The Dictyostelium discoideum RACK1 orthologue has roles in growth and development Inaugural-Dissertation zur
103
0
0
Volltext
(3)
(4)
(5)
(6)
(7)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
(39)
(40)
(41)
(42)
(44)
(48)
(49)
(50)
Abbildung
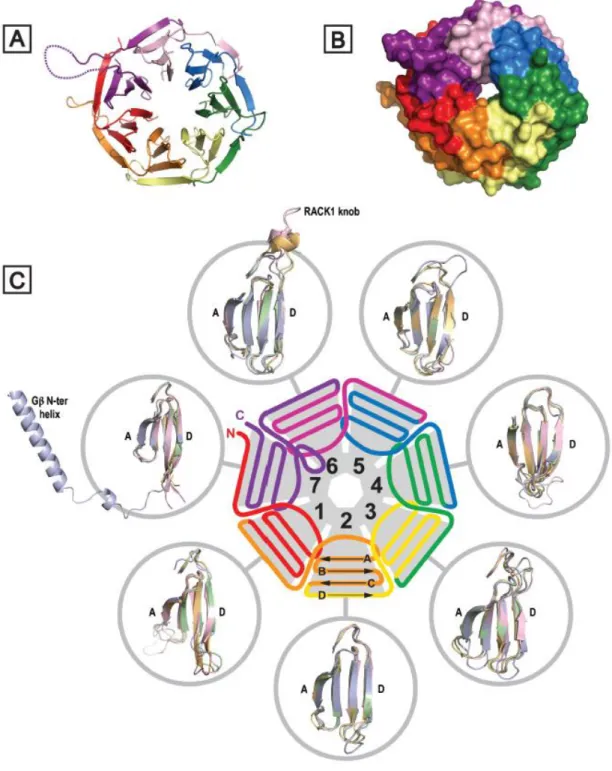
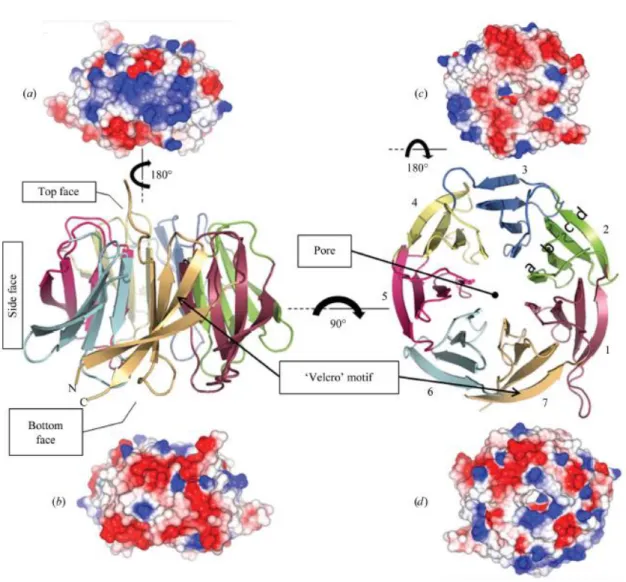
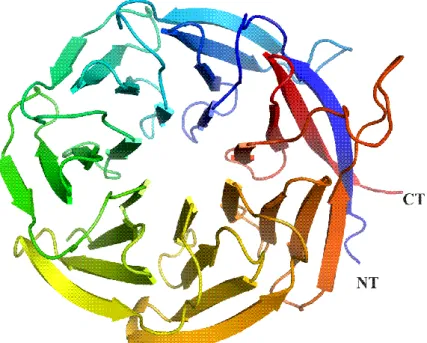
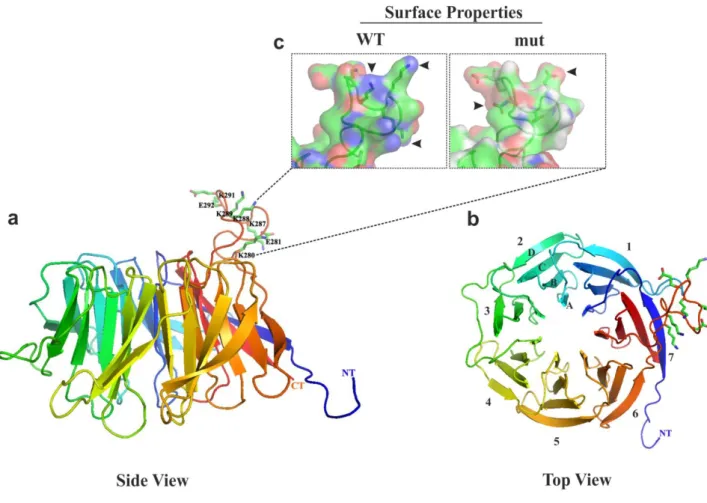
+7
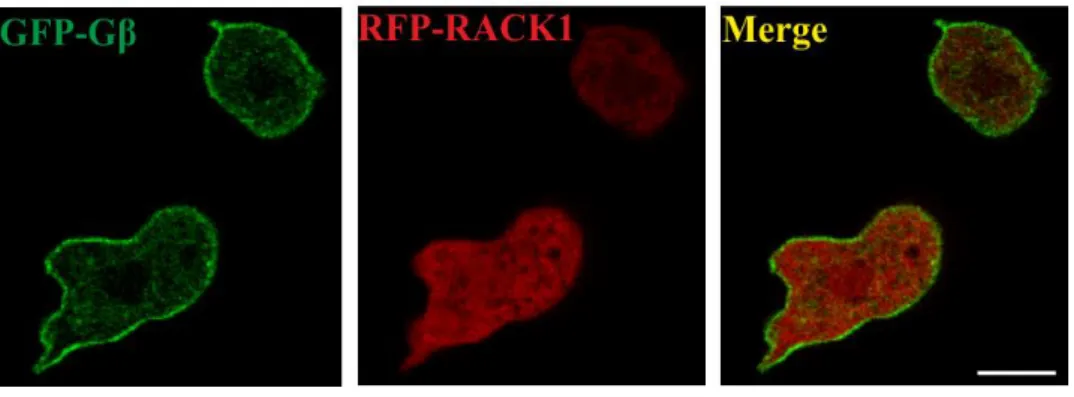
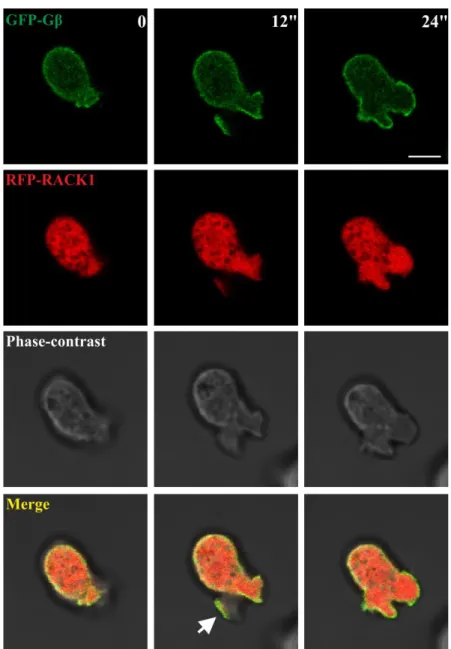

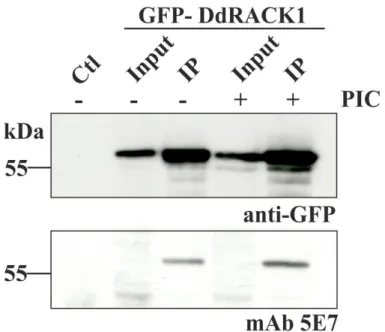

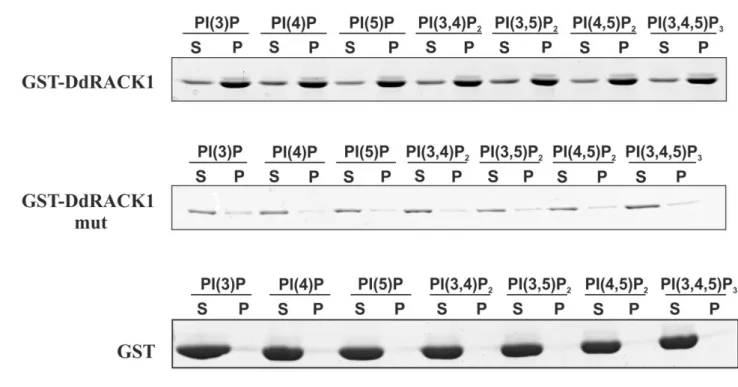
ÄHNLICHE DOKUMENTE
Moreover, although CNG channels are nonselective cation channels in vitro, the currents recorded from these channels are mainly carried by Ca 2+ in vivo (Ohyama et al.,.. Since no
das Hefe 3-Hybrid System in Frage, mit dessen Hilfe Protein-RNA Interaktionen identifiziert werden können (SenGupta et al., 1996). Außerdem ist die Isolierung der Fen1-
Interestingly, we also showed that despite similar binding affinities of DnmA to different RNA substrates, only tRNA Asp(GUC-1) , tRNA Glu(UUC-5) and tRNA Glu(CUA-5)
A minor degree of overexpression was also observed in knock-outs of the gene dicer A (drnA) and the heterochromatin protein gene hcpA (Kaller et al. In contrast,
In addition to the already known chemotaxis defect, we show here that coronin deletion mutants fail to upregulate genes involved in early development and, as a consequence,
This is not surprising, because microtubule plus end- binding proteins such as DdCP224 and DdEB1 have turned out not only to regulate microtubule dynamics but also to
dis- coideum amebae in this chamber with respect to the effects on chemotaxis of varying the midpoint gradient concentra- tion, the gradient steepness, and temporal
Da die mögliche dPMCA in ihrer Cterminalen Kationen ATPAse keine Calmodulinbinde und
