Generierung und Charakterisierung von konditionalen Bdp1-knockout-Mausmodellen - Analysen des Transkriptionsfaktor IIIB-Komplexes
204
0
0
Volltext
(2)(3)
(4)(5)
(6)(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
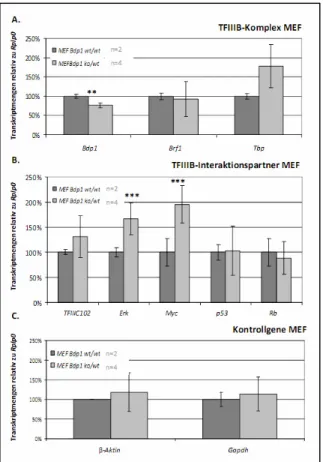
Abbildung

+7
ÄHNLICHE DOKUMENTE
49 Abbildung 10: Immunfluoreszenzdoppelfärbung an Paraffinschnitten von Wildtypnieren nach einer Woche LS/E-Diät: Immunreaktionen Renin (grün), Glattmuskelaktin (rot),
Für eine Amplifikation einer für das AP1S1-(-)-Allel spezifischen DNA-Sequenz wurde der Reverse-Primer s1aR1 nahe des 5’ Ende des inserierten Gene Trap- Vektors
Für Teilnehmende, die ausschließlich eine Maßnahme nach dem Förderbaustein 1 besuchen o- der die sozialversicherungspflichtig beschäftigt sind, wird keine Pauschale für
Anhand dieser Überlegungen und der vorgestellten Ergebnisse kann folgendes Model für das Zusammenspiel der beiden Protein CYP81F2 und PEN2 in der Nichtwirtsresistenz
Eigenschaften holomorpher Funktionen 93 3.1 Satz von LIOUVILLE, Fundamentalsatz
In dem Vortrag werden grundlegende Informationen für die Arbeit mit der Zielgruppe der Geduldeten und Gestatteten im Rahmen von “Durchstarten in Ausbildung und Arbeit”
(3) Das Teilhabemanagement im Rahmen der Initiative „Gemeinsam klappt’s“ startete vor Beginn der Initiative „Durchstarten in Ausbildung und Arbeit“. Aus diesem Grund werden
sind bereits Netzwerk- und Kooperationsstrukturen wie beispielsweise die Bündniskerngruppe etab- liert, welche von den Trägern der Innovationsprojekte zur Gewinnung und
