% und Anthrazit bis zu 95% Kohlensto [5].
1.2 Kohleverbrennung
Die Verbrennung von Kohle deckt zur Zeit mehr als ein Viertel der weltweiten Energieversorgung und erzeugt mehr als ein Drittel des weltweiten Stroms (s.
Abb. 1.1 und 1.2). Da Kohle von allen fossilen Energietragern die groten globalen Vorkommen besitzt, wird sie aller Voraussicht nach auch in naher und mittlerer Zukunft eine bedeutende Rolle bei der Energiegewinnung spielen [8].
Um eine hohere Ezienz der Kohleverbrennung und gleichzeitig eine Verrin- gerung der Schadstoemissionen zu erreichen, ist ein detailliertes Verstandnis der dabei auftretenden physikalischen und chemischen Prozesse notwendig.
Weltweit
Kohle (26 %)
Erdgas (24 %)
Atom (7 %)
Ö l (40 %) Sonst.
(3 %)
Deutschland
Kohle (25 %)
Erdgas (21 %)
Atom (12 %) Ö l
(40 %) Sonst.
(2 %)
Abbildung 1.1: Primarenergieversorgung (Stand: 1998 [9,10]).
Weltweit
Kohle (37 %)
Erdgas (16 %)
Atom (17 %) Ö l (9 %) Regenerative Energie (21%)
Deutschland
Kohle (54 %)
Erdgas (7 %)
Atom (33 %)
Sonst.
(2 %) Regenerative Energie (4 %)
Abbildung 1.2: Stromerzeugung (Stand: 1996/99 [9,10]).
Die Verbrennung von Kohle kann in drei globale Teilprozesse gegliedert werden [1,11]:
1. Pyrolyse. Die Pyrolyse ist ein thermischer Zersetzungsproze, der bei Tem- peraturen uber 573 K einsetzt und die Rohkohle in feste (Koks), ussige (Teer) und gasformige (Fluchtige) Bestandteile aufspaltet. Teere sind da- bei die Bestandteile, die bei Raumtemperatur ussig, wahrend der Pyro- lyse als Dampf vorliegen. Die Fluchtigen bestehen hauptsachlich aus CH4, H2 und CO . Der nach der Pyrolyse ubrigbleibende feste Koks hat einen erhohten Kohlenstogehalt, einen verringerten Sauersto- und Wassersto- gehalt, enthalt aber weiterhin kleine Mengen an Sticksto und Schwefel und den groten Teil der Asche.
2. Koksabbrand. Der verbleibende Koks reagiert an seiner inneren und aue-
ren Oberache mit Gasphasenmolekulen und wird dabei verbraucht. Dabei lassen sich unter anderem folgende globale heterogene Reaktionen unter- scheiden:
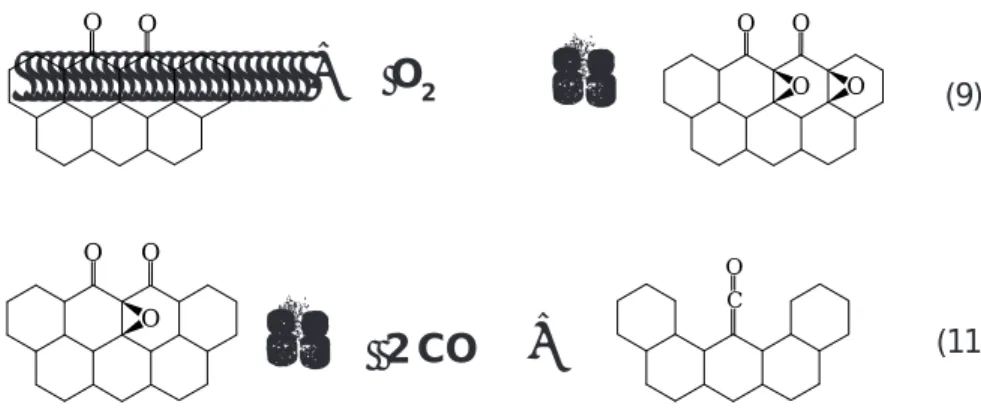
Cfest + O2 !CO2 Cfest + CO2 !2 CO 2 Cfest + O2 !2 CO
3. Abbrand der Fluchtigen. Die Fluchtigen verbrennen in der Gasphase.
Ausfuhrliche Darstellungen der globalen Prozesse bei der Kohleverbrennung n- den sich in der Literatur [12{22].
In Abb. 1.3 symbolisiert CxHyallgemeine Kohlenwasserstoe. Darunter fallen alle vom gasformigen Methan bis zu den Teerdampfen bei der Pyrolyse entga- ste Kohlenwasserstoe. In Abb. 1.3 sind die Pyrolyse und die Abbrandreaktio- nen formal als Folgereaktionen angegeben. Beim Kohleabbrand laufen aber alle drei Teilprozesse parallel ab. Weiterhin entstehen beim Kohle- und Koksabbrand Schadstoe wie z. B. Stickstooxide und Schwefeloxide.
Rohkohle Energie
Koks
CxHy O2
CO, CO2
O2
Asche
Pyrolyse
H2O
Abbildung 1.3: Schematisiertes Kohleabbrandmodell (nach Gorner [11]).
1.3 Zielsetzung der Arbeit
In dieser Arbeit wird ein physikalisch-chemisches Modell des Koksabbrandes auf- gestellt und die bei dem Abbrand auftretenden Vorgange mit Hilfe von Com- putersimulationen untersucht. Als Modell fur die Koksoberache wird eine Gra- phitoberache verwendet. Es werden die elementaren chemischen Prozesse (z. B.
Adsorption und Desorption) analysiert, die den Abbrand der Oberache bestim- men. Weiterhin werden die Reaktionen untersucht, die Stickstooxide (NO und N2O) aus dem im Koks enthaltenen Sticksto bilden.
Im Gegensatz zu anderen Arbeiten mit ahnlicher Zielsetzung [23{29] wird der Reaktionsmechanismus hier mit Hilfe von Elementarreaktionen formuliert. Daher konnen die Geschwindigkeitskoezienten der Oberachenreaktionen aus physika- lischen und chemischen Eigenschaften der miteinander reagierenden Spezies ab- geleitet werden, da diesen Koezienten eine physikalische Bedeutung zukommt.
Bei diesen Eigenschaften handelt es sich um die geometrischen Eigenschaften der Oberache, die Form der bei den Reaktionen gebildeten Oberachenkomplexe und die Wechselwirkung dieser Komplexe mit Gasphasenmolekulen. Um aus die- sen Informationen einen Reaktionsmechanismus aufzustellen, werden Methoden verwendet, die in der mikrokinetischen Analyse von heterogenen Katalysereak- tionen auf Metalloberachen mit Erfolg angewendet werden [30{32].
2 Grundlagen
2.1 Gasphase
2.1.1 Erhaltungsgleichungen
Reaktive Stromungen werden durch ein System von Erhaltunsgleichungen fur verschiedene physikalische Groen beschrieben. Im einzelnen handelt es sich um folgende Groen:
Gesamtmasse
Die Erhaltung der Gesamtmasse wird durch die Kontinuitats- gleichung beschrieben:@
@t + div(~v) = 0: (2.1)
Dabei stelltdie Massendichte, ~v die lokale Stromungsgeschwindigkeit und t die Zeit dar.
Speziesmasse
Fur die Teilchenmassen der einzelnen im Reaktionssystem vor- handenen Spezies gilt folgende Bilanzgleichung:@i
@t + div(i~v) + div(i~Vi) = _!iMi: (2.2) Durchi =Yi ist die Massendichte der Teilchensorte imit dem Massenbruch Yi gegeben. Die lokale Stromungsgeschwindigkeit ~vi setzt sich aus der Stromungs- geschwindigkeit des Massenschwerpunktes ~v und der Diusionsgeschwindigkeit
~Vi der Spezies i zusammen. Weiterhin tritt ein chemischer Quellterm _!iMi auf, der die Umwandlung von Spezies aufgrund chemischer Reaktionen beschreibt; _!i bezeichnet dabei die molare Bildungsgeschwindigkeit und Mi die molare Masse der Teilchensortei.
Impuls
Die Erhaltung des Impulses m~v mit der zugehorigen Impulsdichte ~v lat sich durch folgende Gleichung beschreiben:@(~v)
@t + div(~v ~v) + divp=~g: (2.3) Dabei stellt~v~v die Konvektion undpden Drucktensor dar. Mit~v~v wird das dyadisches Produkt zweier Vektoren bezeichnet. Der Drucktensor p lat sich in zwei Anteile zerlegen; einen fur den hydrostatischen Druckp und einen viskosen
Die Erhaltungsgleichungen werden hier nur kurz aufgefuhrt. Eine ausfuhrliche Darstellung ndet sich z.B. bei Deutschmann [33].
Anteil , der den Impulsu von einem Ort hoherer zu einem Ort niedriger Geschwindigkeit aufgrund der Zahigkeit des Fluids beschreibt:
p=pE+: (2.4)
Hierbei istE der Einheitstensor. Weiterhin verursacht die Gravitation eine Fern- wirkung, die durch ~g gegeben ist, mit~g als Fallbeschleunigung.
Energie
Die spezische Gesamtenergiee des Systems setzt sich zusammen aus spezischer innerer, kinetischer und potentieller Energie:e=u+ 12j~vj2+G; (2.5)
mitu als spezischer innerer Energie undGals Gravitationspotential. Strahlung verursacht einen Fernwirkungsterm sr. Es gilt die Erhaltungsgleichung fur die innere Energie:
@(u)
@t + div(u~v+~jq) +p: grad~v =sr; (2.6) wobei p : grad~v die doppelte Verjungung zweier Tensoren bedeutet. ~jq ist die Warmestromdichte.
Mit der Beziehung h = u+p kann Gleichung (2.6) in eine Gleichung fur die spezische Enthalpie h umgeformt werden:
@(h)
@t ,
@p
@t + div(~vh+~jq) +p: grad~v,div(p~v) =sr: (2.7)
2.1.2 Zustandsgleichung
Zur Schlieung der Erhaltungsgleichungen mussen die Zustandsvariablen Druck, Dichte und Temperatur verknupft werden. In dieser Arbeit konnen die Reaktions- systeme mit hinreichender Genauigkeit als ideale Gase betrachtet und die ideale Gasgleichung als Zustandsgleichung verwendet werden:
p= RM T: (2.8)
Dabei ist R die universelle Gaskonstante und M die mittlere molare Masse der Mischung ( M = 1=PNi=1g MYii mitNg als Anzahl verschiedener Spezies).
2.1.3 Transportprozesse
In den Erhaltungsgleichungen treten die Transportgroen~ji; und~jqauf, welche als Funktionen der abhangigen Variablen bekannt sein mussen, um das Glei- chungssystem zu schlieen. Diese Schlieung erfolgt mit phanomenologischen
Gleichungen (Ficksches Diusionsgesetz, Newtonsches Schubspannungsgesetz und Fouriersches Warmeleitungsgesetz), die empirisch abgeleitet werden. Die zugehori- gen Transportkoezienten werden mit Hilfe der kinetischen Gastheorie aus mo- lekularen Daten abgeleitet.
Diusion
Diusion von Masse aufgrund eines Konzentrationsgradienten be- zeichnet man als Ficksche Diusion. Massendiusion ist aber nicht nur an Kon- zentrationsgradienten gekoppelt. Auch Gradienten von Temperatur und Druck erzeugen einen Massendiusionsstrom, der im allgemeinen klein gegenuber der Fickschen Diusion ist. Druckdiusion spielt fur Systeme mit annahernd kon- stantem Druck, wie sie in dieser Arbeit betrachtet werden, keine Rolle und wird deshalb vernachlassigt. Die Diusionsstromdichte setzt sich somit aus zwei An- teilen zusammen:~ji =~jid+~jiT: (2.9) Ficksche Diusion ~jid und Thermodiusion ~jiT lassen sich mit der kinetischen Theorie verdunnter Gase schreiben als [34,35]:
~jid= Mi M2
X
j6=i
DijPMjgradXj; (2.10)
~jiT =,DiT
T gradT: (2.11)
Dabei sind Xj = MYi=Mj der Molenbruch von Spezies j und DiT der Thermo- diusionskoezient von Spezies i. Die polynaren DiusionskoezientenDijP sind von den Konzentrationen der einzelnen Spezies abhangig, und die Berechnung aus den binaren DiusionskoezientenDijist numerisch aufwendig. Deshalb wird ei- ne Naherungsformel zur Berechnung von~jid verwendet [35]:
~jid = YXiiDiMgradXi; (2.12) mit den eektiven Diusionskoezienten DiM von Spezies i in der Gasmischung.
Diese lassen sich aus der Zusammensetzung und den binaren Diusionskoezi- enten berechnen [35]:
DiM= 1,Yi
P
j6=iXj=Dij: (2.13)
Man mu bei dieser Naherung beachten, da sich die Diusionsstrome nicht mehr notwendigerweise zu Null addieren. Dies wird mit einem Korrekturterm~jcorr =
, P
i~ji kompensiert.
Zur Berechnung der binaren Diusionskoezienten Dij wird die Theorie ver- dunnter Gase von Chapman und Enskog [34,36] herangezogen. Mit ihr lassen sich
die Transportkoezienten in Abhangigkeit von intermolekularen Potentialen(r) berechnen. Als Naherung von (r) werden Lennard-Jones-(6-12)-Potentiale oder Born-Maier-Potentiale verwendet.
Viskositat
Geschwindigkeitsgradienten in einem Gas verursachen Impulsstrom- dichten, die zu diesen proportional sind (Newtonsches Schubspannungsgesetz).Der viskose Drucktensor beschreibt diesen Impultransport und lat sich mit der kinetischen Gastheorie schreiben als [34,37]
=,
(grad~v) + (grad~v)T, 2
3(div~v)E
; (2.14)
wobeidie mittlere dynamische Viskositat der Mischung bezeichnet. Die Visko- sitatskoezienten i der einzelnen Spezies werden wiederum mit der Chapman- Enskog-Theorie aus den intermolekularen Potentialen berechnet. Der mittlere Viskositatskoezient der Gasmischung ergibt sich dann aus der empirischen Naherung
= 12
2
4 X
i
Xii+ X
i
Xi i
!
,1 3
5: (2.15)
Energietransport
Unter Vernachlassigung des Dufour-Eektes setzt sich die Warmestromdichte~jq aus zwei Anteilen zusammen:~jq =,gradT +X
i
hi~ji; (2.16)
wobei der Term (,gradT) die Fouriersche Warmeleitung beschreibt, wahrend der Term (Pihi~ji) durch Diusion von Teilchen unterschiedlicher Enthalpie her- vorgerufen wird. Die Warmeleitfahigkeit eines Gasgemisches wird analog zur Vis- kositat mit der empirischen Formel
= 12
2
4 X
i
Xii+ X
i
Xi i
!
,1 3
5 (2.17)
bestimmt. Die Warmeleitfahigkeitskoezienten i der einzelnen Spezies werden wie die binaren Diusionskoezienten und die Viskositatskoezienten mit der Chapman-Enskog-Theorie aus den intermolekularen Potentialen berechnet.
2.1.4 Thermochemie
Fur ideale Gase ist die spezische Enthalpieh und die spezische Entropies der Gasmischung durch
h=XhiYi; s=XsiYi (2.18)
gegeben. Die Anderungen der spezischen Enthalpiehi bzw. der spezischen Ent- halpiesi des Stoes i beschreiben die totalen Dierentiale:
dhi =
@hi
@T
p
dT+
@hi
@p
T
dp; (2.19)
dsi =
@si
@T
p
dT +
@si
@p
T
dp: (2.20)
Fur ideale Gase ist die spezische Enthalpie und damit auch die spezische Warmekapazitat bei konstantem Druck cp;i unabhangig vom Druck. Es gelten die thermodynamischen Beziehungen
@hi
@T
p
=cp;i;
@hi
@p
T
= 0;
@si
@T
p
= cp;i T ;
@si
@p
T
=, 1
iT : (2.21) Sind die Werte der spezischen Enthalpie und Entropie bei den Standardbedin- gungen (T0 = 298:15 K, p0 = 1 atm) bekannt, dann lassen sich Absolutwerte dieser Groen berechnen:
hi(T) =h0i;T0 +
T
Z
T 0
cp;i(T0)dT0; (2.22) si(T;pi) =s0i;T0 +
T
Z
T 0
cp;i(T0)
T0 dT0, R Mi ln
pi p0
: (2.23)
Dabei bezeichnet pi den Partialdruck der Spezies i im Gasgemisch. Zur nume- rischen Berechnung der thermodynamischen Groen werden experimentelle Da- ten aus den JANAF-Tabellen [38] oder abgeschatzte Werte [39] durch Polyno- mansatze genahert. Man mu beachten, da die folgenden Polynomansatze nicht fur die spezischen, sondern fur die entsprechenden molaren GroenCp;i, Hi, Si gelten:
Cp;i(T) =R,a1+a2T +a3T2+a4T4+a5T5 (2.24) Hi(T) =RT a1 +a2
2T +a3
3T2+ a4
4 T3+ a5
5T4+a6 T
(2.25)
Si0(T) =Ra1ln(T) +a2T +a3
2T2+a4
3T3+ a5
4 T4+a7: (2.26) Die Koezienten a1 bis a5, die Standardbildungsenthalpie Hi;T0 0 und die Stan- dardentropie Si;T0 0 bestimmen die zwei restlichen Koezienten a6 und a7:
a6 = Hi;T0
R ,a1T0,a2
2 T02, a3
3T03, a4
4T04, a5
5 T05 (2.27) a7 = Si;T0
R ,a1ln(T0),a2T0, a3
2T02, a4
3 T03,a5
4 T04: (2.28)
Groere Temperaturbereiche werden in zwei Temperaturintervalle mit jeweils un- terschiedlichen Koezienten eingeteilt, so da man 14 thermodynamische Koef- zienten pro Spezies benotigt.
2.1.5 Reaktionskinetik
Ein Reaktionsmechanismus in der Gasphase bestehend aus Elementarreaktionen lat sich in allgemeiner Form schreiben als
N
g
X
i=1
il0i ! XNg
i=1
il00i (l = 1;::: ;Kg); (2.29) mit den stochiometrischen Koezienten il0 und il0 0 des Stoes i in der Reaktion l, den Teilchensymbolen i undKg als Gesamtzahl der Elementarreaktionen. Die Bildungsgeschwindigkeit _!i der Spezies i ergibt sich dann zu
!_i =
K
g
X
l =1
ilkfl
N
g
Y
j=1
[j]jl0 (2.30)
mitil =il00,il0 und [i] als Konzentration der Spezies i. Die Geschwindigkeits- koezienten kfl sind temperaturabhangig und konnen durch ein modiziertes Arrheniusgesetz beschrieben werden [40]:
kfl =AlTlexp
,
Eal RT
: (2.31)
Dabei bezeichnetAlden praexponentiellen Faktor,lden Temperaturexponenten und Eal die Aktivierungsenergie der Reaktion l.
Aufgrund der mikroskopischen Reversibilitat existiert zu jeder Elementarre- aktion eine Ruckreaktion, deren Geschwindigkeitskoezientkrlsich aus dem Ge- schwindigkeitskoezienten kfl der Hinreaktion und aus der Gleichgewichtskon- stanten Kcl berechnen lat:
krl(T) = kfl
Kcl: (2.32)
Die Gleichgewichtskonstante Kcl wird bestimmt aus der molaren freien Reak- tionenthalpie RG0l bei einem Druck von p0 = 1 atm:
Kcl =
p0 RT
PN
g
i=1
ilexp
,
RG0l RT
: (2.33)
2.2 Oberache
2.2.1 Erhaltungsgleichungen an der Phasengrenze
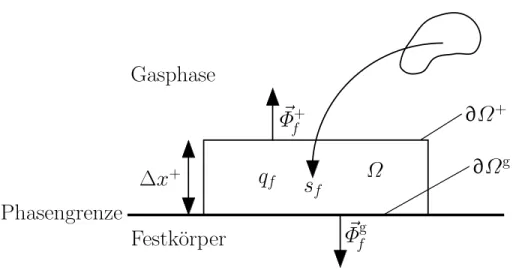
Die Untersuchung heterogener Systeme erfordert die Kopplung der Gasphase mit der reaktiven Oberache. Diese Kopplung lat sich mit Erhaltungsgleichungen beschreiben, die analog zur Gasphase hergeleitet werden konnen [33]. Eine allge- meine Form dieser Erhaltungsgleichungen ergibt sich, wenn man ein kleines, an der Festkorperoberache anliegendes Volumenelement betrachtet. Der Rand vonwird in zwei Teile zerlegt: die Phasengrenze Gas-Festkorper @g des Volu- menelements und den Rand @+ von bezuglich der Gasphase (Abb. 2.1). Die allgemeine Bilanzgleichung fur lautet dann:
Festk¨orper Gasphase
Phasengrenze
∆x+ Ω
∂Ω+
∂Ωg Φ~f+
Φ~fg qf sf
Abbildung 2.1: Anderung der extensiven GroeF an der Phasengrenze.
@F
@t =
Z
@f
@t dV =,
Z
@ +
~f+~ndA, Z
@ g
~fg~ndA+
Z
qfdV +
Z
sfdV : (2.34) Dabei istF eine beliebige extensive Groe, f die zugehorige Dichte, ~f+ der Flu durch die Oberache @+, ~fg der Flu durch die Phasengrenze @g, qf die
Anderung durch Produktion undsf die Fernwirkung.
Massenstrome an der Phasengrenze
Die Bilanz fur die Teilchenmassen der Gasphasenspezies im Volumenelement ergibt sich aus Gleichung (2.34):Z
@Yi
@t dV =,
Z
@ +
(~ji+~uYi)~ndA+
Z
@ g
s_iMidA+
Z
!_iMidV : (2.35) Der Term (~ji +Yi~u) bezeichnet die Diusions- und Konvektionsstrome in der Gasphase, _siMi~n ist der Teilchenu an der Phasengrenze aufgrund von Adsorp- tion und Desorption und _!iMi ist der chemische Quellterm aufgrund von Gas- phasenreaktionen. Verhalt sich das System an der Phasengrenze stationar und fuhrt man in (2.35) den Grenzubergang x+ !0 durch, so gelangt man zu
(~ji+~uYi)~n= _siMi: (2.36) Dieser Grenzubergang lat sich auch fur instationare Prozesse durchfuhren, aller- dings nur wenn man voraussetzt, da Diusionsprozesse einerseits und Adsorpti- ons- und Desorptionsprozesse andererseits gleiche Zeitskalen besitzen. Summiert man Gleichung (2.36) uber alleNg Gasphasenspezies so erhalt man
~n~u= 1
N
g
X
i=1
s_iMi: (2.37)
Diese Gleichung sagt aus, da die Konvektionsgeschwindigkeit an festen Wan- den nicht notwendigerweise verschwindet. Tritt aufgrund von Adsorption oder Desorption ein Netto-Massenstrom an der Oberache auf, so induziert dies eine Stromungsgeschwindigkeit ~u normal zur Oberache, die sogenannte Stefan-Ge- schwindigkeit.
Oberachenbedeckung
Die maximale Anzahl der zur Adsorption zur Verfu- gung stehenden Platze pro Flache ist durch die Oberachenplatzdichte , mit der Einheit [molm,2] gegeben. Durch Adsorption konnen die Oberachenplatze belegt werden und es entstehen sogenannte Oberachenspezies bzw. Oberachen- komplexe. Allen Oberachenspezies, wobei auch freie Platze als Oberachenspezi- es deniert werden, lat sich ein Bedeckungsgradizuordnen, der angibt, welcher Anteil der Oberache mit der Oberachenspeziesibedeckt ist. Oensichtlich mu dann immer die BedingungNs
X
i=1
i = 1 (2.38)
erfullt sein, mit Ns als Anzahl der Oberachenspezies. Durch die Bedeckungen i ist der chemische Zustand der reaktiven Oberache deniert. In dieser Arbeit wird die Oberache nulldimensional modelliert und die Bedeckungsgrade stellen Mittelwerte uber die gesamte Oberache dar (mean eld approximation). Dies
bedeutet, da geometrische Strukturen auf der Oberache nur indirekt modelliert werden konnen, indem zusatzliche Oberachenspezies deniert werden, die die Eigenschaften der entsprechenden Strukturen beschreiben.
Die zeitliche Anderung der Bedeckungsgrade ist durch
@i
@t = _sii
, (2.39)
gegeben. Hierbei ist _sidie molare Bildungsgeschwindigkeit der Oberachenspezies i und i bezeichnet die Anzahl der Oberachenplatze, die die Spezies i belegt.
Dadurch wird berucksichtigt, da eine Spezies auf der Oberache mehr als einen Platz belegen kann.
2.2.2 Thermochemie
Fur die thermodynamischen Groen der Oberachenspezies wird ein analoger Formalismus wie fur die Gasphasenspezies angewendet. Allerdings liegen die ther- modynamischen Eigenschaften von Adsorbaten nicht in Tabellenwerken vor. Die Temperaturabhangigkeit dieser Eigenschaften lat sich mit Hilfe der statistischen Physik aus der Zustandsumme berechnen [39]. Dazu mussen insbesonders die Schwingungsfrequenzen der betrachteten Spezies bekannt sein, was in der Regel nicht der Fall ist. Testrechnungen mit abgeschatzten Frequenzen haben keine be- friedigen Ergebnisse erbracht. Deshalb werden fur den Oberachenmechanismus in der vorliegenden Arbeit die Geschwindigkeitskoezienten der Ruckreaktionen nicht uber die Gleichgewichtskonstanten bestimmt, sondern es werden jeweils die Ruckreaktionen explizit angegeben.
Die Bildungsenthalpie Hfs einer Oberachenspezies lat sich bei Kenntnis der Adsorptionsenthalpie Hads und der Bildungsenthalpie Hfg der Spezies in der Gasphase bestimmen aus [41]
Hfg,Hfs = Hads: (2.40)
2.2.3 Reaktionskinetik
Geschwindigkeitsgesetze
Die Reaktionskinetik auf der Oberache wird ana- log zur Gasphase durch Elementarreaktionen beschrieben. Man mu allerdings beachten, da die chemischen Symbolei in Gleichung (2.29) fur Gasphasenspe- zies und Oberachenspezies stehen. Oberachenspezies sind sowohl adsorbierte Spezies aus der Gasphase, als auch unbedeckte Oberachenplatze. Oberachen- spezies konnen einen oder mehrere Oberachenplatze belegen. Es wird angenom- men, da die Oberachenplatzdichte, konstant ist; dann mussen in jeder der Ks Oberachenreaktionen die Anzahl der Platze konstant bleiben:N
g +N
s
X
i=N +1
ili = 0 (l= 1;::: ;Ks): (2.41)
Analog zu Gleichung (2.30) ist die Bildungsgeschwindigkeit _si von Speziesidurch Oberachenreaktionen gegeben durch
s_i =XKs
l =1
ilkflNg+NsY
j=1
[j]jl0 (i= 1;::: ;Ng+Ns): (2.42) Die Konzentrationen der Gasphasenspezies sind z.B. in [molm,3] gegeben, die der Oberachenspezies z.B. in [molm,2].
Die Reaktionsgeschwindigkeit einer Oberachenreaktion hangt nicht nur von der Temperatur, sondern auch von der Bedeckung der Oberache ab. Grund hierfur sind anziehende und abstoende Wechselwirkungen zwischen den Ober-
achenspezies. Der entsprechende Geschwindigkeitskoezient kfl wird deshalb durch einen modizierten Arrheniusansatz beschrieben:
kfl=AlTlexp
,
Eal RT
fl(1;::: ;Ns): (2.43) Dabei wird die Bedeckungsabhangigkeit durch die Funktion fl(1;::: ;Ns) be- schrieben. In dieser Arbeit wird folgende funktionale Form gewahlt [33]:
fl=YNs
i=1
exp
"ili RT
: (2.44)
"il ist der Wert, um den sich die Aktivierungsenergie Eal bei vollstandiger Be- deckung mit Speziesi andert.
Arrhenius-Parameter
Die Arrhenius-Parameter der meisten Oberachenre- aktionen sind noch nicht experimentell bestimmt. Deshalb mussen Abschatzun- gen fur diese Werte durchgefuhrt werden. Diese Abschatzungen werden mit Me- thoden der mikrokinetischen Analyse von heterogenen Katalysereaktionen durch- gefuhrt.Der praexponentielle Faktor lat sich mit Hilfe der Theorie des Ubergangszu- stands zumindest groenordnungsmaig abschatzen [30,42]. Dazu werden physi- kalisch sinnvolle Annahmen uber die moglichen Freiheitsgrade des im Ubergangs- zustand vorliegenden aktivierten Komplexes der betrachteten Reaktion und der miteinander reagierenden Spezies gemacht. Hiermit ergibt sich fur jede Art der Elementarreaktion (z. B. Adsorption, Desorption etc.) ein bestimmter Groen- ordnungsbereich, in dem der jeweilige praexponentielle Faktor liegen kann (siehe Tabelle 2.1).
Um zu einer Abschatzung der Aktivierungsenergie zu gelangen, mu erst eine Abschatzung der jeweiligen Reaktionsenthalpie durchgefuhrt werden. Die Enthal- pie einer Adsorptionsreaktion ist die Netto-Anderung aller Bindungsenergien, die
Reaktion nach Zhdanov [42] nach Dumesic [30]
Molekulare Adsorption 6109 ::: 61013 8:3107T ::: 8:3109T Dissoziative Adsorption 2:21015 ::: 2:21022 3:11016T ::: 3:11018T Langmuir-Hinshelwood 61019 ::: 61027 3:71016 ::: 3:71021 Eley-Rideal 6109 ::: 61013 8:3107T ::: 8:3109T Molekulare Desorption 1013 ::: 1019 1013 ::: 1016
Assoziative Desorption 61019 ::: 61027 3:71016 ::: 3:71024
Tabelle 2.1: Groenordnungsintervalle der praexponentiellen Faktoren fur ver- schiedenen Oberachenreaktionen. Einheiten sind (cm, mol, s, K).
an der betrachteten Reaktion beteiligt sind. Fur die atomare Adsorption eines Atoms A auf einer Oberache mit Atomen Si gilt dann [31]:
Hads =XD(Si,A) +XD(Si,Sj) (2.45) Summiert wird jeweils uber alle Oberachenatome.D(Si,A) gibt die Bindungs- energie zwischen A und den Oberachenatomen an, D(Si,Sj) ist die Anderung der Bindungen der Oberachenatome untereinander. Geht man von der haug gemachten Annahme aus [31], da sich letztere nicht andert (frozen lattice ap- proximation) und nimmt man weiter an, da A nur mit einem Oberachenatom S eine Bindung eingeht, vereinfacht sich Gl. (2.45) zu:
Hads =D(S,A) (2.46)
Wenn ein zweiatomiges Molekul AB adsorbiert, wird die Adsorptionsenthalpie durch die Bildung der S{A und der S{B Bindungen sowie die Schwachung der A{B Bindung bestimmt [31]:
Hads =D(S(A,B)),D(A,B) +
X[D(Si,A) +D(Si,B)] +XD(Si,Sj) (2.47) Summiert wird wieder uber alle Oberachenatome. Die ersten beiden Terme in Gl.
(2.47) stellen die Dierenz zwischen der A{B Bindungsenergie im adsorbierten Molekul, D(S(A,B)), und der Bindungsenergie im freien Gasphasenmolekul, D(A,B), dar. Im dritten Term sind die Bindungsenergien der Molekule A und B mit den einzelnen Oberachenatomen zusammengefat. Geht man davon aus, da A und B jeweils nur mit einem Oberachenatom eine Bindung eingehen und wendet man weiterhin die frozen lattice approximation an, vereinfacht sich Gl.
(2.47) zu:
Hads =D(S(A,B)),D(A,B) +D(SA,A) +D(SB,B) (2.48)
Diese und die folgenden Gleichungen geben den Betrag der Adsorptionsenthalpie an.
Mit den so bestimmten Adsorptionsenthalpien und den tabellierten Bildungs- enthalpien der entsprechenden Gasphasenspezies kann man aus Gleichung (2.40) die Bildungsenthalpien der Oberachenspezies berechnen. Damit ergeben sich fur Oberachenreaktionen die Reaktionsenthalpien Hr nach der Formel
Hr =Ng+NsX
l =1
l(Hf)l: (2.49)
Hieraus wiederum bekommt man mit Hilfe des Postulats von Hammond [43,44] ei- ne grobe Abschatzung fur die Aktivierungsenergien der Hin- und Ruckreaktionen (Eaf und Ear). Dieses Postulat besagt, da der aktivierte Komplex einer exother- men Reaktion stark den Reaktanden gleicht, wahrend der aktivierte Komplex einer endothermen Reaktion mehr den Produkten gleicht. Aus der Beziehung
Hr =Eaf ,Ear (2.50)
ergibt sich dann, da bei exothermen Reaktionen die Aktivierungsenergie fur die Ruckreaktion deutlich hoher ist als die der Hinreaktion, wahrend bei endothermen Reaktionen die Aktivierungsenergie der Ruckreaktion deutlich geringer ist als diejenige der Hinreaktion.
Haftkoezienten
Bei Adsorptionsprozessen kann man anstatt der Geschwin- digkeitskoezienten kfl auch sogenannte Haftkoezienten Sl verwenden. Der Haftkoezient gibt die Wahrscheinlichkeit (0 Sl 1) an, mit der ein Teil- chen l, das mit der Oberache kollidiert, adsorbiert wird. Haftkoezienten sind im allgemeinen temperatur- und bedeckungsabhangig. Die Bedeckungsabhangig- keit von Sl ist durchSl=Sl0gl(1;::: ;Ns) (2.51) gegeben. Der AnfangshaftkoezientSl0 ist die Adsorptionswahrscheinlichkeit bei vollig unbedeckter Oberache. Die Funktiongl(1;::: ;Ns) spiegelt die Bedek- kungsabhangigkeit wieder. Bei sogenanntem Langmuirschen Verhalten [45] hat sie folgende Form:
gl(1;::: ;Ns) = (frei)l; (2.52) wobei frei die Bedeckung der Oberache mit freien Platzen darstellt und l deniert ist durch
l =
N
g +N
s
X
i=N
g +1
il0: (2.53)
Fur Oberachenreaktionen ist die Aktivierungsenergie gleich der Aktivierungsenthalpie [45].
Das heit, bei molekularer Adsorption ist l = 1 und bei dissoziativer Adsorp- tion gilt l = 2. Man kann die Haftkoezienten entsprechend der kinetischen Gastheorie in Geschwindigkeitskoezienten transformieren:
kflads = Sl0
1, S20l(frei)l 1 (,)l
r RT
2Ml: (2.54)
Diese Formel gilt nur bei Langmuirschen Verhalten. Der Term 1, S2l0(frei)l ist eine Korrektur aufgrund der nicht-Maxwellschen Geschwindigkeitsverteilung nahe der Oberache [46].
2.3 Simulation und Numerik
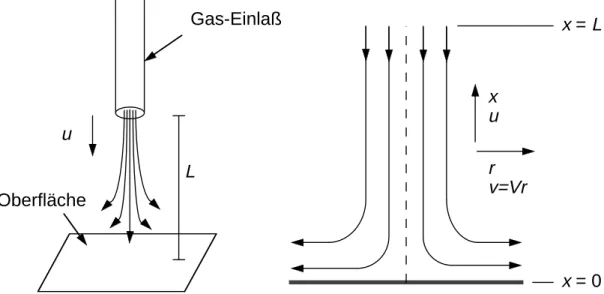
2.3.1 Staupunktstromung
Grundlage der numerischen Simulation in dieser Arbeit bildet die Staupunkt- stromung auf eine chemisch reaktive Platte (Abb. 2.2). Das dazugehorige Pro- grammpaket wurde ursprunglich von Behrendt [47] zur Simulation laminarer Ge- genstromdiusionsammen entwickelt und von Behrendt und Deutschmann [33]
fur die vorliegende Konguration erweitert.
u
L
Gas-Einlaß
x u r v=Vr
x = 0 x = L
Oberfläche
Abbildung 2.2: Staupunktstromung auf eine chemisch reaktive Platte.
Falls der Durchmesser der Platte und des Gaseinlasses gro gegenuber dem Ab- stand L zwischen Platte und Gaseinla sind, so kann man das zweidimensiona- le Problem auf ein eindimensionales zuruckfuhren [48]. Die Gleichungen (2.55){
(2.58) sind die entsprechenden Bilanzgleichungen fur Masse:
@
@t =,@(u)
@x ,2V; (2.55)
radialen Impuls:
@V
@t = @@x
@V
@x
,u@V
@x ,V2,; (2.56)
Spezies:
@Yi
@t =,
@ji
@x ,u@Yi
@x +Mi!_i (i= 1;::: ;Ng) (2.57) und Energie:
cp@T
@t =
@
@x
@T
@x
,ucp@T
@x ,
N
g
X
i=1
cpiji@T
@x ,
N
g
X
i=1
!_iMihi: (2.58) Das partielle Dierentialgleichungssystem wird durch die Gleichungen fur den diussiven Massenu:
ji =,DMi Yi Xi@Xi
@x ,DTi T @T
@x; (2.59)
fur den radialen Druckgradienten:
= 1r@p
@r; @
@x = 0 (2.60)
und die ideale Gasgleichung:
p=
M RT (2.61)
geschlossen. Die zeitliche Entwicklung der Oberachenbedeckungen ist durch
@i
@t = _sii
, (i=Ng+ 1;::: ;Ng +Ns) (2.62) gegeben. Der Abstand zur Platte x und die Zeit t bilden die unabhangigen Va- riablen im obigen Gleichungssystem. Die abhangigen Variablen sind die axia- le Massenstromdichte u, die skalierte radiale Geschwindigkeit V = v=r, die TemperaturT, die Massenbruche Yi, der radiale Druckgradient und die Ober-
achenbedeckungen i. Der thermodynamische Druck pwird raumlich konstant angenommen, allerdings erfordert die Impulserhaltungsgleichung einen kleinen radialen Druckgradienten . Die Annahme eines annahernd konstanten thermo- dynamischen Drucksp gilt also fur 12r2p.
2.3.2 Ortsdiskretisierung und Losungsverfahren
Die Ortsdiskretisierung erfolgt mit der Methode der niten Dierenzen. In das Integrationsintervall [0;L] werden eine Anzahl von ng Stutzstellen gelegt. Die Losung des partiellen Dierentialgleichungssystems wird durch die Losung des diskreten Problems genahert. Die in den Gleichungen enthaltenen Ortsableitun- gen werden durch eine Dierenzenapproximation ersetzt. Nach der Diskretisie- rung ergibt sich ein System aus ng(Ng + 4) +Ns gewohnlichen Dierentialglei- chungen und algebraischen Gleichungen.
In dem verwendeten Programmpaket ist eine nicht aquidistante, statische Git- teranpassung implementiert [47]. Bei Bedarf, d.h. bei groen bzw. kleinen Gra- dienten und Krummungen der abhangigen Variablen, wird das alte Gitter durch Hinzufugen bzw. Entfernen von Gitterpunkten modiziert. Die Losung des letz- ten Zeitschrittes wird auf das modizierte Gitter interpoliert und die Integration neu gestartet.
Die numerische Losung des nach der Diskretisierung erhaltenen Systems aus gewohnlichen Dierentialgleichungen und algebraischen Gleichungen erfolgt mit einem semi-impliziten Extrapolationsverfahren, das von Deuhardt, Hairer, No- wak und Zugk entwickelt und im Programmpaket LIMEX realisiert wurde [49,50].
Eine ausfuhrliche Darstellung des Losungsverfahrens ndet sich in [33].
2.3.3 Sensitivitats{ und Reaktionsuanalyse
Sensitivitatsanalysen identizieren die geschwindigkeitsbestimmenden Reaktions- schritte eines Mechanismus. Dazu wird untersucht, wie sich die Konzentrationen der betrachteten Spezies andern, wenn die Geschwindigkeitskoezienten der che- mischen Reaktionen variiert werden [40].
Reaktionsuanalysen ermitteln die charakteristischen Reaktionspfade eines Mechanismus. Hierbei wird untersucht, welcher Prozentsatz eines Stoes in einer bestimmten Reaktion verbraucht oder gebildet wird [40]. Damit lat sich fest- stellen, welche Elementarreaktionen besonderen Einu auf globale Groen wie z. B. die Produktzusammensetzung haben. Fur die vorliegenden Arbeit wurde ein existierendes Programm zur Reaktionsuanalyse von Gasphasenreaktionen auf Oberachenreaktionen erweitert.
2.3.4 Struktur des Programmpaketes
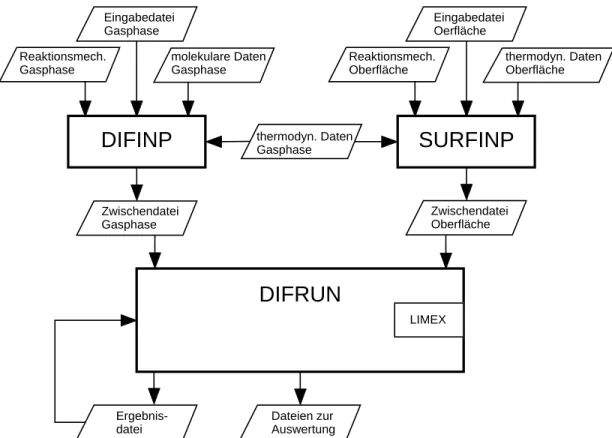
Die zur Simulation benotigten Eingabedateien und die Verbindungen zwischen den einzelnen Programmteilen sind in Abbildung 2.3 schematisch wiedergegeben.
Im Programmteil DIFINP werden die physikalischen Bedingungen, der Reakti- onsmechanismus in der Gasphase und die thermodynamischen bzw. molekularen Daten fur die Gasphasenspezies eingelesen. Die Daten werden auf Vollstandigkeit und Konsistenz uberpruft. Auerdem berechnet DIFINP die Geschwindigkeits- koezienten der Ruckreaktionen und Polynomts fur die Transportkoezienten.
Eingabedatei Gasphase
Eingabedatei Oerfläche Reaktionsmech.
Gasphase
molekulare Daten Gasphase
Reaktionsmech.
Oberfläche
thermodyn. Daten Oberfläche
Zwischendatei Gasphase
Zwischendatei Oberfläche
DIFINP thermodyn. Daten SURFINP
Gasphase
DIFRUN
LIMEX
Dateien zur Auswertung Ergebnis-
datei
Abbildung 2.3: Struktur des Programmpakets.
Schlielich wird eine Zwischendatei ausgegeben, die direkt als Eingabe fur das eigentliche Simulationsprogramm DIFRUN dient. Der Programmteil SURFINP ist ahnlich aufgebaut wie DIFINP. Es werden die physikalischen Bedingungen der Oberache, der Reaktionsmechanismus auf der Oberache und die thermo- dynamischen Daten fur die Oberachenspezies eingelesen. Zum Schlu wird eine Zwischendatei angelegt, die als Eingabe fur DIFRUN verwendet wird.
In DIFRUN werden die Zwischendateien eingelesen, die Anfangs- und Rand- bedingungen festgelegt und die Erhaltungsgleichungen numerisch gelost. In die- sem Programmteil ist das Programmpaket LIMEX implementiert. Die Ergebnisse werden in Dateien geschrieben, die auch als Anfangsbedingungen fur einen erneu- ten Programmstart dienen konnen.
3 Koksmodell
3.1 Graphit als Koksmodell
Koks besteht uberwiegend aus Kohlensto, der in Form von Graphitkristalliten vorliegt. Diese Kristallite sind zufallig in Form und Groe verteilt[52]. Weiterhin enthalten Koksteilchen eine Reihe von geometrischen Defekten (Fehlstellen) und Verunreinigungen (Sauersto, Wassersto, Sticksto, Schwefel und verschiedene Metalle). Die Oxidation der Kokskritallite ist also prinzipiell bestimmt durch ih- re graphitische Struktur, allerdings modiziert durch die Eekte der Fehlstellen und Verunreinigungen [52]. Der Einu dieser Eekte ist zwar von groer prak- tischer Bedeutung, tragt aber nicht zum mikrokinetischen Verstandnis der Re- aktion zwischen der Koksoberache und den Gasphasenmolekulen bei [53]. Als erstes einfaches Koksmodell wird deshalb in dieser Arbeit eine ideale Graphit- oberache verwendet, d. h. der Einu der Fehlstellen und Verunreinigungen wird vernachlassigt.
In der Literatur nden sich experimentelle und theoretische Hinweise, da Aussagen uber Graphit auch auf ungeordnetere Formen von festem Kohlensto
ubertragen werden konnen:
Messungen von Kelemen und Freund [54] haben ergeben, da Graphit- oberachen und Oberachen von weniger geordnetem Kohlensto (glassy carbon) ein ahnliches Chemisorptionsverhalten zeigen. Die Autoren folgern daraus, da diese beiden physikalisch unterschiedlichen Oberachen auf mi- kroskopischer Ebene chemische Ahnlichkeiten aufweisen.
Stein et al. [55, 56] haben mit Hilfe von theoretischen Untersuchungen (Huckelsche Molekulorbitaltheorie) entdeckt, da die Reaktivitat eines ge- gebenen Platzes auf einer Kohlenstooberache hauptsachlich von den be- nachbarten chemischen Strukturen abhangt. Sie folgern, da es eine enge Verbindung zwischen dem chemischen Verhalten von Graphit und weniger geordneten Kohlenstoen gibt.
Somit stellt das in dieser Arbeit verwendete einfache Koksmodell keine Uberver- einfachung dar. Auerdem lat es sich dahingehend erweitern, da auch Verun- reinigungen eingebaut werden. In Kapitel 5 wird hiervon Gebrauch gemacht.
3.2 Eigenschaften von Graphit
Das Grundelement der Graphitstruktur ist der planare Kohlensto-Sechsring mit sechs sp2-hybridisierten Atomen. Durch allseitige Kondensation entstehen Schich- ten, die als Graphene bezeichnet werden (siehe Abbildung 3.1). Zwischen diesen
Generell gilt fur die meisten Arten von ungeordneten Kohlenstoen, da sie sich aus der Graphitstruktur ableiten lassen [51].
0.142 nm Abstand 0.335 nm
Abstand
Abbildung 3.1: Struktur von Graphit (nach [58]).
Schichten sind nur schwache van der Waals-Krafte wirksam. Ein Graphitkri- stall stellt somit eine schichtformige Aufeinanderfolgen acher Riesenmolekule dar [5,57].
Die Begrenzungen der Graphen-Schichten ergeben unterschiedliche Muster in unterschiedlichen Richtungen. Entlang der niedrigst indizierten Flachen (Miller- Index (100)) tritt ein Zickzackmuster auf, entlang der Flachendiagonale (Miller- Index (110)) tritt ein als Sessel bezeichnetes Muster auf [57]. Diese Randachen werden allgemein Prismenachen genannt. Die Flache mit dem Index (001) wird als Basisache bezeichnet (siehe Abbildung 3.2).
3.3 Oberachenkomplexe
3.3.1 Ubersicht
Die Randatome der Graphen-Schichten, die die Prismenachen bilden, sind ko- ordinativ ungesattigt. Im Ultrahochvakuum liegen an solchen Stellen ungepaar- te Elektronen vor. Diese Oberachenplatze sind hochreaktiv und reagieren mit fast jedem angebotenen Atom oder Molekul unter Bildung einer Kohlensto- Heteroatom-Bindung. Wassersto, Sauersto und Stickstoverbindungen sind die wesentlichen Reaktanten fur den Kohlensto. Da haug mehrere Hetero- atome oder Atome der Kohlenstooberache an der Bildung der Kohlensto- Heteroatom-Verbindungen beteiligt sind, werden diese allgemein unter dem Be- gri Oberachenkomplexe zusammengefat [57].
Auf der Graphitoberache bilden sich wahrend der Verbrennung eine Reihe
Basis- fläche
Prismenfläche (Zick-Zack)
Prismenfläche (Sessel)
Abbildung 3.2: Schematische Darstellung der Basisache (nach [59]).
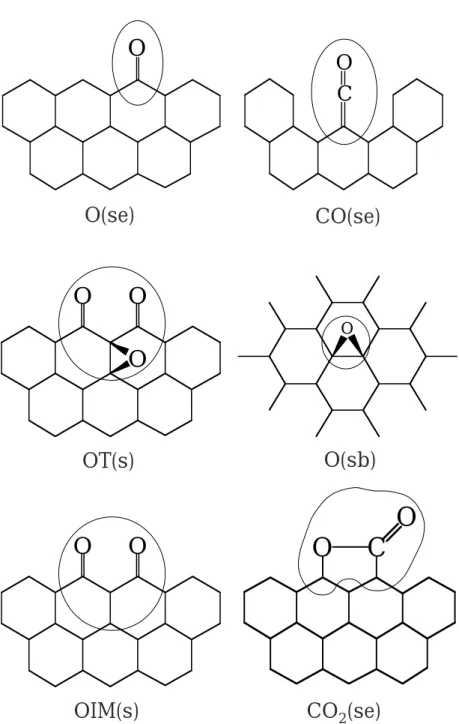
dieser Komplexe, die den Verbrennungsvorgang entscheidend pragen [60{75]. Zur Beschreibung des Grundmechanismus (Reaktion mit Sauersto) werden in dieser Arbeit folgende Oberachenkomplexe eingefuhrt:
freie Platze: Prismenache (se), Basisache (sb)
Semichinon O(se)
Carbonyl CO(se)
Lacton CO2(se)
Basisachenkomplex O(sb)
o-plane-Komplex OT(s)
Zwischenspezies OIM(s)
Eine grasche Darstellung der Komplexe ndet sich in Abbildung (3.3).
Unter 700 K existieren noch wesentlich mehr Komplexe auf der Graphit- oberache. Diese zerfallen aber bei hoheren Temperaturen und sind somit fur die Verbrennung nicht von Bedeutung.
Zusatzliche Oberachenspezies zur Beschreibung der Stickstoreaktionen werden in Kapitel Funf dargestellt.
Es wird nicht zwischen Zick-Zack- und Sesselplatzen unterschieden.
O(se) CO(se)
OT(s) O(sb)
OIM(s) CO2(se)
O
O
O O
C O
O O O
O C O
Abbildung 3.3: Darstellung der verwendeten Oberachenkomplexe.
3.3.2 Eigenschaften
Semichinon O(se)
Ein Semichinon-Komplex O(se) entsteht unter anderem durch direkte Adsorption eines Sauersto-Atoms auf einem Prismenplatz (se).Als Bindungsenergie D(C-O) der dabei entstehenden C-O-Bindung wird in der Literatur zwei Drittel einer C-O-Doppelbindung angegeben [76]. Der Wert fur D(C-O) betragt damit 499 kJ/mol. Nach Gleichung (2.46) ist dies dem Betrag nach die Adsorptionsenthalpie. Mit der Bildungsenthalpie eines Sauerstoatoms in der Gasphase (Hfg = 249:2 kJ/mol) ergibt sich mit Gleichung (2.40) die Bildungsenthalpie von O(se) zu Hfs=,250 kJ/mol.
Carbonyl CO(se)
Beim Carbonyl wird davon ausgegangen, da wahrend des Adsorptionsprozesses von CO auf Graphit eine dreifache C-O-Bindung im Gas- phasen-CO in eine zweifache umgewandelt wird. Dabei wird eine Energie von un- gefahr 321 kJ/mol frei. Das Sauerstoatom geht mit einem Kohlenstoatom auf der Graphitoberache eine Bindung ein, deren Energie wieder als zwei Drittel ei- ner C-O-Doppelbindung abgeschatzt wird. Das Kohlenstoatom im CO-Molekul geht keine Bindung mit der Oberache ein. Nach Gleichung (2.48) ergibt sich dann als Betrag der Adsorptionsenthalpie 86 kJ/mol. Mit der Bildungsenthal- pie von CO in der Gasphase (,110:5 kJ/mol) folgt mit Gleichung (2.40) die Bildungsenthalpie von CO(se): Hfs=,197 kJ/mol.Lacton CO
2(se)
Beim Lactonkomplex verbindet sich jeweils ein Kohlensto- atom auf der Oberache mit einem Kohlenstoatom und dem Sauerstoatom des CO2-Molekuls. Das zweite Kohlenstoatom im CO2 geht keine Bindung mit der Oberache ein. Weiterhin wird eine C-O-Doppelbindung im CO2 in eine C-O- Einfachbindung umgewandelt, wobei eine Energie von ca. 387 kJ/mol frei wird.Nach Gleichung (2.48) folgt daraus mit den entsprechenden Bindungsenergien fur C-O und C-C: jHadsj= 319 kJ/mol. Mit der Bildungsenthalpie von CO2 in der Gasphase (,393:5 kJ/mol) folgt mit Gleichung (2.40) die Bildungsenthalpie von CO2(se): Hfs =,713 kJ/mol.
Basisachenkomplex O(sb)
Ein Basisachenkomplex O(sb) entsteht unter anderem durch direkte Adsorption eines Sauersto-Atoms auf einem Basisplatz (sb). Die Moglichkeit dieser Adsorption wird beschrieben in [62,77{79]. Die Bin- dungsenergie D(C-O) der dabei entstehenden C-O-Bindung wird mit Hilfe von quantenchemischen Methoden zu 352 kJ/mol abgeschatzt [80]. Analog zum Se- michinon ergibt sich mit Gleichung (2.40) die Bildungsenthalpie von O(sb) zu Hfs =,103 kJ/mol.O-Plane-Komplex OT(s)
Die Existenz des o-plane Komplexes wird von Chen et al. [81{83] vorgeschlagen, ndet sich aber auch bei anderen Autoren[84{86]. Chen et al. haben mit Hilfe der Molekulorbitaltheorie die Moglichkeit eines solchen Komplexes nachgewiesen. Indirekte experimentelle Hinweise nden sich ebenfalls in der Literatur [71]. Im o-plane-Komplex schwacht das Sauersto- atom auf der Basisache die benachbarten C-C-Bindungen im Graphit, so da diese leichter brechen konnen als zum Beispiel die Bindungen im Semichinon.
Aufgrund der noch sehr ungenauen Kenntnisse uber die Eigenschaften des o- plane-Komplexes kann die Bildungsenthalpie nur sehr grob uber die entsprechen- den Werte des Semichinons und des Basiachenkomplexes abgeschatzt werden:
Hfs =,584 kJ/mol.

![Abbildung 3.1: Struktur von Graphit (nach [58]).](https://thumb-eu.123doks.com/thumbv2/1library_info/5539614.1688164/26.918.226.690.80.449/abbildung-struktur-von-graphit-nach.webp)
![Abbildung 3.2: Schematische Darstellung der Basisache (nach [59]).](https://thumb-eu.123doks.com/thumbv2/1library_info/5539614.1688164/27.918.268.601.89.445/abbildung-schematische-darstellung-der-basis-ache-nach.webp)
![Abbildung 4.2: Abbrandgeschwindigkeit in Abhangigkeit von der Oberachen- Oberachen-temperatur (Mewerte: Nagle und Strickland-Constable [89]) .](https://thumb-eu.123doks.com/thumbv2/1library_info/5539614.1688164/36.918.207.672.92.477/abbildung-abbrandgeschwindigkeit-angigkeit-temperatur-mewerte-nagle-strickland-constable.webp)

