photophysikalischen Eigenschaften von Terephthalonitril-basierten
Donor-Akzeptor-Fluorophoren
Institut für Theoretische Chemie und Computerchemie
Masterarbeit
zur Erlangung des akademischen Grades Master of Science
vorgelegt von
Yusuf Acar
geboren am 06.08.1990 in Saarbrücken
im November 2017
Erstprüferin: Frau Prof. Dr. Christel Marian
Ich, Yusuf Acar, Matrikel-Nr. 2088295, versichere hiermit, dass ich meine Mas- terarbeit mit dem Thema
“Quantenchemische Untersuchung der photophysikalischen Eigen- schaften von Terephthalonitril-basierten Donor-Akzeptor-Fluorophoren”
“Quantum chemical Investigation of the photophysical properties of terephthalonitrile-based donor-acceptor fluorophores”
selbständig verfasst und keine anderen als die angegebenen Quellen und Hilfs- mittel benutzt habe, wobei ich alle wörtlichen und sinngemäßen Zitate als solche gekennzeichnet habe. Die Arbeit wurde bisher keiner anderen Prüfungs- behörde vorgelegt und auch nicht veröffentlicht.
Mir ist bekannt, dass ich meine Masterarbeit zusammen mit dieser Erklärung fristgemäß nach Vergabe des Themas in zweifacher Ausfertigung und gebunden im Prüfungsamt der Heinrich-Heine-Universität abzugeben oder spätestens mit dem Poststempel des Tages, an dem die Frist abläuft, zu senden habe.
Düsseldorf, den 9. November 2017
Yusuf Acar
An dieser Stelle möchte ich mich bei allen Personen bedanken, die mir während dieser Masterarbeit zur Seite standen.
Ein besonderer Dank gilt Frau Prof. Dr. Christel Marian für das interessante Thema und vor allem für Ihre stetige Unterstützung und Hilfe beim Auswerten der Daten.
Ich danke allen Mitgliedern des Instituts der Theoretischen Chemie und Com- puterchemie für die Hilfe bei der Durchführung der quantenchemischen Be- rechnungen und für die schöne Zeit.
Herrn Prof. Dr. T. J. J. Müller danke ich herzlichst für die Übernahme des Zweitgutachtens.
Für die Bereitstellung der experimentellen Daten danke ich Herrn Prof. Dr.
Claus A. M. Seidel und Dr. Denis Dörr.
Nicht zuletzt möchte ich mich bei meiner Famile und meinen Freunden bedan- ken, die immer hinter mir stehen und mir Kraft gibt, auch an Tagen an denen nicht alles nach Plan verläuft. Arigato.
In dieser Arbeit wurde ein auf Terephthalonitril-basierendes Molekül auf seine photophysikalischen Eigenschaften hin untersucht, um einschätzen zu können ob sich das Molekül als TADF-Emitter in OLEDs eignen könnte. Dazu wur- den Relaxed-Scans und DFT/MRCI-Rechnungen, sowohl im Grundzustand als auch in den angeregten ZuständenS1,T1undT2 durchgeführt. Zusätzlich wur- de das Molekül mittels Kontinuumsmodellen in verschiedenen Lösungsmitteln simuliert. Die daraus resultierenden Effekte auf Absorptionsspektren wurden mit experimentellen Daten verglichen.
Abstract
In this thesis, a terephthalonitril-based molecule was examined for its photo- physical properties to assess whether the molecule could serve as a TADF emit- ter in OLED. For this purpose, relaxed scans and DFT/MRCI-Calculations were performed in both the ground state and the excited states S1, T1 and T2. In addition, the molecule was simulated in different solvents using conti- nuum models. The resulting effects on absorption spectra were compared with experimental data.
Inhaltsverzeichnis
Abbildungsverzeichnis III
Tabellenverzeichnis VI
Abkürzungsverzeichnis 1
1 Einleitung 2
2 Theorie 5
2.1 Photophysikalische Prozesse . . . 5
2.2 Organische Lichtdiode, OLED . . . 6
2.3 Thermally Activated Delayed Fluorescence . . . 6
2.4 Dichtefunktionaltheorie . . . 9
2.5 Kohn-Sham-Funktionen . . . 10
2.6 TDDFT . . . 11
2.7 DFT/MRCI . . . 13
2.8 Lösungsmitteleinfluss . . . 15
2.8.1 Conductor-Like-Screening Model . . . 16
2.8.2 The Polarizable Continuum Model . . . 16
3 Details der quantenchemischen Berechnungen 18 3.1 Grundzustandsoptimierung . . . 18
3.2 Optimierung der angeregten Zustände . . . 18
3.3 DFT/MRCI . . . 19
3.4 COSMO . . . 19
3.5 PCM/cLR . . . 19
3.6 Diagramme und Abbildungen . . . 20
4 Ergebnisse 21 4.0.1 DFT-Optimierung des Grundzustandes . . . 21
4.0.2 DFT/MRCI-Energien des Grundzustandes . . . 23
4.1 Optimierung der angeregten Zustände im Vakuum . . . 26
4.1.1 Vergleich der optimierten Geometrien im Vakuum . . . . 27
4.1.2 TDDFT-Optimierung der angeregten Zustände im Va- kuum . . . 28
4.2 Absorptionsspektren im Vakuum und Lösungsmittel . . . 37
4.3 Optimierung in verschiedenen Lösungsmitteln . . . 41
4.3.1 COSMO . . . 41
4.3.2 PCM . . . 45
5 Zusammenfassung und Ausblick 48 Literaturverzeichnis 50 A Anhang Abbildungen i A.1 Differenzdichten . . . ii
A.2 Absorptionsspektren . . . vi
B Tabellen viii B.1 Bindungslängen und -winkel . . . x
Abbildungsverzeichnis
1 Jablonski-Diagramm der möglichen Übergänge. 1: Absorption, 2 und 6: Innere Konversion (engl. internal conversion, IC), 3: In- terkombination (engl. Intersystem Crossing, ISC), 4: Reverse Interkombination (engl reverse ISC), 5: Fluoreszenz, 6: Phos- phoreszenz . . . 6 2 Vereinfachte Darstellung der im TADF-Mechanismus vorkom-
menden Zustände und Ratenkonstanten. . . 7 3 Schematische Darstellung der einzelnen Schritte zur Berechnung
von Absorption und Fluoreszenz mittels cLR. . . 17 4 Die einzelnen Schritte der PCM/cLR-Optimierung. Schwarz = Mo-
lekül, Blau = Lösungsmittelumgebung des Grundzustands, Rot
= Lösungsmittel umgebung des angeregten Zustands. . . 20 5 S0-Struktur der Methylverbindung mit Atomnummern. Kohlen-
stoff in grau, Stickstoff in blau und Wasserstoff in weiß . . . 21 6 Relaxed-Scan an der Grundzustandsgeometrie. Aufgetragen sind
die DFT-Energien in eV, bezogen auf das Minimum bei 240°, gegen den Torsionswinkel. Bild im Bild zeigt ungeschnittenen Verlauf. . . 22 7 Differenzdichten von a)S0−S1 an derS1-Geometrie, b)S0−T1
an derT1-Geometrie und c) S0−T2 an der T2-Geometrie bei 90°. 23 8 Molekülorbitale von HOMO und LUMO an der Grundzustands-
geometrie bei 90°. . . 24 9 Die Dipolmomente der einzelnen Zustände an der Grundzu-
standsgeometrie. . . 25 10 Differenzdichte zwischen T1 und S0 bei a) 50° und b) 90° im
Grundzustand. . . 26 11 Differenzdichte zwischen T2 und S0 bei a) 50° und b) 90° im
Grundzustand. . . 26
12 Die TDDFT-Energien in eV der optimierten angeregten Zustän- de, bezogen auf dasS0-Minimum der TDDFT-Rechnungen, bei festgehaltenem Diederwinkel. . . 29 13 Alle Molekülorbitale, die an den Übergängen aus Tabelle 3 be-
teiligt sind. . . 31 14 DFTCI-Energien der jeweiligen Zustände an der optimiertenS1-
Geometrie bezogen aufS0 der Grundzustandsgeometrie. . . 32 15 DFTCI-Energien der jeweiligen Zustände an der optimiertenT1-
Geometrie bezogen aufS0 der Grundzustandsgeometrie. . . 33 16 DFTCI-Energien der jeweiligen Zustände an der optimierten
T2-Geometrie bezogen aufS0 der Grundzustandsgeometrie. . . . 33 17 Alle Molekülorbitale, die an den Übergängen aus Tabelle 4 be-
teiligt sind. . . 35 18 Die energetische Lage der Zustände und die Zusammensetzung
ihrer Übergänge an dem jeweiligen Minimum (links der Barrie- re) der optimierten S0-,S1-, T1- und T2-Geometrie. Alle Werte sind aufS0 des optimierten Grundzustandes bezogen
(CT = Charge-Transfer-Charakter, LA = Lokale Anregung, TPA = Triphenylamin-Rest, TPN = Terephthalonitril-Rest und gem = gemischter Charakter). . . 37 19 Die Absorptionsspektren im Vakuum ohne Normierung im Win-
kelbereich von 40° bis 150°. . . 39 20 Absorptionsspektren bei 60° (rot) und 240° (blau) im Vakuum
und den dazugehörigen Oszillatorstärken. . . 40 21 Absorptionsspektrum des Grundzustandes in DMSO, n-Hexan
und Ethylacetat bei 60°. . . 41 22 Absorptionsspektrum des Grundzustandes in DMSO, n-Hexan
und Ethylacetat bei 240°. . . 42 23 Vergleich der Absorptionsspektren mit experimentellen Daten
in a) Vakuum, b) n-Hexan c) Ethylacetat und d) DMSO. . . 44 24 Differenzdichten an der Grundzustandsgeometrie bei 0, 90 und
140°. . . ii 25 Differenzdichten an den optimierten, angeregten Zuständen S1,
T1 und T2 bei 0° . . . iii 26 Differenzdichten an den optimierten, angeregten Zuständen S1,
T1 und T2 bei 90° . . . iv
27 Differenzdichten an den optimierten, angeregten Zuständen S1, T1 und T2 bei 140° . . . v 28 Die energetische Lage der Zustände und die Zusammensetzung
ihrer Übergänge an dem jeweiligen Minimum (rechts der Barrie- re) der optimierten S0-,S1-, T1- und T2-Geometrie. Alle Werte sind aufS0 des optimierten Grundzustandes bezogen
(CT = Charge-Transfer-Charakter, LA = Lokale Anregung, TPA = Triphenylamin-Rest, TPN = Terephthalonitril-Rest und gem = gemischter Charakter). . . vi 29 Die Absorptionsspektren ohne Normierung im Winkelbereich
von 220° bis 320° . . . vii
Tabellenverzeichnis
1 Diederwinkel der Minima der optimierten Geometrien links und rechts der Barriere. . . 27 2 Unterschiede der Bindungslängen und -winkel zwischen den op-
timierten S1-, T1-, T2-Zustand und dem Grundzustand des Te- rephthalonitril-Restes links der Barriere. . . 28 3 Zusammensetzung der Übergänge und ihre Charakter der
TDDFT-optimierten Zustände an derT1- bzw.T2-Geometrie bei festgehaltenem Diederwinkel. (LA = Lokale Anregung;
TPA = Triphenylamin-Rest; TPN = Terephthalonitril-Rest;
CT = Charge-Transfer) . . . 30 4 Zusammensetzung und Charakter der DFT/MRCI-optimierten
Zustände T1 bzw. T2 an der T2-Geometrie bei festgehaltenem Diederwinkel. (LA = Lokale Anregung; TPA = Triphenylamin- Rest; TPN = Terephthalonitril-Rest; CT = Charge-Transfer) . . 34 5 Die Differenz der Bindungslängen und -winkel zwischen dem
Molekül in DMSO und in Vakuum (RDM SO−RV ac). . . 46 6 Aufgelistet sind die Energieunterschiede der im Vakuum und
der mittels cLR-berechneten vertikalen Emission in eV. Sowohl links als auch rechts der Rotationsbarriere. . . 46 7 Die Maxima der exp. Emissionsspektren und die berechneten
vertikalen Emissionsenergien (links und rechts der Barriere) in nm. . . 47 8 Bindungslängen der Minima in verschiedenen Lösungsmittel nach
der PCM-Optimierung in nm. . . x 9 Bindungswinkel der Minima in verschiedenen Lösungsmittel nach
der PCM-Optimierung. . . xi 10 Bindungslängen der Minima in verschiedenen Lösungsmittel nach
der COSMO-Optimierung in nm. . . xii
11 Bindungswinkel der Minima in verschiedenen Lösungsmittel nach der COSMO-Optimierung. . . xiii 12 Bindungslängen an den Minima der optimierten Zustände im
Vakuum. Alle Werte in nm. . . xiv 13 Bindungswinkel an den Minima der optimierten Zustände im
Vakuum. . . xv
Abkürzungsverzeichnis
Abb. Abbildung Tab. Tabelle
HOMO Highest Occupied Molecular Orbital LUMO Lowest Unoccupied Molecular Orbital EE Ethylacetat
DFT Dichtefunktionaltheorie
BH-LYP Becke Half and Half Lee-Yang-Parr IC Internal Conversion
ISC Intersystem Crossing
rISC reverse Intersystem Crossing MO Molecular Orbitall
OLED Organic Light Emitting Diode
TADF Thermally Activated Delayed Fluorescence TDDFT Time Dependant Density Functional Theory TPA Triphenylamin-Rest
TPN Terephthalonitril-Rest CT Charge-Transfer
COSMO Conductor-Like-Screening Model PCM Polarizable Continuum Model cLR corrected linear response
1 Einleitung
Licht und seine Erzeugung oder Nutzung spielt eine wichtige Rolle im Leben, ob nun für Tiere oder Pflanzen. Am Anfang wurde schlicht etwas verbrannt, um Licht zu erzeugen, später durch die Erfindung der Glühbirne und der Nutz- barmachung von Strom gewann man Licht durch das Erhitzen eines Drahtes.
Die Glührbirne war lange Zeit die weit verbreitetste und unangefochtene Me- thode zur Lichterzeugung. Da diese aber einen sehr schlechten Wirkungsgrad besitzt, also den Großteil des verwendeten Stroms bloß als Wärme abgibt und gleichzeitig die Nachfrage nach Strom bzw. Energie aber steigt, wird intensiv nach effektiveren Alternativen gesucht.
Einen Grundstein dafür legte 1936 Destriau, der die Lumineszenz einiger Leucht- stoffe mit Hilfe von elektrischen Feldern angeregt hat, was er Jahre später Elektrolumineszenz nannte[1]. Diese wurde an verschiedenen Materialien un- tersucht, so auch an organischen Substanzen[2], um diesen Effekt für verschie- dene Anwendungen zugänglich zu machen. Die ersten brauchbaren OLEDs, die zur Lichterzeugung verwendet werden konnten, wurden 1987 von C.W. Tang und S. A. VanSlyke vorgestellt[3]. Der Durchbruch lag in Höhe der verwen- deten Spannung, welche mit 10 V sehr viel niedriger lag als die bei den bis dahin bekannten OLEDs. Seit dem werden sie stetig weiterentwickelt, denn sie besitzen eine Reihe von Vorteilen gegenüber anderer Lichtquellen. So haben sie z.B. einen höheren Kontrast, sind leichter, flexibler und dünner gegenüber LCDs, was ihren Anwendungsbereich stark vergrößert. Außerdem sind sie effi- zienter. Circa 20% der weltweit verbrauchten Energie wird zur Lichterzeugung genutzt[4]. Durch einen Umstieg auf OLEDs könnte somit ein Großteil der Energie eingespart werden.
Die elektrische Anregung eines Moleküls verläuft über eine Elektronen-Loch- Rekombination. Dabei werden Exzitonen erzeugt, welche sich zu 1/4 in Singu- lett- und zu 3/4 in Triplett-Zuständen befinden. OLEDs werden in verschie- dene Klassen, sogenannte Generationen unterteilt. In der ersten Generation wird zur Lichterzeugung lediglich die Fluoreszenz ausgenutzt, sodass nur 25%
der erzeugten Exzitonen genutzt werden, während die restlichen 75% nicht- strahlend zerfallen. Durch den Einsatz von Schwermetallen können diese 75%
Triplett-Exzitonen zugänglich gemacht werden zur Lichterzeugung, da dadurch das Spinverbot aufgehoben wird und die Phosphoreszenz (schneller) stattfin- det. OLEDs, die die Phosphoreszenz nutzen, bilden die zweite Generation. Da der Einsatz von Schwermetallen teuer ist und sie gifitig für die Umwelt sind, wurde nach anderen Wegen gesucht. Heute spricht man von OLEDs der dritten Generation, diese sind aus rein organischen Materialien aufgebaut. Sie nutzen Fluoreszenz zur Lichterzeugung, aber nutzen 100% der Exzitonen, da durch den reverse Intersystem Crossing-Prozess die Triplett-Exzitonen in Singulett- Exzitonen umgewandelt werden und es so zu einer verzögerten Fluoreszenz kommt.
Das erste Mal, dass TADF, früher „e-type“-Fluoreszenz genannt, in orga- nischen Molekülen nachgewiesen wurde, war 1961 in Eosin von Parker und Hatchard[5]. Diese glaubten aber, es sei bloß eine verzögerte Fluoreszenz aus demselben Singulett-Zustand. Dieser Effekt trat nur selten auf, da Eosin ei- ne große Singulett-Triplett-Aufspaltung besitzt und wurde daher nicht weiter verfolgt.
Adachi et al. erkannten 2009, dass der Mechanismus des TADF-Prozesses in OLEDs verwendet werden kann[6]. 2011 stellten sie dann den ersten rein-orga- nischen auf TADF-basierenden OLED-Emitter vor[7]. Das generelle Vorgehen war, eine kleine Singulett-Triplett-Aufspaltung ∆EST zu erreichen, während die Fluoreszenz-Rate groß genug blieb. Ihr Lösungsansatz war dem Molekül einen Charge-Transfer-Charakter zu geben um Donor und Akzeptor elektro- nisch weitgehend zu entkoppeln.
In den aktuellsten System (auch Generation 3.5 genannt) wer den TADF- Emitter als Zusatz verwendet. So wird die Photonen die beim TADF-Prozess entstehen dazu genutzt, den eigentlichen Fluoreszenz-Emitter anzuregen. Da es sich dabei um eine Photonenanregung handelt, wird lediglich der Singulett- Zustand angereichert und es ist eine 100%ige Quantenausbeute möglich. So werden weiterhin nach neuen Systemen und Molekülgeforscht, idealerweise Sys- teme die im kurzwelligen, blauen Wellenlängenbereich emittieren. Aufgrund der hohen Energie die von diesen emittiert werden, ist ihre Lebensdauer sehr gering.
Das in dieser Arbeit betrachtete Molekül ist bisher in keinem Paper veröf- fentlicht, so ist wenig über seine photophysikalischen Eigenschaften bekannt.
Jedoch behinaltet es einen Terephthalonitril-Rest. Derivate davon werden un- ter anderem zum Bio-Imaging von Proteinen[8].
2 Theorie
2.1 Photophysikalische Prozesse
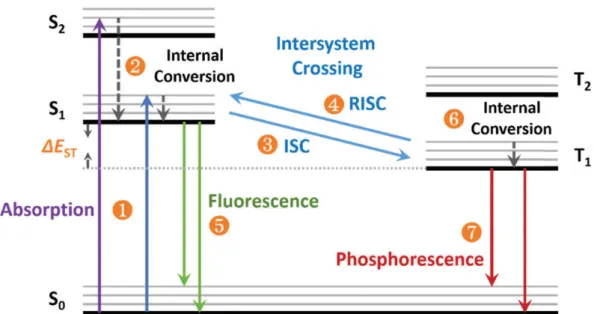
Wird die Energie eines Photons von einem Molekül aufgenommen, so gelangt ein Elektron in einen elektrisch angeregten Zustand, welcher energetisch höher liegt. Um diese Energie abzugeben und wieder in den Grundzustand zu ge- langen (= Relaxation), hat das angeregte Elektron mehrere Optionen. Einige dieser Optionen werden in Abbildung 1 schematisch dargestellt.
Erfolgt die Abgabe der Energie unter der Aussendung von Photonen, so spricht man von Lumineszenz (Emission), wobei zwischen zwei Arten unterschieden wird. Zum einen die Fluoreszenz, die Emission aus einem Singulett-Zustand und zum anderen die Phosphoreszenz, die Emission aus einem Triplett-Zustand.
Die Kasha-Regel besagt, dass die Emissionen fast ausschließlich aus dem S1- bzw. T1-Zustand stattfinden[9]. Grund dafür sind die energetischen Abstän- de zwischen den angeregten Zuständen, denn mit steigender Energie, werden diese immer kleiner. Somit ist der Abstand zwischen dem Grundzustand und dem S1-Zustand am größten und die Übergänge der angeregten Zustände un- tereinander (internal conversion, IC) sind viel schneller. Die Phosphoreszenz und der ISC-Prozess sind aus quantenmechanischer Sicht verboten, da sich der Spin des Elektrons dabei ändern muss. Daher verlaufen diese Übergänge nur sehr langsam.
Ein Übergang zwischen einem Singulett- und einem Triplett-Zustand (intersys- tem crossing, ISC) erfolgt strahlungslos. Ist die Energielücke ∆EST zwischen demS1- und demT1-Zustand klein genug, so kann der umgekehrte Prozess er- folgen (reverse intersystem crossing, rISC). Die Schwingungsrelaxation (engl.
vibrational relaxation, VR) ist ein weiterer strahlungsloser Prozess, welcher die Desaktivierung schwingungsangeregter Zustände in den Schwingungsgrundzu- stand des jeweiligen elektronischen Zustand beschreibt.
Abbildung 1: Jablonski-Diagramm der möglichen Übergänge. 1: Absorpti- on, 2 und 6: Innere Konversion (engl. internal conversion, IC), 3: Interkombination (engl. Intersystem Crossing, ISC), 4: Re- versible Interkombination (engl reverse ISC), 5: Fluoreszenz, 6: Phosphoreszenz[10]
2.2 Organische Lichtdiode, OLED
In OLEDs (engl. organic light emitting diode) erfolgt die Anregung nicht über Photonen, sondern auf elektrischem Wege. Dabei werden durch Elektronen- Loch-Rekombination Exzitonen erzeugt, die sich zu 75% in Triplett-Zustände und zu 25% in Singulett-Zustände aufteilen. Die möglichen Relaxationsprozes- se sind gegenüber der Photonenanregung unverändert. Je nach Mechanismus zur Lichterzeugung werden OLEDs in verschiedene Generationen unterteilt.
Die aktuellste Generation der OLEDs nutzt den TADF-Mechanismus.
2.3 Thermally Activated Delayed Fluorescence
Wie der Name bereits vermuten lässt, handelt es sich hierbei um Fluoreszenz- Emitter. Abbildung 2 ist eine einfache Darstellung der im TADF-Prozess vor- kommenden Zustände und Ratenkonstanten[11]. Damit ein Molekül TADF- tauglich ist, muss es folgende Bedingung erfüllen:
• Die Ratenkonstanten des reverse Intersystem Crossing krISC und der Fluoreszenz kF müssen groß und die des strahlungslosen Zerfalls des Triplett-Zustandes kTISC und der Phosphoreszenz kP H klein sein.
Abbildung 2: Vereinfachte Darstellung der im TADF-Mechanismus vorkom- menden Zustände und Ratenkonstanten[11].
Somit ist die Ratenkonstante krISC für den TADF-Prozess sehr wichtig. Vor- ausgesetzt die RatenkISCT und kP H sind sehr klein und es herrscht ein Gleich- gewicht zwischen ISC und rISC, kann krISC über die Boltzmann-Verteilung bestimmt werden[11]:
k
rISC= A exp (− ∆E
STkT ) (1)
mit der Boltzmannkonstante k und der Temperatur T. Um krISC zu maxi- mieren muss also ∆EST minimiert werden. Zum Berechnen von ∆EST werden die Energien desS1- bzw.T1-Zustands benötigt. Diese sind gegeben durch die Orbitalenergie E, die der Ein-Elektronenenergie in einer festen Umgebung wi- dergibt, der Elektronen-Abstoßungsenergie K und dem Austauschterm J. Da es sich dabei um die selbe Zusammenstellung an Elektronen im selben Molekül handelt, sind diese Terme für Singulett und Triplett gleich. Da es sich aber im Triplett um zwei ungepaarte Elektronen mit gleichem Spin und im Singulett
mit ungekehrtem Spin handelt, führt der Austauschterm im Triplett zu einer Absenkung und im Singulett zu einer Erhöhung der Zustände. Der Unterschied zwischen ES und ET entspricht somit zweimal dem Austauschterm.
ES =EOrb+K +J (2)
ET =EOrb+K−J (3)
∆EST =ES−ET = 2J (4)
Bei S1 und T1 sind die ungepaarten Elektronen hauptsächlich auf die Gren- zorbitale, also HOMO und LUMO verteilt und besitzen so ein gleiches J (den Spin außer Acht lassend). Dadurch lässt sich die Austauschenergie wie folgt berechnen[12]:
J =
Z Z
φL(1)φH(2)( e2
r1−r2)φL(2)φH(1)dr1dr2 (5) Wobei φH und φL die Wellenfunktion von HOMO und LUMO sind und e der Elektronenladung entspricht. Daraus folgt, dass eine kleines ∆EST durch ein kleines Überlappungintegral von hφH|φLi erreicht wird.
Ebebfalls wichtig für den TADF-Prozess ist Fluoreszenzrate. Diese kann über folgende Gleichung wiedergegeben werden, wenn für|iidie Wellenfunktion des S1-Zustands und für hf| die des Grundzustands angenommen wird[13].
kF = 4e2 3c3~4
(Ei−Ef)3| hf|~r|ii |2 (6) Der letzte Term ist der Ausdruck für den Übergangsdipolmoment, welcher kleiner wird je kleiner die Überlappung der Dichten von HOMO und LUMO sind. Das bedeutet, dass das Minimieren von ∆EST und das Maximieren von kF sich gegenseitig stören.
2.4 Dichtefunktionaltheorie
In der Dichtefunktionaltheorie (engl. density functional theory, DFT) wird vorausgesetzt, dass die Dichte, über den gesamten Raum integriert, die Anzahl der Elektronen im System ergibt:
Z
ρ(~ r)d~ r = N (7)
Mittels der Einelektronen-Dichte eines Systems lassen sich Rückschlüsse auf alle zum Beschreiben des Systems notwendigen Informationen ziehen. Norma- lerweise müssten 3N (N = Anzahl der Teilchen im System) Variablen berück- sichtigt werden, aber da die Dichte lediglich von den Ortskoordinaten abhängt, wird die Rechnung stark vereinfacht. Damit können selbst große Systeme ohne gro”sen Rechenaufwand bewältigt werden. Grundlage bilden die Hohenberg- Kohn-Theoreme [14].
• Jede Grundzustandsdichte ρ0 ist eindeutig einer Grundzustandsenergie E0 zugeordnet (oder die Grundzustandsenergie ist ein Funktional der Grundzustandsdichte):
E
0= E
0[ρ
0] (8)
• Die Energie der wahren Grundzustandsdichte E0(ρ0) bildet eine untere Schranke (Variationsprinzip):
E(ρ) ≥ E
0(ρ
0) (9)
Somit existiert eindeutig eine GrundzustandsenergieE0, welche mittels Varia- tionsprinzip ermittelt werden kann. Daraus folgt für die Energie des Systems:
E[ρ] =
Z
ρ(~ r )υ(~ r)d~ r + T [ρ] + V
ee[ρ] (10)
mit einem äußeren Potenzialυ(~r), der kinetischen EnergieT[ρ] und der Elektron- Elektron-WechselwirkunsgenergieVee[ρ]. Die kinetische Energie T[ρ] exakt zu berechnen ist sehr aufwändig, daher werden Näherungen benötigt.
2.5 Kohn-Sham-Funktionen
Ein mögliches Näherungsverfahren bietet der Kohn-Sham-Formalismus. Da- bei wird von einem fiktiven System mit n Fermionen (meistens Elektronen) ausgegangen, welche keine Wechselwirkungen untereinander zeigen und kei- ne Ladung oder Spin besitzen. Diese sollen aber die selbe Dichte besitzen wie das gleiche System mit wechselwirkenden Elektronen. Um nun die Dichte zu berechnen, werden N Einelektronen-Wellenfunktionen (sog. Kohn-Sham- Funktionen) angesetzt, welche N Lösungen der Schrödingergleichung in einem (fiktiven) effektiven Potenzial υs(~r) sind. Dies vereinfacht die Rechnungen, da es sich nun um voneinenander unabhängige Lösungen einer Schrödingerglei- chung handelt. Diese Einelektronen-Schrödingergleichungen werden auch als Kohn-Sham-Gleichungen bezeichnet:
[− 1
2 ∇
2+ υ
ef f(~ r)]φ
i(~ r) = ε
iφ
i(~ r) (11)
Damit ist es möglich, die Elektronendichte dieses Systems exakt zu berech- nen:
ρ(~ r) =
n X
i
|φ
i(~ r)|
2(12)
Dabei ist das effektive Potenzialυef f(~r) wiederum von der Dichteρ(~r) abhän- gig:
υ
ef f(~ r) = υ(~ r) +
Z
ρ(~ r
0)
|~ r − ~ r
0| d
3r
0+ υ
xc(~ r) (13)
Der erste Term υ(~r) steht für das externe Potenzial, welches im Grunde die Anziehung der Elektronen durch die Atomkerne beschreibt. Der zweite Term beschreibt die elektrostatische Wechselwirkung der Elektronen untereinander.
Der dritte Termυxc(~r), das sogenannte Austausch-Korrelationspotenzial sorgt für die korrekte Behandlung des Vielelektronensystems. Da υef f(~r) sowohl in den Kohn-Sham-Gleichungen vorkommt, als auch von der Dichte ρ(~r) und so- mit von den Lösungen dieser Gleichungen abhängt, können die Lösungen nur iterativ gefunden werden. Ein gefundenes Potenzialυef f(~r) wird zur Lösung der Kohn-Sham-Gleichungen eingesetzt um damit ein neues Potenzial zu berech- nen, welches wieder zum Lösen benutzt wird. Dieser Vorgang wird mehrmals wiederholt, bis eine sile (selbstkonsistente) Lösung gefunden wird.
2.6 TDDFT
Mit der (zeitunabhängigen) Dichtefunktionaltheorie lassen sich lediglich Grund- zustände berechnen. Zur Optimierung von angeregten Zuständen wird oft die zeitabhängige Dichtefunktionaltheorie (engl. Time-Dependent Density Func- tional Theory, TDDFT) benutzt. Startpunkt ist die zeitabhängige Schrödin- gergleichung[15]:
i∂Φ(t)
∂t = ˆH(t)φ(t) (14)
mit
H(t) = ˆˆ T + ˆV(t) + ˆW (15)
wobei ˆT die kinetische Energie, ˆV ein zeitabhängiges, lokales und spin-un- abhängiges Ein-Teilchen-Potenzial ist und ˆW für spin-unabhängige Teilchen- Wechselwirkungen steht. Durch Lösen der Schrödinger-Gleichung (Gl. 14) mit verschiedenen Potenzialen und einer festen Start-Wellenfunktion Φ0 werden
verschiedene Wellenfunktionen Φt erhalten und zu jeder dieser Wellenfunktio- nen Φt werden wiederum Dichten ρ(~r, t) berechnet. Zwei verschiedene Poten- ziale unterscheiden sich lediglich durch eine zeitabhängige Phase, sodass die daraus erhaltenen Dichten identisch sind:
Φ(t) =e e−iα(t)Φ(t)⇒ρ(~e r, t) = ρ(~r, t) (16) Daraus folgt, dass jede Observable Funktionale der zeitabhängigen Dichte sind:
hOiˆ (t) =hΦ(t)|Oˆ|Φ(t)i=O[ρ(t)] (17) Das HK-Theorem basiert auf dem Rayleigh-Ritz-Prinzip, was ein Problem für zeitabhängige Systeme darstellt, da das RR-Prinzip hier nicht anwendbar ist.
Statt der Energie ist das Aktionsintegral A ein eindeutiges Funktional der zeitabhängigen Dichte:
A =
Z t1
t0
dthΦ(t)|i∂
∂t−H(t)ˆ |Φ(t)i ⇒A[ρ(t)] (18) Die stationären Punkte des Aktionsintegrals A liefern die exakte zeitabhängige Dichte des Systems. Diese können durch die Euler-Formel erhalten werden:
∂A[ρ]
∂ρ(r, t) = 0 →ρ(r, t) (19) Analog zur DFT kann mit Hilfe des Kohn-Sham-Ansatzes, die zeitabhängi- ge Dichte des wechselwirkenden Systems durch die zeitabhängige Dichte eines nicht-wechselwirkenden Systems mit lokalem Potenzial beschrieben werden.
Die Elektronen dieses nicht-wechselwirkenden Systems folgen ebenfalls der zeit- abhängigen Schrödinger-Gleichung, wobei der Kohn-Sham-Hamiltonoperator wie folgt definiert ist:
HˆKS(r, t) = −∇2
2 +υKS[ρ](r, t) (20)
mit dem lokalen externen PotenzialυKS. Somit lässt sich die Dichte des wech- selwirkenden Systems mittels Kohn-Sham Orbitalen berechnen:
ρ(~r, t) =X
i
fi|ϕKSi (~r, t)|2 (21)
Auch hier kann das zeitabhängige Kohn-Sham Potenzial υKS als Summe von drei Termen aufgefasst werden:
υKS[ρ](r, t) = υext(r, t) +υHartree[ρ](r, t) +υXC[ρ](r, t) (22) Der erste Term steht für das externe Potenzial und der zweite Term berücksich- tigt die klassische elektrostatische Wechselwirkung der Elektronen. Der dritte Term ist das Austausch-Potenzial, welches alle nicht-trivialen Mehrteilchen- Effekte beinhaltet[16].
2.7 DFT/MRCI
Eine weitere Methode, um DFT zu erweitern und somit auch angeregte Zu- stände zugänglich zu machen, ist die Kombination von MRCI (Multireference Configuration Interaction) mit DFT. Die DFT-Methode liefert schnell und günstig Informationen über die dynamische Korrelation der Elektronen, wel- che in der MRCI-Methode eingesetzt werden können, wohingegen die MRCI- Methode einen guten Rückschluss über die statische Korrelation liefert. In der MRCI-Methode wird berücksichtigt, dass angeregte Zustände Anteile von mehreren Elektronenkonfigurationen besitzen:
φ
0= c
0ψ
0+
bes X
i vir X
r
c
riψ
ir+
bes X
i<j vir X
r<s
c
rsijψ
ijrs+ . . . (23)
Dazu wird die Vielelektronenwellenfunktion φ0 durch eine Linearkombination aus gewichteten Anteilen verschiedener Konfigurationen dargestellt (siehe 23).
Der erste Summand steht für den Grundzustand, der zweite für die Einfach- anregung, der dritte für die Zweifachanregung usw. Werden alle Anregungen
berücksichtigt, spricht man von einer Full CI. Dies sprengt für größere Mo- leküle jedoch den Rahmen der Rechenleistung und Speicherkapazität. Daher beschränkt man sich auf Einfach- bzw. Zweifachanregungen und limitiert eben- falls die Anzahl an Orbitalen, aus denen bzw. in die angeregt werden kann. In jeder MRCI-Rechnung wird die Referenzwellenfunktion ψ0 neu berechnet und als neue Referenzwellenfunktion in der nächsten Rechnung verwendet.
Die Hamilton-Matrix lässt sich in Diagonal- und Außerdiagonal-Elemente ein- teilen und separat behandeln. Die Diagonalelemente (Einträge zwischen zwei Konfigurations-Zustandsfunktionen, CSFs, mit gleichem Raumteil und glei- cher Spin-Kopplung) werden aus Hartree-Fock-Ausdrücken und einem DFT- spezifischen Korrekturterm zusammengesetzt.
hωw|HˆDF T −EDF T |ωwi=hωw|Hˆ −EHF |ωwi
−
nexc
X
c
Fcc[HF −FccKS+
nexc
X
a
FaaHF −FaaKS
+ 1 nexc
nexc
X
a nexc
X
c
pj(aa|cc)−p[N0](ac|ac)
(24)
In den ersten beiden Summanden werden alle HF Orbitalenergien durch ih- re KS-Gegenstücke ersetzt, da Teile der exakten Matrix-Elemente als Summe über alle Orbitalenergien ausgedrückt werden können. Die letzten beiden Sum- manden sind Korrekturterme in die empirische Parameter einfließen.
Da es durch die Kombination von DFT und MRCI zu doppelten Wertun- gen der Korrelationseffekte in den Außer-Diagonalen kommen kann, wird der Hamilton-Matrix ein Skalierungsfaktor ρ1 hinzugefügt[17] und es findet eine Selektion der Wechselwirkungen statt, die dies verhindert:
hωw| H
DF T|ω
0w
0i = hωw| H |ω
0w
0i ρ
1e
−ρ2∆E4ww0(25)
∆Eww4 0 ist die Energiedifferenz der Diagonalelemente zwischen zwei CSFs. Durch die e-Funktion und die Energiedifferenz werden nur Wechselwirkungen zwi- schen zwei CSFs betrachtet, die energetisch sehr nah beieinander liegen. Durch die vierte Potenz der Energiedifferenz werden viele Wechselwirkungen bereits
im Vorfeld aussortiert und viele Außer-Diagonal-Einträge gehen gegen 0, was die Methode so schnell und kosteng´’unstig macht, selbst für große Molekü- le. Durch die Anpassung der Parameter an experimentelle Daten, erreich- ten Grimme und Waletzke einen mittleren Fehler der relativen Energien von 0.2 eV[17].
2.8 Lösungsmitteleinfluss
Reaktionen von Stoffen sind nicht bloß von den Ausgangsstoffen abhängig, son- dern auch vom verwendeten Lösungsmittel. Bereits 1862 hat Marcelin Bert- helot erkannt, dass das verwendete Lösungsmittel z.B. die Reaktionsgeschwin- digkeit beeinflussen kann[18]. Aber auch auf das Absorptions- oder Emissi- onsspektrum hat das Lösungsmittel einen großen Einfluss. So kann sich das Spektrum einer Substanz im Gaszustand/Vakuum vom Spektrum derselben Substanz in Lösung charakteristisch in Lage und Intensität der Banden unter- scheiden. Dieses Phänomen wird Solvatochromie genannt.
Je polarer das Molekül oder der Zustand (also je größer das Dipolmoment), umso mehr wird es durch ein polares Lösungsmittel stabilisiert. Umgekehrt bedeutet das auch, dass je unpolarer das Molekül oder der Zustand ist, desto schlechter wird es durch ein polares Lösungsmittel stabilisiert. Der Lösungsmit- teleffekt ist sehr gering, wenn das Molekül oder die Zustände neutral/unpolar sind oder wenn Anfangs- und Endzustand des Übergangs ähnliche Dipolmo- mente besitzen.
Als Maß für die Polarität des Lösungsmittels wird allgemein die Dielektrizitäts- konstanteε angesehen, wohingegen für das gelöste Molekül das Dipolmoment betrachtet wird.
Zum Berechnen von Lösungsmitteleffekten gibt es zwei Möglichkeiten. Zum einen kann unter expliziter Einbeziehung von Lösungsmittel-Molekülen gerech- net werden und zum anderen gibt es Modelle, die den Lösungsmitteleinfluss nä- herungsweise beschreiben. Dabei ist zu beachten, dass nicht jeder Lösungsmit- teleffekt mit diesen Modellen beschreibbar ist, wie z.B.Wasserstoffbrücken.
2.8.1 Conductor-Like-Screening Model
Eines dieser Modelle ist das Conductor-Like Screening Model (COSMO), wel- ches zu den Kontinuumsmodellen geh´’ort. In diesen wird das Lösungsmittel als ein kontinuierliches Medium mit einer Dielektrizitätskonstante ε betrach- tet, indem das Solvat in einem „Loch“ (einer Kavität) platziert wird. Zum Erzeugen dieser Kavität wird um jedes Atom im Molekül ein Kreis gezogen, welcher durch den van-der-Waals-Radius definiert ist. So wird ein Hohlraum mit einer „Grenzfläche“ zwischen Lösungsmittel und Solvat geschaffen. Weiter wird vorausgesetzt, dass das Lösungsmittel nur bis an diese Grenzfläche reicht und es somit in keinen direkten Kontakt mit dem Solvat tritt. Das vereinfacht die Rechnung, da nun lediglich elektrostatische Wechselwirkungen betrachten werden müssen.
In COSMO wird das Lösungsmittel als idealer elektrischer Leiter (ε = ∞, im Kontinuum) angesehen. Somit muss das elektrische Feld auf der „Grenz- fläche“ verschwinden. Bei bekannter Ladungsverteilung im Molekül lässt sich die Abschirmladung an der Fläche bestimmen[19] und so auf die Energie der Wechselwirkung zwischen Solvat und Lösungsmittel schließen.
2.8.2 The Polarizable Continuum Model
Ein weiteres Model zur näherungsweisen Beschreibung des Lösungsmitteleffek- tes ist PCM, welches ebenfalls zu den Kontinuumsmodellen zählt. Im Gegen- satz zu COSMO wird das Lösungsmittel durch das gelöste Molekül, welches sich ebenfalls in einer Kavität befindet, polarisiert, was seinerseits wiederum die Ladungsverteilung des Moleküls beeinflusst.
Mittels einer linearen Antwort (linear response) und TDDFT in PCM[20] ist es möglich auch angeregte Zustände in Lösung zu optimieren. In diesen Rech- nungen wird explizit ein Lösungsmitteloperator (oft „Reaktionsfeld“-Operator) zum Hamilton-Operator hinzugefügt. Dieser Lösungsmitteloperator ist eine Funktion der Ladungsverteilung des gelösten Moleküls, womit nicht-lineare Effekte zum Hamilton-Operator hinzugefügt werden, was zu Fehlern führt.
Um diese zu Umgehen haben Caricato et al. eine Methode entwickelt, die sie
„corrected linear response, cLR“ nennen.[21]
In dieser Methode teilt man die Rechnung in einzelne Schritte auf (siehe Abb. 3).
Abbildung 3: Schematische Darstellung der einzelnen Schritte zur Berech- nung von Absorption und Fluoreszenz mittels cLR.[22]
Im ersten Schritt wird der Grundzustand in Gegenwart des Lösungsmittels op- timiert. Dann wird das Molekül angeregt, wobei sich das Lösungsmittel aber noch im Gleichgewicht mit dem Grundzustand befindet. Schritt 3 ist die Lö- sungsmittelreorganisation (das Lösungsmittel passt sich der Struktur des an- geregten Zustandes an). Zum Schluss wird die vertikale Relaxation berechnet, nachdem das Reaktionsfeld des angeregten Zustands ermittelt wurde.
3 Details der quantenchemischen Berechnungen
3.1 Grundzustandsoptimierung
Die Startgeometrie wurde mit Hilfe des Programms MOLDEN 5.6 erstellt.
Die Geometrie-Optimierungen wurden mit DFT [23] und dem TURBOMOLE- Programm Version 7.0 durchgeführt[24]. Als Funktional wurde das parame- terfreie PBE0-Hybridfunktional benutzt[25],welches das PBE-Funktional[26]
mit einem HF-Austauschterm mit vordefinierten Koeffizienten erweitert. Als Basissatz wurde der TZVP-Basissatz[27] des Turbomole-Programms verwen- det. Das Konvergenzkriterium wurde auf 6 Nachkommastellen gelassen und m3 als Gridsize ausgewählt. Die Koordinaten der Moleküle für den Relaxed- Scan wurden mit TmoleX14[28] erstellt, dabei wurde der Diederwinkel zwi- schen C17-C18-C20-C21 (siehe Abb. 5) festgehalten. Nach jeder 10°-Drehung wurden Geometrie-Optimierungen und anschließend DFT/MRCI-Rechnungen durchgeführt
3.2 Optimierung der angeregten Zustände
Als Startgeometrien wurden die bereits DFT-optimierten Geometrien des Grundzustands genommen. Die Geometrie der angeregten Zustände wurden mit Hilfe von Turbomole und TDDFT optimiert (ohne festgehaltenem Win- kel), dabei wurden weder Funktional, Basissatz, Konvergenzkriterium noch Gridsize verändert. Die Koordinaten des Relaxed wurden ebenfalls dem opti- mierten Grundzustand entnommen, diesmal jene mit festgehaltenem Winkel.
Ein Relaxed-Scan wurde ebenfalls durchgeführt, mit dem Unterschied, dass nur einige ausgewählte Winkel genommen wurden.
3.3 DFT/MRCI
Als Koordinaten dienten die optimierten Koordinaten aus DFT bzw. TDDFT.
Auch hier wurden die selben Einstellungen wie zur Optimierung der Geome- trie bei Turbomole verwendet, außer dass diesmal das Hybridfunktional BH- LYP verwendet wurde. Zunächst wurde eine DSCF-Rechnung gestartet, um die Einteilchenbasis und die Orbitalenergien für die nachfolgende DFT/MRCI- Rechnung zu erhalten. Es wurden sowohl für die Grundzustandsgeometrie, als auch für die der angeregten Zustände die untersten 30 und die obersten 229 (al- le mit einer Energie>2Eh) Orbitale eingefroren. Gerechnet wurde mit einem ESEL-Wert von 0.8 Eh, der tight Parametereinstellung, sowie mit der neu- en Parametrisierung nach Lyskov[29] und mit 30 Singulett-/Triplett-Wurzeln.
Zuerst wurde manuell der aktive Raum bestimmt. Hierzu wurden aus den obersten, doppelbesetzten und den untersten, unbesetzten Orbitalen diejeni- gen ausgewählt, die eine ähnliche Energie besitzen . Nach dem ersten Durchlauf der Rechnung wurden aktive Räume von Singulett und Triplett angeglichen und eine zweite Rechnung gestartet.
3.4 COSMO
Die Lösungsmittel wurden unter anderem mithilfe des Conductor-Like Scree- ning Model (COSMO) berücksichtigt. Die benutzten Dielektrizitätskonstan- ten lauten: ε(H2O) = 78.3553, ε(n−Hexan) = 1.8819, ε(DM SO) = 46.826, ε(Ethylacetat) = 5.9867 [gaussian]. Nach Erzeugung der Kavität wurde eine DFT-Optimierung und anschließend eine DFT/MRCI-Optimierung durchge- führt.
3.5 PCM/cLR
Zusätzlich zu COSMO wurden die Lösungsmitteleffekte mit Hilfe des Polariz- able Continuum Model (PCM) und der corrected linear response-Methode mit einbezogen. Zunächst wurde der S1-Zustand des Moleküls in Lösung, ausge- hend von den Koordinaten der TDDFT-optimiertenS1-Struktur im Vakuum,
mittels TDDFT optimiert. Anschließend wurde das Lösungsmittel an diese Geometrie angepasst und daraufhin die vertikale Emission berechnet.Dies ent- spricht dem Grundzustand mit der f´’ur dieS1-Geometrie angepasste Lösungs- mittelumgebung. Zusätzlich wurden DFT/MRCI-Rechnungen nach den letz- ten beiden Schritten durchgeführt, dazu wurde das Lösungsmittel als Punkt- ladungen in die Rechnungen mit einbezogen. In Abbildung 4 ist eine einfache Veranschaulichung der Schritte dargestellt.
(a) 1. Schritt (b) 2. Schritt (c) 3. Schritt
Abbildung 4: Die einzelnen Schritte der PCM/cLR-Optimierung.
Schwarz = Molekül, Blau = Lösungsmittelumgebung des Grundzustands, Rot = Lösungsmittel umgebung des angereg- ten Zustands.
3.6 Diagramme und Abbildungen
Alle Diagramme wurden mit Hilfe von Gnuplot oder Excel erstellt. Abbildun- gen von Orbitalen, Differenzdichten und das Messen von Bindungslängen oder -winkel wurden mit Jmol erzeugt.
4 Ergebnisse
4.0.1 DFT-Optimierung des Grundzustandes
Die Geometrieoptimierung des Grundzustandes ergab folgende Struktur (. 5), in der alle Phenylringe zueinander verdreht sind.
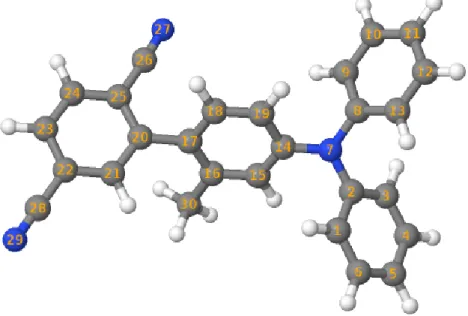
Abbildung 5: S0-Struktur der Methylverbindung mit Atomnummern. Koh- lenstoff in grau, Stickstoff in blau und Wasserstoff in weiß
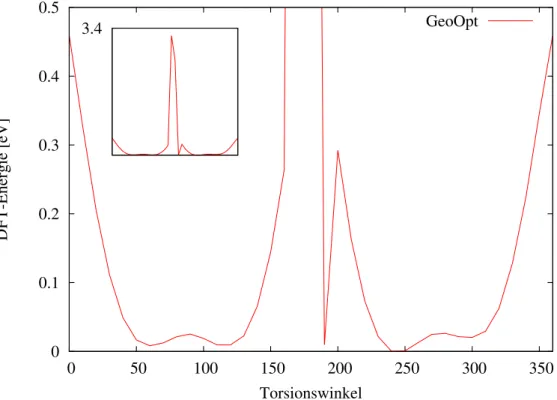
In Abb. 6 ist der Relaxed-Scan dargestellt, in der die DFT-Energien, bezogen auf das Minimum, gegen den Torsionswinkel aufgetragen sind. Das Bild im Bild oben links zeigt den gesamten Verlauf ohne Zoom. Auffällig ist der hohe Anstieg der Energie um ∼160° auf ca. 3.4 eV. Aufgrund der räumlichen Nähe des Substituenten Methylgruppe C30 und der Cyanogruppe C26N27, kommt es im Winkelbereich 160°-200° zur Ausbildung neuer Bindungen. Da diese neue Verindungen energetisch so hoch liegen, bilden sie eine Rotationsbarriere, die ein „Durchdrehen“ des Moleküls verhindern. Der größte energetische Abstand
im Winkelbereich 50°-130° bzw. 230°-310° ist mit ca. 0.02 eV bzw. 0.03 eV so gering, dass das Molekül diese Winkel thermisch erreichen kann und sich somit bevorzugt in diesen aufhalten wird. Um dies zu berücksichtigen, wur- den alle weiteren Rechnungen jeweils links und rechts der Barriere ausgeführt.
Die niedrigsten Energien des optimierten Grundzustandes liegen bei 60° mit -1203.488674Eh und bei 240° mit -1203.488875 Eh.
0 0.1 0.2 0.3 0.4 0.5
0 50 100 150 200 250 300 350
DFT-Energie [eV]
Torsionswinkel
GeoOpt 3.4
Abbildung 6: Relaxed-Scan an der Grundzustandsgeometrie. Aufgetragen sind die DFT-Energien in eV, bezogen auf das Minimum bei 240°, gegen den Torsionswinkel. Bild im Bild zeigt ungeschnit- tenen Verlauf.
Die Bindungslängen der Kohlenstoff-Kohlenstoff-Bindungen liegen alle im Be- reich von 0.139 nm, was der normalen Bindungssituation in Benzolringen entspricht[30]. Leichte Abweichungen zeigen die Atome C22 und C25 (C22- C23 0.14 nm, C25-C20 0.141 nm), da diese mit der Cyanogruppe verbunden sind und somit eine elektronenziehende Wirkung erfahren. Beide Minima sind in Bezug auf Bindungslängen und -winkel nahezu gleich (siehe Tab. 12 im An- hang).
Da es zwischen den Winkeln ca. 150°-200° zu starken Abweichungen, aufgrund
der neu gebildeten Bindungen kommt, wird dieser in allen folgenden Diagram- men weggelassen.
4.0.2 DFT/MRCI-Energien des Grundzustandes
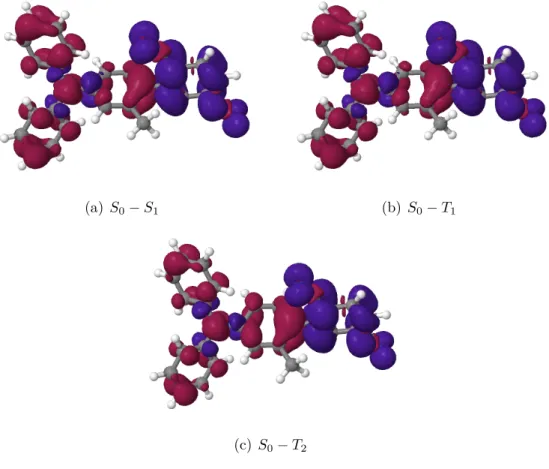
Anhand von Differenzdichten ist leicht zu erkennen, wo bei einer Anregung zwischen zwei Zuständen Elektronendichte abgezogen (rote Flächen) und wo Elektronendichte hinzukommt (blaue Flächen). Betrachtet man die Differenz- dichten bei 90° zwischenS0und dem jeweiligen angeregten und optimierten Zu- standS1,T1undT2, so wird deutlich, dass Elektronendichte am Triphenylamin- Rest verringert wird und im Terephthalonitril-Rest zunimmt (Abb. 7).
(a) S0−S1 (b)S0−T1
(c) S0−T2
Abbildung 7: Differenzdichten von a)S0−S1 an derS1-Geometrie, b) S0−T1 an derT1-Geometrie und c)S0 −T2 an der T2-Geometrie bei 90°.
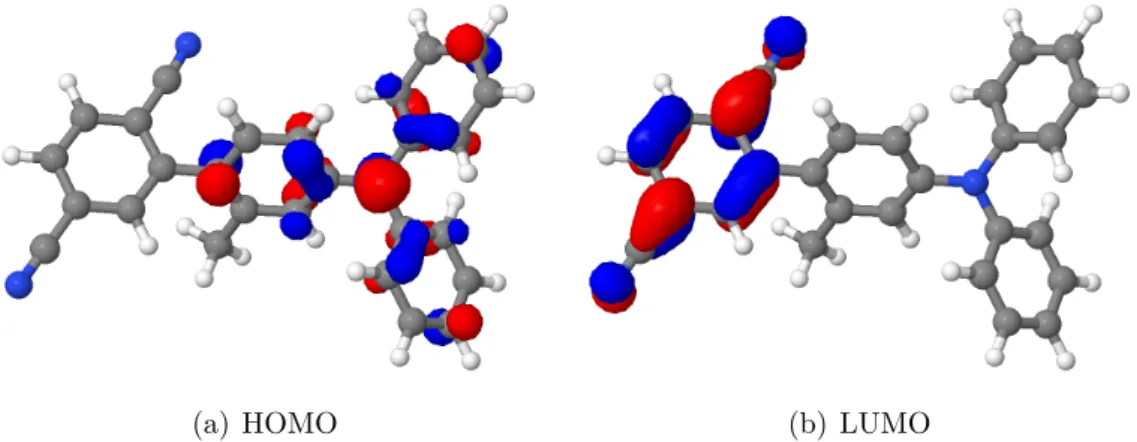
Vergleicht man diese Differenzdichten mit den Anregungen und den dazuge- hörigen Molekülorbitalen aus den DFT/MRCI-Rechnungen bei 90°, stellt man
fest, dass es sich hierbei hauptsächlich um HOMO-LUMO Übergänge handelt.
In Abb. 8 sind HOMO und LUMO im Grundzustand bei 90° gezeigt. Diese sind identisch mit denen aus den angeregten Zuständen bei 90°.
(a) HOMO (b) LUMO
Abbildung 8: Molekülorbitale von HOMO und LUMO an der Grundzu- standsgeometrie bei 90°.
Diese Separation von HOMO und LUMO führt zu einem gewollten Charge- Transfer-Charakter (CT-Charakter), welcher für TADF-Moleküle von Vorteil ist. Durch das Einstellen des Winkels zwischen Donor und Akzeptor kann so Einfluss auf den energetischen Abstand vonS1 und T1 genommen werden.
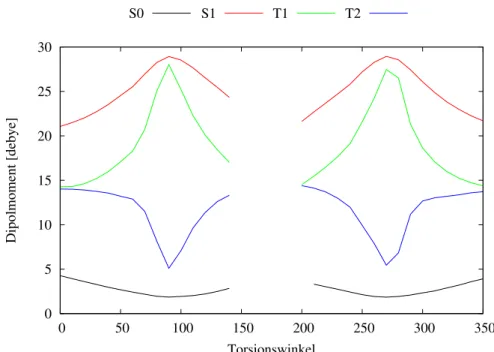
Ein Vergleich der Dipolmomente der jeweiligen Zustände an der Grundzu- standsgeometrie (siehe Abb. 9) bestätigt den CT-Charakter. Der Grundzu- stand hat ein sehr geringes Dipolmoment, wohingegen die angeregten Zustände viel größere Dipolmomente besitzen. Auffallend ist das gegenläufige Verhalten der T1- und T2-Dipolmomente.
0 5 10 15 20 25 30
0 50 100 150 200 250 300 350
Dipolmoment [debye]
Torsionswinkel
S0 S1 T1 T2
Abbildung 9: Die Dipolmomente der einzelnen Zustände an der Grundzu- standsgeometrie.
Betrachtet man die Anregungen desT1- bzw.T2-Zustandes vor und an 90°, er- kennt man, dass der T1-Zustand bei 50° mit 0.59 und 0.12 aus einem HOMO- LUMO- bzw. HOMO-LUMO+1 Übergang besteht. Das Molekülorbital LU- MO+1 hat Elektronendichte auf dem Methyl-substituierten Phenylring des Triphenylamin-Restes, was bedeutet, dass der T1-Zustand neben einer CT- Anregung auch Anteile einer lokalen Anregung besitzt. Bei 90° besteht dieser jedoch zu 0.85 aus dem HOMO-LUMO-Übergang. Dieser Wechsel von einem gemischten Zustand in einen reinen CT-Zustand erklärt den starken Anstieg des Dipolmomentes bei 90°.
Der T2-Zustand ist vor 90° ebenfalls gemischt, aber bei 90° dominieren die lokalen Anregungen. Anhand der Differenzdichten in Abb. 10 und 11 ist der Wechsel der einzelnen Zustände gut zu erkennen.
(a) 50° (b) 90°
Abbildung 10: Differenzdichte zwischenT1 und S0 bei a) 50° und b) 90° im Grundzustand.
(a) 50° (b) 90°
Abbildung 11: Differenzdichte zwischenT2 und S0 bei a) 50° und b) 90° im Grundzustand.
4.1 Optimierung der angeregten Zustände im Vakuum
Die Geometrien der angeregten Zustände wurden mittels TDDFT optimiert.
Die daraus erhaltenen Koordinaten wurden für DFT/MRCI-Rechnungen wei- ter verwendet.
4.1.1 Vergleich der optimierten Geometrien im Vakuum
Zunächst wurden die Geometrien der angeregten Zustände mit TDDFT op- timiert, ohne dass der Diederwinkel festgehalten wurde. Dazu wurden Start- koordinaten mit einem Diederwinkel von 60° und 270° erstellt, um so Opti- mierungen links und rechts der Rotationsbarriere durchzuführen. Als Minima wurden folgende Diederwinkel erhalten:
Tabelle 1: Diederwinkel der Minima der optimierten Geometrien links und rechts der Barriere.
S0 S1 T1 T2 links 60° 101° 41° 91°
rechts 240° 250° 236° 270°
Von den Startkoordinaten aus hat sich derT1-Zustand am stärksten verändert.
Die Unterschiede der Bindungslängen und -winkel der Minima links und rechts eines Zustands sind sehr gering, weshalb Vergleiche nur für eins der Minima erfolgen.
Wie zu erwarten sind die Unterschiede zwischen den Geometrieparametern des Grundzustands und de angeregten Zustände sehr groß. Am stärksten be- troffen sind die Bindungen im Terephthalonitril-Rest, was aufgrund des CT- Charakters zu erwarten war. In Tabelle 2 sind die Änderungen der Bindungs- längen im Terephthalonitril-Rest aufgeführt.
Tabelle 2: Unterschiede der Bindungslängen und -winkel zwischen den opti- miertenS1-, T1-, T2-Zustand und dem Grundzustand des Tereph- thalonitril-Restes links der Barriere.
Atom S1-S0[pm] T1-S0[pm] T2-S0[pm]
C20-C21 -3 -4 0
C21-C22 4 0 -4
C22-C23 3 0 7
C23-C24 -2 4 7
C24-C25 3 -2 -4
C25-C20 3 2 7
C25-C26 -3 6 7
C26-N27 2 -3 -5
C22-C28 -3 1 2
C28-N29 1 -2 -5
Es sind große Veränderungen sichtbar, vorallem an der optimiertenT2-Geometrie mit bis zu 7 pm. Um diese Unterschiede nachzuvollziehen muss man sich die Molekülorbital der Zustände an ihren Minima betrachten. So entspricht der S1-Zustand bei 100° einem HOMO→LUMO-Übergang wie sie bereits in Ab- bildung 8 zu sehen sind. Leicht zu erkennen ist, dass die Bindungen an denen sich Dichte im LUMO befindet, kürzer und die Bindungen an denen sich Kno- ten befinden, länger geworden sind.
4.1.2 TDDFT-Optimierung der angeregten Zustände im Vakuum
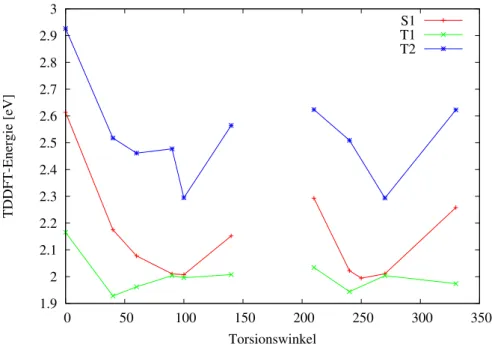
Abbildung 12 zeigt einen Relaxed-Scan der angeregten Zustände, d.h. an je- dem Schritt wurde der Diederwinkel festgehalten. Die Energien wurden auf das S0-Minimum der TDDFT-Rechnungen bezogen und gegen den Torisons- winkel aufgetragen. Die Makierungen in den Kurven repräsentieren die Winkel an denen Rechnungen durchgeführt wurden.
1.9 2 2.1 2.2 2.3 2.4 2.5 2.6 2.7 2.8 2.9 3
0 50 100 150 200 250 300 350
TDDFT-Energie [eV]
Torsionswinkel
S1 T1 T2
Abbildung 12: Die TDDFT-Energien in eV der optimierten angeregten Zustände, bezogen auf das S0-Minimum der TDDFT- Rechnungen, bei festgehaltenem Diederwinkel.
Der Verlauf, insbesondere der starke Abfall der Energie bei 100° an der T2- Geometrie ist unüblich für eine Geometrieoptimierung. Die Zusammensetzung der einzelnen Zustände an den jeweiligen Winkeln könnte Rückschlüsse liefern.
Die S1-Zustände an der S1-Geometrie sind in Tabelle 3 nicht aufgeführt, da diese an jedem Punkt aus einer H→L-Anregung bestehen. Die T1-Zustände an der T1-Geometrie besitzen oft einen CT-Charakter, welche vereinzelt mit anderen Anregungen gemischt werden. Diese tragen jedoch nur einen kleinen Anteil der Zustände bei. Bei 90 °, 100° und 270° ist derT1-Zustand nahezu ein reiner CT-Zustand.
Die T2-Zustände an der T2-Geometrie sind sehr gemischt. An den meisten Winkeln sind es die Übergänge H→L+1 und H→L aus denen der T2-Zustand besteht. Bei 100° und 270°, dominieren aber plötzlich lokale Anregungen auf dem Terephthalonitril-Rest. Das bedeutet an diesen Winkeln ist ein, sonst hö- her liegender Zustand energetisch unter denT2-Zustand gefallen, sodass dieser fälschlicherweise alsT2-Zustand angeommen und optimiert wurde.
Tabelle 3: Zusammensetzung der Übergänge und ihre Charakter der
TDDFT-optimierten Zustände an der T1- bzw. T2-Geometrie bei festgehaltenem Diederwinkel. (LA = Lokale Anregung;
TPA = Triphenylamin-Rest; TPN = Terephthalonitril-Rest;
CT = Charge-Transfer) T1-Zustand an der T1-Geometrie
T2-Zustand an der T2-Geometrie Übergang
(Koeff.) Charakter Übergang
(Koeff.) Charakter
40° H→L (76) CT H→L+1 (54) gemischt
H-1→L (10) gemischt H→L (26) CT
60°
H→L (84) CT H→L+1 (54) gemischt
H→L (19) CT
H→L+3 (13) gemischt
90° H→L (99) CT H→L+2 (55) gemischt
H→L+1 (26) LA TPA
100° H→L (99) CT H-4→L (63) LA TPN
H-6→L (14) CT
140° H→L (79) CT H→L+1 (50) gemischt
H→L (22) CT
210° H→L (75) CT H→L+1 (44) gemischt
H-1→L (10) gemischt H→L (27) CT
240°
H→L (90) CT H→L+1 (53) gemischt
H→L (15) CT
H→L+3 (15) gemischt
270° H→L (99) CT H-4→L (91) LA TPN
330° H→L (73) CT H→L+1 (53) gemischt
H-1→L (13) LA TPA H→L (27) CT
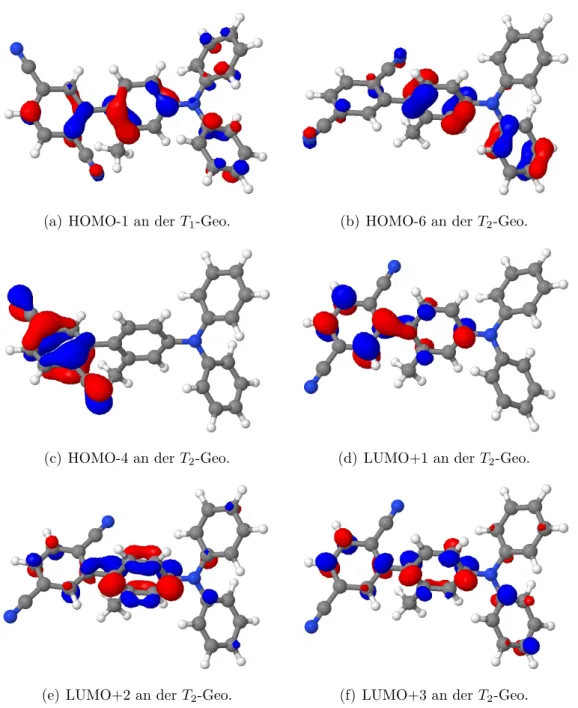
Die Molekülorbital von HOMO und LUMO sehen aus wie an der Grundzu- standsgeometrie (siehe Abb. 8), alle weiteren sind in Abbildung 13 darge- stellt.
(a) HOMO-1 an derT1-Geo. (b) HOMO-6 an derT2-Geo.
(c) HOMO-4 an der T2-Geo. (d) LUMO+1 an derT2-Geo.
(e) LUMO+2 an derT2-Geo. (f) LUMO+3 an derT2-Geo.
Abbildung 13: Alle Molekülorbitale, die an den Übergängen aus Tabelle 3 beteiligt sind.
In den Abbildungen 14, 15 und 16 sind die DFTCI-Energien an den optimierten S1-, T1- und T2-Geometrien, bezogen auf das S0-Minimum am Grundzustand dargstellt. Die untersten Zustände sind S1 und T1, welche sich bei 90°, 100°
und 270° sehr nahe kommen. Mit etwas größerem Abstand folgen dann die weiteren Triplett-Zustände.
2.8 3 3.2 3.4 3.6 3.8 4 4.2 4.4
0 50 100 150 200 250 300 350
DFTCI-Energie [eV]
Torsionswinkel
S1 T1 T2 T3 T4 T5
Abbildung 14: DFTCI-Energien der jeweiligen Zustände an der optimierten S1-Geometrie bezogen auf S0 der Grundzustandsgeometrie.
Die Abhängigkeit der Singulett-Triplett-Aufspaltung ∆EST vom Torsionswin- kel wird in Abbildung 15 nochmal deutlich. ∆EST ist klein, wenn Akzeptor und Donor im rechten Winkel zu einander stehen und groß, wenn sie in einer Ebene sind.
Dieser Abstand ist für einen TADF-Emitter sehr wichtig, da der reverse-ISC- Prozess mit steigendem ∆EST immer unwahrscheinlicher wird. Ab einer Ener- gie von ca. ∆EST<0.1 eV[31] tritt rISC verstärkt auf. Berechnet man ∆EST mit den Energien aus den jeweiligen optimierten Zuständen, so erhält man einen Abstand von 0.062 eV bei 90° und 0.059 eV bei 270°.
2.4 2.6 2.8 3 3.2 3.4 3.6 3.8 4 4.2 4.4
0 50 100 150 200 250 300 350
DFTCI-Energie [eV]
Torsionswinkel
S1 T1 T2 T3 T4 T5
Abbildung 15: DFTCI-Energien der jeweiligen Zustände an der optimierten T1-Geometrie bezogen aufS0 der Grundzustandsgeometrie.
Der Verlauf der DFTCI an der T2-Geometrie (Abb. 16) weicht stark von den vorherigen DFTCI-Diagrammen ab. Zum einen sind alle Energien um ca. 0.2 eV größer und zum anderen liegen S1 und T1 energetisch sehr nah.
2.8 3 3.2 3.4 3.6 3.8 4 4.2 4.4 4.6
0 50 100 150 200 250 300 350
DFTCI-Energie [eV]
Torsionswinkel
S1 T1 T2 T3 T4 T5
Abbildung 16: DFTCI-Energien der jeweiligen Zustände an der optimierten T2-Geometrie bezogen auf S0 der Grundzustandsgeometrie.
In Tabelle 4 sind alle Übergänge, aus denen die Zustände T1 bzw. T2 an der T2-Geometrie an den verschiedenen Winkeln bestehen, aufgelistet.
Tabelle 4: Zusammensetzung und Charakter der DFT/MRCI-optimierten Zustände T1 bzw. T2 an der T2-Geometrie bei festgehaltenem Die- derwinkel. (LA = Lokale Anregung; TPA = Triphenylamin-Rest;
TPN = Terephthalonitril-Rest; CT = Charge-Transfer) T1-Zustand
an der T2-Geometrie
T2-Zustand an der T2-Geometrie Übergang
(Anteil) Charakter Übergang
(Anteil) Charakter
40° H→L (0.48) CT H→L+1 (0.42) gemischt
H→L+1 (0.25) gemischt H→L (0.28) CT
60° H→L (0.37) CT H→L (0.43) CT
H→L+1 (0.32) gemischt H→L+1 (0.27) gemischt
90° H→L+2 (0.44) LA TPA H→L (0.85) CT
H→L+1 (0.33) gemischt
100° H-3→L (0.48) CT H→L (0.83) CT
H-4→L (0.34) CT
140° H→L (0.54) CT H→L+1 (0.45) gemischt
H→L+1 (0.19) gemischt H→L (0.23) CT
210° H→L (0.54) CT H→L+1 (0.47) gemischt
H→L+1 (0.21) gemischt H→L (0.21) CT 240°
H→L (0.52) CT H→L+1 (0.37) gemischt
H→L+1 (0.19) gemischt H→L (0.28) CT H→L+3 (0.13) gemischt
270° H-3→L (0.6) CT H→L (0.57) CT
H→L (0.29) CT H-3→L (0.32) LA TPN
330° H→L (0.51) CT H→L+1 (0.46) gemischt
H→L+1 (0.22) gemischt H→L (0.23) CT
An der T2-Geometrie ist der T1-Zustand gemischter als an der optimierten T1-Geometrie. Auffallend sind die beiden lokalen Anregungen bei 90° im T1- Zustand und bei 270° imT2-Zustand. Die Übergänge beider Zustände sind an fast jedem Winkel umgedreht. In Abbildung 17 sind alle an den Übergängen beteiligten Molekülorbitale dargestellt. Alle bereits dargestellten Molekülorbi- tale, wurden der Übersicht halber weggelassen. Die Molekülorbitale wurden an jedem Winkel miteinanader verglichen um sicherzustellen, dass sie an jedem
Winkel gleich sind. Daher wird jeweils beispielhaft ein Molekülorbital an einem beliebigen Winkel dargstellt.
(a) HOMO-3 an derT1 (b) HOMO-4 an derT1
(c) LUMO+1 an derT1 (d) LUMO+2 an der T1
(e) HOMO-3 an derT2
Abbildung 17: Alle Molekülorbitale, die an den Übergängen aus Tabelle 4 beteiligt sind.
HOMO-3 war in der TDDFT-Optimierung HOMO-4 und hat somit seine ener- getische Lage verändert. Um einen besseren Einblick zu erhalten werden in Abbildung 18 die energetischen Lagen der einzelnen Zustände am jeweiligen Minimum der optimierten Geometrien links der Rotationsbarriere, relativ zum
![Abbildung 2: Vereinfachte Darstellung der im TADF-Mechanismus vorkom- vorkom-menden Zustände und Ratenkonstanten[11].](https://thumb-eu.123doks.com/thumbv2/1library_info/4532311.1596360/18.892.175.735.214.537/abbildung-vereinfachte-darstellung-mechanismus-vorkom-vorkom-zustände-ratenkonstanten.webp)
![Abbildung 3: Schematische Darstellung der einzelnen Schritte zur Berech- Berech-nung von Absorption und Fluoreszenz mittels cLR.[22]](https://thumb-eu.123doks.com/thumbv2/1library_info/4532311.1596360/28.892.279.619.121.499/abbildung-schematische-darstellung-einzelnen-schritte-berech-absorption-fluoreszenz.webp)