Generierung und Charakterisierung von konditionalen knock-outs des Spleißfaktors Sfrs10 im Mausmodell
213
0
0
Volltext
(2)(3)
(4)(5)
(6)(7)
(8)
(9)
(10)
(11)
(12)(13)
(14)
(15)
(17)
(18)
(20)
(21)
(22)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(33)
(34)
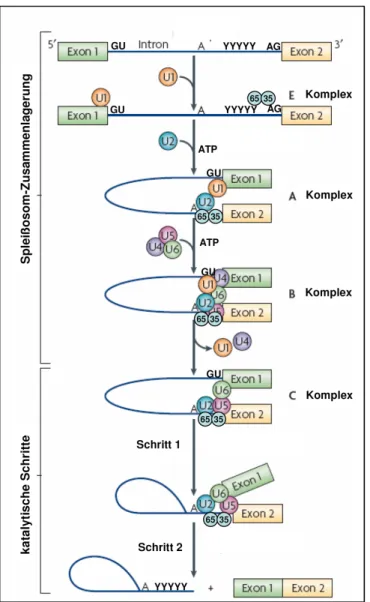
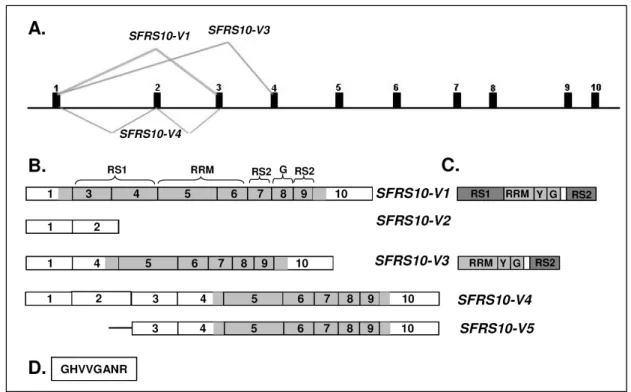
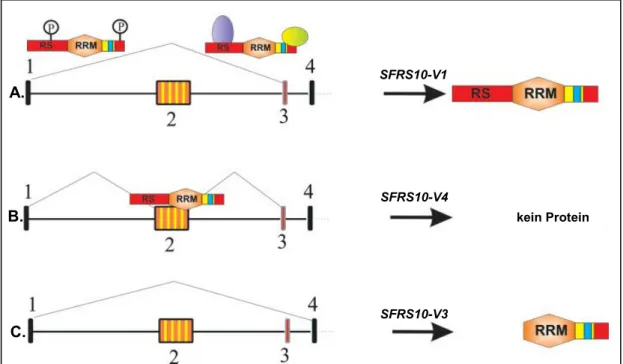
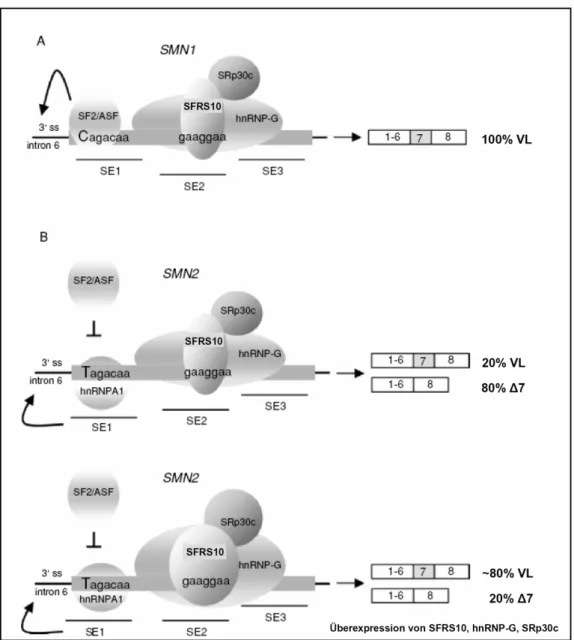
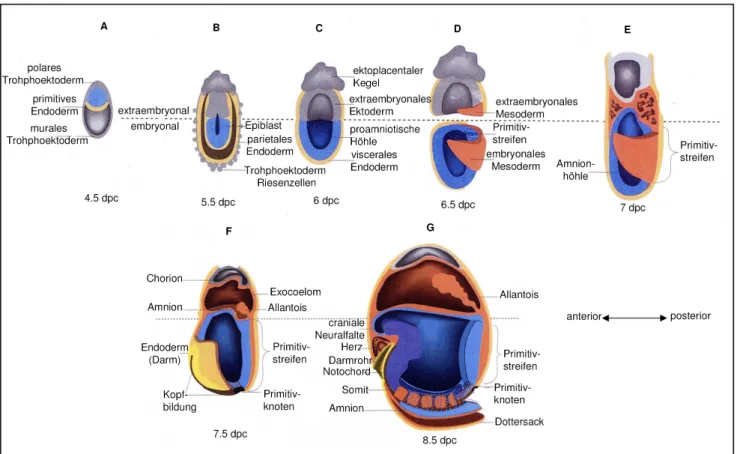
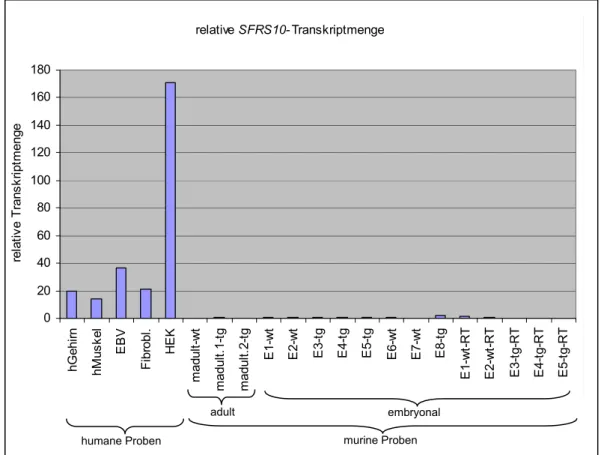
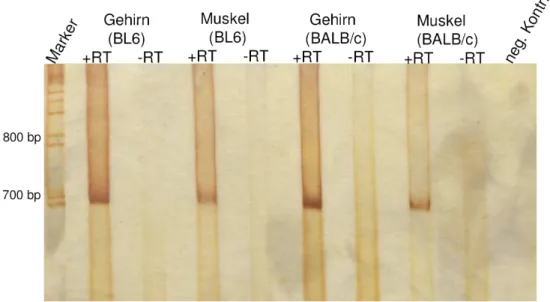
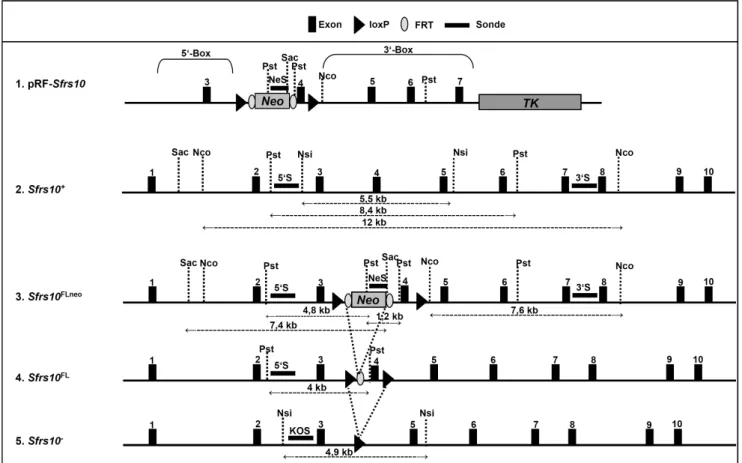
Abbildung

+7

ÄHNLICHE DOKUMENTE
[r]
‘ eines Dihe'xaeders der Hauptreihe entweder sechsfl. derEndecken, oder Zusch. der schärferen Endkanten, oder vierfl. der spitzeren Seitenecken, die Zusp. auf die Fl. aufgesetzt; \
Summ.: On macroscopic changes in the organs of thy- roidectomized guinea pigs in experimental tuberculosis... Serum proteinogram as the index of the reactivity of the
Für die Aufreinigung von T-Zellen aus der Milz wurde das entnommene Organ zunächst steril über ein Zellsieb (Cell Strainer 100µm, BD Falcon) in einer Petrischale (Easy Grip
N¨ahere das Fl¨achenst¨uck durch ein Parallelo- gramm mit etwa gleicher Fl¨ache, entnimm der Karte die entsprechenden Maße, rechne im Maßstab um und berechne damit die
Zeichne auf ein kariertes Papier einen Kreis mit Radius 3,5 cm und bestimme damit n¨aherungsweise (ohne eine Fl¨achenformel f ¨ur Kreisfl¨achen) den Fl¨acheninhalt des
Quelle: VOGIS - Daten Politische Grenzen: BEV, Wien. Abb.: Gauß-Krüger (3° breite Meridianstreifen),
bei der Kassenverwaltung. so dürfte dieser Einnahme Posten für das Jahr 1882 auf dieselbe Summe zu veranschlage» sein. Zufolge einer ministeriellen Verordnung vom 15. September
