Quantenchemische Charakterisierung der photophysikalischen Eigenschaften des
Zn(dtsq)(batho)-Komplexes
Institut für Theoretische Chemie und Computerchemie Mathematisch-Naturwissenschaftliche Fakultät
Heinrich-Heine-Universität Düsseldorf
Abschlussarbeit
zur Erlangung des akademischen Grades Bachelor of Science (B. Sc.)
vorgelegt von Markus Putscher
August 2020
Erstprüferin: Prof. Dr. Christel M. Marian Zweitprüfer: Prof. Dr. Christian Ganter
Eidesstattliche Erklärung
Hiermit erkläre ich an Eides statt, dass ich die vorliegende Arbeit mit dem Titel "Quanten- chemische Charakterisierung der photophysikalischen Eigenschaften des Zn(dtsq)(batho)- Komplexes" selbstständig und nur mit den angegebenen Quellen erstellt habe.
Wörtliche oder inhaltliche Stellen sowie Abbildungen und Formeln, die aus anderen Wer- ken entnommen worden sind, sind als solche gekennzeichnet.
(Markus Putscher)
Düsseldorf, 30 August 2020
Danksagung
Bedanken möchte ich mich im Zuge dieser Arbeit zuallererst bei Prof. Dr. Christel Mari- an. Vielen Dank für die Vorbereitung dieses interessanten Themas meiner Bachelorarbeit, sowie die Hilfe bei Problemen und Fragen während der Bearbeitung. Trotz der erschwer- ten Umstände durch die SARS-CoV-2-Pandemie konnte ein reibungsloser, sicherer und angenehmer Ablauf der Arbeit gewährleistet werden.
Auch bei Professor Ganter bedanke ich mich für die Übernahme der Zweitkorrektur.
Für das Gelingen dieser Arbeit sind ebenfalls die Mitarbeiterinnen und Mitarbeiter des Ar- beitskreises der Theoretischen Chemie und Computerchemie verantwortlich. Vielen Dank auch an euch für die Beantwortung all meiner Fragen und umgehende Hilfe bei sämtlichen Problemen.
Zuletzt gilt mein Dank meiner Familie, meiner Freundin Emma und meinen Freunden, die mich auf dem gesamten Weg meines bisherigen Studiums begleitet haben. Ohne eure Unterstützung wäre alles so nicht ansatzweise möglich gewesen.
Inhaltsverzeichnis 1
Inhaltsverzeichnis
1 Abkürzungsverzeichnis 3
2 Einleitung 5
3 Theorie 9
3.1 Lumineszenz . . . 9
3.2 Franck-Condon-Prinzip . . . 10
3.3 Dichtefunktionaltheorie . . . 10
3.4 Multireferenzkonfigurationswechselwirkung . . . 13
3.5 DFT/MRCI . . . 13
3.6 TDDFT . . . 14
3.7 Lösungsmitteleffekte . . . 14
4 Technische Details der Rechnungen 17 5 Ergebnisse und Auswertung 19 5.1 Grundzustand . . . 19
5.1.1 Geometrie . . . 19
5.1.2 Anregung und Absorption . . . 21
5.2 Geometrien der angeregten Zustände . . . 24
5.3 Adiabatische Anregungsenergien . . . 27
5.3.1 Vakuum . . . 27
5.3.2 PCM . . . 28
5.3.3 PCM+EtOH . . . 29
5.4 Emissionsspektren . . . 32
5.4.1 Raumtemperatur . . . 32
5.4.2 77 Kelvin . . . 33
6 Fazit 37 7 Literatur 39 8 Anhang 41 8.1 Bindungslängen und -winkel . . . 41
8.2 Absorptionsspektrum . . . 47
8.3 DFT/MRCI-Rechnung . . . 48
3
1 Abkürzungsverzeichnis
batho Bathocuproin
BH-LYP Becke-Half-and-Half-Lee-Yang-Parr CI Konfigurationswechselwirkung COSMO Conductor-like Screening Model DFT Dichtefunktionaltheorie
dtsq Dithioquadratsäure EtOH Ethanol
ECP Effektives Kernpotential
GGA Verallgemeinerte Gradientennäherung HF Hartree-Fock
HOMO Höchstes besetztes Molekülorbital LCD Flüssigkeitsbildschirm
LDA Lokale Dichtenäherung LED Leuchtdiode
LLCT Ligand-zu-Ligand-Charge-Tranfser LUMO Niedrigstes unbesetztes Molekülorbital MRCI Multireferenzkonfigurationswechselwirkung OLED Organische Leuchtdiode
PCM Polarisierbares Kontinuumsmodel RISC Reverse Intersystem Crossing
TADF Thermisch aktivierte verzögerte Fluoreszenz TDDFT Zeitabhängige Dichtefunktionaltheorie TTA Triplett-Triplett-Auslöschung
5
2 Einleitung
Organische Leuchtdioden (OLED) sind dünne, selbstleuchtende Bauelemente, die über elektrische Anregung geeigneter Emittermaterialien und anschließender Emission farbiges Licht abgeben. Besonders für die Verwendung in Displays von Smartphones oder Fern- sehern und in der Beleuchtungstechnik werden sie immer beliebter. Dies liegt vor allem daran, dass OLEDs keine Hintergrundstrahlung benötigen, stromsparender sind und über ein höheres Kontrastverhältnis und eine höhere Auflösung im Vergleich zu herkömmlichen Leuchtdioden (LED) oder Flüssigkristallbildschirmen (LCD)verfügen. Zudem können sie auf transparenten und formbaren Oberflächen angebracht werden, was beispielsweise eine Faltbarkeit von Displays oder die Beleuchtbarkeit von Fensterscheiben ermöglicht.1 Zentrales Element der OLED-Forschung ist eine Effizienzsteigerung der Lichtausbeute.
Diese ist unter anderem mit der inneren Quantenausbeute verknüpft, welche von der Ver- wertbarkeit der in OLEDs entstehenden Exzitonen abhängt. Die Exzitonen entstehen in den OLEDs, indem Löcher und überschüssige Elektronen durch Anode und Kathode inji- ziert werden und ein gebundenes Elektron-Loch-Paar gebildet wird. Aufgrund des Spins der Elektronen können sie in Singulett- und Triplett-Exzitonen unterteilt werden, die im Verhältnis 1:3 auftreten. Triplett-Exzitonen zerfallen im Regelfall strahlungslos und nur bei Singulett-Exzitonen entsteht sichtbares Licht durch Fluoreszenz.2 Können nur die Singulett-Exzitonen zur Erzeugung von Licht genutzt werden, ist die Effizienz der organi- schen Leuchtdiode stark limitiert, da eine Quantenausbeute von 25 % nicht überschritten werden kann.
Um den Energieverlust in OLEDs durch den strahlungslosen Zerfall der Triplett-Exzito- nen zu minimieren, ist es von großem Interesse, Triplett-Exzitonen in Singulett-Exzitonen umwandeln zu können. Diese Umwandlung kann über zwei verschiedene Mechanismen er- folgen. Zum einen kann es zur einer Triplett-Triplett-Auslöschung (TTA) kommen, bei der zwei Triplett-Exzitonen zusammenstoßen und sich gegenseitig auslöschen. Dabei entsteht ein Teilchen im S0-Zustand und ein weiteres, welches in den Sn-Zustand angeregt wird und daraufhin in den S1-Zustand relaxiert. Dadurch kann die Quantenausbeute auf bis zu 62,5 % erhöht werden. Der zweite Mechanismus ist das sogenannte thermisch aktivierte reverse Intersystem Crossing (RISC). Dabei erfolgt ein Übergang vom Triplett-Zustand in den Singulett-Zustand unter Spinumkehr. Knüpft im Anschluss an das thermisch aktivier- te RISC eine Energieabgabe in Form von Fluoreszenz an, wird dieser Prozess als thermisch aktivierte verzögerte Fluoreszenz (TADF) bezeichnet. Moleküle, in denen dieser Vorgang beobachtbar ist, werden TADF-Emitter genannt. Durch diesen Prozess ist eine Quanten- ausbeute von 100 % möglich. Die dadurch mögliche Effizienz macht TADF-Emitter zu vielversprechenden, forschungsrelevanten Molekülen.3
Von ähnlicher Relevanz sind phosphoreszente Emitter, welche ebenfalls eine interne Quan-
6 2 Einleitung tenausbeute von 100 % vorweisen können. Bei diesen Emittern handelt es sich meist um metallorganische Komplexe. Aufgrund geeigneter photophysikalischer Eigenschaften ist hier ein quantenchemisch verbotener, strahlungsreicher Übergang aus demT1-Zustand in denS0-Zustand der bevorzugt ablaufende Prozess. Phosphoreszente Emitter bilden dabei eine Ausnahme, indem nicht nur Singulett-Exzitonen strahlungsreich in den Grundzu- stand übergehen, sondern auch die Triplett-Exzitonen durch Phosphoreszenz direkt zur Umwandlung von Energie in Licht beitragen.4
Als Emittermaterialien für OLEDs werden häufig Iridium- oder Platin-Komplexe ver- wendet. Probleme bereiten hier jedoch solche Emitter, die blaues Licht produzieren. Sie sind gegenüber den roten und grünen Emittern deutlich weniger stabil und effizient. Einen großen Einfluss auf die Verringerung der Effizienz können metallzentrierte dd*-Übergänge haben. Sie rufen strahlungslose Desaktivierungsprozesse hervor, die die interne Quanten- ausbeute veringern.5 Derartige Probleme treten bei d10-Übergangsmetallen nicht auf, da sie keine niederenergetischen metallzentrierten dd*-Übergänge besitzen. Aus diesem Grund wurden für die OLED-Forschung vor einigen Jahren Kufer(I)-Komplexe interessant. Sie bieten nicht nur eine kostengünstigere Alternative gegenüber den selten vorkommenden Elementen Iridium und Platin, sondern bringen auch aufgrund einer kleinen Energielücke zwischen dem niedrigsten Singulett- und Triplett-Zustand geeignete photophysikalische Eigenschaften für TADF-Emitter mit.6 Aber auch andered10-Elemente, die ein ähnliches Verhalten zeigen, werden für die OLED-Forschung immer interessanter. Dabei zeigen vor allem Gold(I), Silber(I), Cadmium(II) und Zink(II) vielversprechende Eigenschaften. Bei den Zink-Komplexen wird zur Zeit nach solchen gesucht, die phosphoreszente Strahlung emittieren.7
Für einen solchen phosphoreszierenden Emitter könnte der in der vorliegenden Arbeit untersuchte Zink-Komplex in Frage kommen. Die beiden Liganden des Zinks sind dabei eine Dithioquadratsäure und ein Bathocuproin. Die genaue Struktur ist Abbildung 1 zu entnehmen. Synthetisiert und untersucht wurde das Molekül erstmals von P. J. Gronlund et al..8 Die heute bekannte Kristallstruktur konnte mittels einer Röntgenstrukturanalyse bestimmt werden. Es zeigte sich, dass im Kristall das Molekül im Verhältnis 1:1 mit einem wasserstoffbrückengedundenem Ethanolmolekül vorliegt. Zusätzlich zur Strukturanalyse erstellten die Autoren Emissionspektren des Moleküls. Bei Raumtemperatur zeigt sich ei- ne breite, strukturlose Bande im Bereich von 500 nm, die von den Autoren des Artikels einem Ligand-zu-Ligand-Charge-Transfer (LLCT) zugeordnet wird. Bei tieferer Tempera- tur (77 K) erweitert sich die Emission zu einer dualen Lumineszenz. Hierbei postulieren die Autoren, dass zusätzlich zum LLCT-Übergang auch ein lokaler 3ππ∗-Übergang auf dem Bathocuproin-Liganden stattfindet, dessen Emission im Spektrum sichtbar wird. Im Emissionsspektrum sind nun Schwingungsstrukturen zu erkennen. Es zeichnen sich drei scharfe Banden ab, die ebenfalls im Bereich von 500 nm liegen und die breite Bande des
7
vermuteten LLCT-Übergangs überlagern.
Im Rahmen dieser Arbeit sollen diese photophysikalischen Eigenschaften des Zn(dtsa)- (batho)-Komplexes quantenchemisch untersucht und charakterisiert werden. Dafür werden Berechnungen des Moleküls im Vakuum, in Lösemittelumgebung mit dem polarisierba- ren Kontinuumsmodell (PCM) und mit einem expliziten Ethanol-Molekül zusätzlich zur Lösemittelumgebung mit PCM für die niedrig liegenden elektronisch angeregten Zustän- de durchgeführt. Zur Charakterisierung des 3ππ∗- und LLCT-Zustandes werden Franck- Condon-Emissionsspektren erstellt.
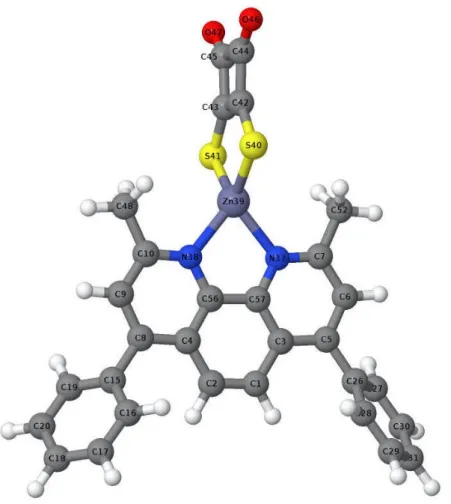
Abb. 2.1: Struktur des Zn(dtsq)(batho)-Komplexes.
9
3 Theorie
3.1 Lumineszenz
Lumineszenz beschreibt einen komplexen Vorgang, der sich durch Emission sichtbaren Lichts nach erfolgter Elektronenanregung kennzeichnet. Er kann aufgeteilt werden in den Prozess der Fluoreszenz (Relaxation vonS1zuS0) und Phosphoreszenz (Relaxation vonT1 zuS0). Der Ablauf von Absorption bis Emission kann gut über das sogenannte Jablonski- Schema dargestellt werden.
Abb. 3.1: Diese Abbildung zeigt ein Jablonski-Schema, welches im nachfolgenden Textab- schnitt erläutert wird. Erstellt wurde es von Prof. Dr. Ziegenbalg.9
Die Einleitung des Vorgangs erfolgt dabei über die im Schema grün eingezeichnete Ab- sorption. Im Fall der Photolumineszenz trifft ein Photon auf ein Elektron und regt dieses in einen energetisch höher liegenden Zustand an. Dabei ist im Schema nur die Anregung in einen höherliegenden Singulett-Zustand eingezeichnet, da die Anregung unter Spinumkehr deutlich unwahrscheinlicher ist (ausgenommen bei großer Spin-Bahn-Kopplung) und Sys- teme im Grundzustand meist mit Singulett-Multiplizität vorliegen.
Das System ist nun bestrebt, wieder in den energetisch günstigsten Zustand zurück zu fallen. Dabei findet zunächst eine Schwingungsrelaxation (SR) zur Nullpunktsschwingung
10 3 Theorie desS1-Zustandes statt. Dieser Vorgang läuft im Normalfall so schnell ab, dass keine Kon- kurrenzprozesse relevant sind. Das beschriebene Verhalten wird durch das photochemische Dogma, auch Kasha-Regel genannt, zusammengefasst. Hierbei ist es nicht von Bedeutung, wie hoch der angeregten Zustand des Systems liegt, aus dem zur Nullpunktsschwingung desS1-Zustandes relaxiert wird.
Ausgehend vomS1-Zustand können nun verschiedene Prozesse ablaufen. Eine Möglichkeit ist die in der Abbildung 3.1 rot eingezeichnete Fluoreszenz (F). Dabei wird Energie in Form eines Photons abgeben, um in den S0-Zustand zurück zu gelangen. Dieser strahlungsrei- che Vorgang ist einer von zweien, die zur Lumineszenz führen. Der andere strahlungsreiche Vorgang ist die Phosphoreszenz (P). Dabei wird ein Photon abgegeben, wobei das Elek- tron unter Spinumkehr aus demT1-Zustand in denS0-Zustand übergeht.
Keine Lumineszenz tritt auf, wenn die Relaxation aus demS1- oderT1-Zustand strahlungs- los erfolgt. Bei diesen Konkurenzprozessen zu Lumineszenz sind beispielsweise eine erneute Schwingungsrelaxationen oder das strahlungslose Intersystem Crossing zu nennen.10
3.2 Franck-Condon-Prinzip
Elektronen bewegen sich deutlich schneller als Atomkerne, da sie um ein Vielfaches leichter sind. Dieses Verhalten wirkt sich auch auf die Übergänge unterschiedlicher Zustände im Rahmen elektronischer Anregung und anschließender Relaxion aus.
Durch die Quantisierung kann ein Molekül nur Zustände diskreter Energiewerte, die an eine bestimmte Geometrie gekoppelt sind, annehmen. Aufgrund der Trägheit der Kerne ist ein Übergang in einen anderen energetischen Zustand umso wahrscheinlicher, je ähnlicher sich die Geometrien des neuen Zustandes und des Ausgangszustandes sind, da dabei die Kernanodnung nur geringfügig geändert werden muss. Die Übergangswahrscheinlichkeit PT, auch Franck-Condon-Faktor genannt, kann aus dem Betragsquadrat der Ausgangs- φm und Endwellenfunktionφn berechnet werden.11
PT =| hφm|φni |2 (3.1)
3.3 Dichtefunktionaltheorie
Bei der Dichtefunktionaltheorie (DFT) wird die Energie eines Systems, anders als bei der Hartree-Fock-Theorie, nicht über eine Wellenfunktion bestimmt, sondern über die Elektro- nendichte. In Bezug auf diese stellten Hohenberg und Kohn zwei Theoreme auf. Das erste Theorem besagt, dass die Energie eines Systems von der Einelektronendichte abhängig ist und der Zusammenhang dieser beiden Größen über ein systemunabhängiges, univer- selles Funktional beschrieben werden kann. Das zweite Theorem sagt aus, dass für die Einelektronendichte das Variationsprinzip gilt. Das heißt, je besser die Beschreibung der Elektronendichte des Systems ist, desto kleiner und näher am realen Wert ist die Energie, die durch das Funktional erhalten wird.
3.3 Dichtefunktionaltheorie 11 Vorteil der DFT ist, dass die Einelektronendichte nur von 3 Koordinaten abhängt und nicht von 4N Koordinaten, wie es bei der Erstellung einer Wellenfunktion aus einer Sla- terdeterminanten, unter Berücksichtigung des Spins, der Fall ist. Ebenfalls wird durch das Funktional die Elektronenkorrelation direkt berücksichtigt und muss nicht wie bei Post-Hartree-Fock-Verfahren über aufwendige Korrekturen nachträglich berechnet wer- den. Problem der Dichtefunktionaltheorie ist jedoch, dass das benötigte Funktional nicht bekannt ist. Es kann nur näherungsweise bestimmt werden, da nicht alle Teile des Funk- tionals exakt bestimmbar sind.
Zentrales Element der DFT ist demzufolge, ein geeignetes Funktional zu finden, um Syste- me möglichst gut beschreiben zu können. Da auch, wie beim Erhalten des elektronischen Hamilton Operators die Born-Oppenheimer-Näherung auf das Funktional anwendbar ist, setzt dieses sich grundlegend aus drei Teilen zusammen. Es besteht aus jeweils einem Term für die kinetische Energie der ElektronenTe[ρ], die potentielle Energie der Elektron-Kern- WechselwirkungEek[ρ]und die potentielle Energie der Elektron-Elektron-Wechselwirkung Eee[ρ].
E[ρ] =Te[ρ] +Eek[ρ] +Eee[ρ] (3.2)
Dabei kann der Elektron-Elektron-Term in einen Coulomb-J[ρ]und einen Austauschterm K[ρ] unterteilt werden.
E[ρ] =Te[ρ] +Eek[ρ] +J[ρ] +K[ρ] (3.3)
Die Energie der Elektron-Kern-Wechselwirkung und der Coulombterm lassen sich analog zu ihren klassische Ausdrücken formulieren, die kinetische Energie der Elektronen und der Austauschterm jedoch nicht.
Eine Möglichkeit, einen großen Teil der kinetischen Energie des Funktionals bestimmen zu können, bietet die Kohn-Sham-DFT. Dabei wird zunächst ein Teil der kinetischen Energie TSD über ein Referenzsystem berechnet, bei dem die Elektronen untereinander nicht wechselwirken. Den Energiewert des Systems erhält man über das Lösen der Schrö- dingergleichung, wofür eine Slaterdeterminante als Wellenfunktion eingesetzt wird. Zur Erstellung dieser Slaterdeteminanten werden die sogenannten Kohn-Sham-Orbitale einge- führt, die durch die Kohn-Sham-Gleichung erhalten werden.
12 3 Theorie
TSD =
N
X
i=1
hφi| − 1
2∇2|φii (3.4)
Die zur Berechnung des Funktionals benötigte Ladungsdichte ρ wird ebenfalls aus den Orbitalen ermittelt.
ρ=
N
X
i
|φi|2 (3.5)
Der nicht berechenbare Teil der kinetischen Energie(Te[ρ]−TSD[ρ]), auch Korrekturterm genannt, kommt durch die fehlende Berücksichtigung der Wechselwirkung der Elektronen zustande. Er wird mit dem Austauschterm, sowie einem Term zur Berücksichtigung der Elektronenkorrelation zum AustauschkorrelationstermExc zusammengefasst.
Exc = (Te[ρ]−TSD[ρ]) +K[ρ] (3.6)
E[ρ] =TSD[ρ] +Eek[ρ] +J[ρ] +Exc[ρ] (3.7)
Der Austauschkorrelationsterm wird näherungsweise über das Dichtefunktional erfasst. Die verschiedenen Dichtefunktionale unterscheiden sich in ihrer Qualität und ihrem Einsatzbe- reich. Grundlegend lassen sie sich in fünf Kategorien einteilen: die lokale Dichtenäherung (LDA), verallgemeinerte Gradientennäherung (GGA), meta-GGA, Hybride und Doppel- Hybride; wobei die Qualität von links nach rechts zunimmt.12
Ein Beispiel für ein Dichtefunktional ist das BH-LYP13 14. Es gehört zu den Hybriden und wurde in dieser Arbeit für jede der durchgeführten Rechnungen verwendet. Dabei setzt sich der Austauschterm zur einen Hälfte aus dem Hartree Fock (HF)- und zur anderen aus dem LDA/B88-Austauschfunktional zusammen. Um das Austauschkorrelationsfunktional zu vervollständigen, kommt ein LYP-Korrelationsterm hinzu.15
Exc = 0,5ExHF + 0,5ExLDA/B88+EcLY P (3.8)
3.4 Multireferenzkonfigurationswechselwirkung 13
3.4 Multireferenzkonfigurationswechselwirkung
Gerade bei Berechnungen elektronischer Anregungen oder Prozessen der Bindungsspal- tung müssen Elektronenkorrelationen berücksichtigt werden. Der Beitrag der Elektronen- korrelation in Bezug auf die Gesamtenergie ist zwar gering, jedoch liegt er hinsichtlich der Größenordnung im Bereich von chemischen Bindungsenergien und kann bereits bei gerin- gen Abweichungen Ergebnisse grundlegend verändern. Um korrekte Resultate zu erhalten, muss die Elektronenkorrelation somit möglichst exakt bestimmt werden. Dafür reicht nur der Ansatz der Dichtefunktionaltheorie nicht aus.16
Eine geeignete Methode, bei der eine gute Berücksichtigung der Elektronenkorrelations- energie erfolgen kann, ist die MRCI-Methode. Sie beruht auf der Konfigurations-Wechsel- wirkungs-Methode (CI).17Bei der CI-Methode handelt es sich um ein Mehrdeterminanten- Verfahren, bei dem jeder Zustand durch eine Linearkombination von Slaterdeterminanten dargestellt wird.
|Ψi =c0|Ψ0i+X
a,r
cra|Ψrai+ X
a,b,r,s
crsab|Ψrsabi+ X
a,b,c,r,s,t
crstabc|Ψrstabci+... (3.9)
Werden in der CI-Wellenfunktion alle für das System möglichen Anregungen betrachtet, wird eine Full-CI-Entwicklung erhalten. Sie berücksichtigt die exakte Elektronenkorrela- tion und ist mit einem hohen Rechenaufwand verknüpft. Um die Berechnung auch für größere Moleküle praktikabel zu machen, wird die Entwicklung der Wellenfunktion oft- mals frühzeitig, zum Beispiel nach der zweiten Anregung, abgebrochen. In diesem Fall wird von einer SDCI-Entwicklung gesprochen.
Bei der MRCI-Methode wird die CI-Entwicklung nun nicht mehr nur ausgehen von der De- terminante des Grundzustandes entwickelt. Es wird ein sogenannter „Activ Space“ festge- legt, der Orbitale um das höchste besetzte Molekülorbital (HOMO)/niedrigste unbesetzte Molekülorbital (LUMO) beinhaltet. Ausgehend von unterschiedlichen Elektronenbeset- zung diese Raumes werden nun möglichen Referenzdeterminanten erstellt. Dies verhilft zu einer besseren Beschreibung der Korrelationseffekte.18
3.5 DFT/MRCI
Eine recheneffiziente Methode zur Bestimmung elektronischer Anregungen stellt die DFT/- MRCI-Methode17 19 dar. Sie kombiniert die Vorteile von MRCI und DFT und beinhaltet eine gute Beschreibung der Elektronenkorrelation.
Dabei wird die statische Korrelation über die MRCI-Methode erfasst, da diese schon bei kleiner CI-Entwicklung, also ohne großen Rechenaufwand, gut wiedergegeben wer-
14 3 Theorie den kann. Die dynamische Korrelation würde erst bei größerer Entwicklung hinreichend erfasst werden, da die Werte bedeutend langsamer konvergieren. Eine deutlich effizientere Berechnung der dynamischen Korrelation kann über die Dichtefunktionaltheorie erfol- gen.17 Damit durch die DFT/MRCI die dynamische Korrelation nicht doppelt gezählt wird, müssen empirische Parameter eingeführt werden. Sie werden im Hamilton-Operator berücksichtigt. Dadurch handelt es sich beim DFT/MRCI-Verfahren streng genommen um eine semiempirische Methode.
Bei der DFT/MRCI-Rechnung besteht die Wahl zwischen verschiedenen Hamilton-Ope- ratoren, die an bestimmte Problemstellungen angepasst sind. In dieser Arbeit wurde der von Adrian Heil entwickelte Hamilton-Operator R2018 verwendet, der eine modifizierte Dämpfungsfunktion für die Außerdiagonalelemente des Hamilton-Operators enthält und sich besonders gut für die Berechnung von Übergangsmetallverbindungen eignet.20
3.6 TDDFT
Die zeitabhängige Dichtefunktionaltheorie eignet sich für die Berechnung der Eigenschaf- ten von Systemen im angeregten Zustand.21In dieser Arbeit wird die Methode vorrangig dazu genutzt, Geometrien bestimmter angeregter Zustände zu bestimmen.
Grundlange der TDDFT bildet das Runge-Gross-Theorem. Es kann als Analogon zum Hohnberg-Kohn-Theorem, welches bei der zeitunabhängigen DFT angewandt wird, gese- hen werden und ermöglicht die Erweiterung zur Zeitabhängigkeit.22
Die in dieser Arbeit ebenfalls verwendete Tamm-Dancoff-Näherung bildet eine Erweiterung der TDDFT und verkürzt die Rechenzeit, indem Matrixelemente der Eigenwertgleichung der TDDFT vernachlässigt werden.
Problematisch ist jedoch die Berechnung von Charge-Transfer-Zuständen, aufgrund fal- scher Beschreibung langreichweitiger Wechselwirkungen bei der Benutzung lokaler Dich- tefunktionale. Um diese Problem möglichst gering zu halten, wird in der vorliegenden Arbeit das BH-LYP-Funktional mit einem 50 %igen Anteil nicht lokaler Austauschwech- selwirkungen verwendet.21
3.7 Lösungsmitteleffekte
Lösemittel haben in vielen Fällen einen erheblichen Einfluss auf Moleküle. Sie können die Struktur und das chemische Verhalten dieser maßgeblich verändern. Daher ist es auch in der theoretischen Chemie oftmals unerlässlich, bei der Berechnung von Molekül- eigenschaften Lösemitteleffekte mit einzubeziehen, um sinnvolle Ergebnisse zu erhalten.
Besonders bei polaren Molekülen und Lösemitteln kann es bei der Berücksichtigung die- ser Effekte zu starken Abweichungen im Vergleich zur Betrachtung im Vakuum kommen.
Diese Abweichungen übertragen sich auch auf Absorptions- und Emissionspektren. Die
3.7 Lösungsmitteleffekte 15 Beeinflussung der Spektren durch ein Lösemittel wird als Solvatochromie bezeichnet.
Auch bei der Betrachtung von anregerten Zuständen spielen Lösemitteleinflüsse eine wich- tige Rolle, da sich das Dipolmoment durch die Anregung stark verändern kann. In polaren Lösemitteln werden beispielsweise Zustände mit hohem Dipolmoment deutlich besser sta- bilisiert und somit im Vergleich zu Zuständen mit geringerem Dipolmoment energetisch stärker herabgesetzt. Das kann vor allem bei Charge-Transfer-Zuständen zu einer Umver- teilung der energetischen Lage von anregten Zuständen führen.23
Die Berücksichtigung von Lösemitteleffekten kann über explizit hinzugefügte Lösemittel- moleküle erfolgen. Das Hinzufügen zusätzlicher Atome ist jedoch meist mit einem starken Anstieg der Rechenzeit verbunden. Um die genannten Effekte dennoch mit angemessenem Rechenaufwand berücksichtigen zu können, wurden Lösemittelmodelle eingeführt.
Ein häufig verwendetes Beispiel sind die dielektrischen Kontinuumsmodelle. Dabei wird ein Hohlraum aufgebaut, indem jedes Atom von einer Kugel eingeschlossen wird. Der Ra- dius der Kugeln wird dabei über experimentell gefittete Parameter bestimmt. Der durch die Kugeln eingeschlossene Hohlraum stellt den Bereich dar, den das Solvat für sich bean- sprucht und in den das Solvens nicht eindrigen kann. Die Oberfläche des umschlossenen Volumens wird nun in viele kleine Bereiche unterteilt, an denen über die Poisson-Gleichung Punktladungen berechnet werden.
∇2φ(r) =−4πρ(r)
(3.10)
Dabei istφ(r)das elektrostatische Potential,ρ(r) die Ladungsdichte unddie Dielektri- zitätskonstante des Lösemittels. Durch Einsetzten der Punktladungen in die quantenche- mische Rechnung wird der Einfluss des Solvens auf das Solvat berücksichtigt. Die wich- tigsten Vertreter der dielektrischen Kontinuummodelle sind das Polarizable Continuum Model (PCM)24 und das Conduktor-like Screening Model (COSMO).25
17
4 Technische Details der Rechnungen
Die nachfolgend beschriebenden Berechnungen wurden für das untersuchte Molekül mit drei unterschiedlichen Parametern für die Umgebungseinflüsse durchgeführt. Es wurde das Molekül im Vakuum, in PCM-Ethanol-Umgebung und PCM-Ethanol-Umgebung mit ex- plitzitem Ethanol-Molekül berechnet.
Die Ausgangsgeometrien der Moleküle wurden mithilfe des Programms Avogadro26 er- stellt. Die anschließende Minimabestimmung der Grundzustände erfolgte mittels DFT über Gaussian 16.27 Dabei wurde für die Elemente Wasserstoff, Kohlenstoff, Stickstoff und Sauerstoff eine def2-SV(P)-Basis28verwendet, für Schwefel der Basissatz def2-SVPD29 und für Zink der Basissatz 10-mdf 6s5p3d in Kombination mit dem ECP10MDF.30 Das verwendete Funktional in dieser und auch allen darauf folgenden Berechnungen war das BH-LYP-Funktional13.14Für die Berechnungen im Ethanol wurde das in Gaussian imple- mentierte Polarizable Continuum Model (PCM) verwendet. Die Dielektrizitätskonstante des Ethanol betrug 24,852. Schwingungsfrequenzanalysen zur Überprüfung der stationären Punkte wurden ebenfalls mit Gaussian 16 durchgeführt.
Auch die Geometrien der angeregten Zustände wurden über Gaussian 16 berechnet. Für die Berechnung der Singulett-Zustände wurde die TDDFT verwendet, für Triplett-Zustände die TDA.
DieTCT- undTLE(batho)-Geometrie der angeregten Zustände bei der Berechnung mit expli- zitem Ethanol konnten nur ausgehend von den zugehörigen Singulettzuständen gefunden werden. Da für nachfolgende Spin-Bahn-Kopplungsberechnungen eine f-Funktion benötigt wurde, wurde bei der Berechnung für das Zink-Atom eine f-Funktion zum Pseudopoten- tial hinzugefügt. Testrechnungen ergaben, dass sich durch das zusätzliche f-Potential die Anregungsenergie um weniger als 0,01 eV ändern. Die Rechnungen wurden von Frau Prof.
Dr. Marian übernommen.
An den gefundenen Minima der jeweiligen Zustände wurden DFT/MRCI-Rechnungen17 19 durchgeführt. Dafür wurden für das Zinkatom der Basissatz def2-TZVP31 als Auxiliarba- sis eingesetzt. Für die übrigen Atome wurden zu den normalen Basissätzen gleichnamige Auxiliarbasen32 verwendet.
Die Berechnungen wurden mit dem Hamiltonoperator R201820durchgeführt. Dabei wur- den jeweils 21 Singulett- und 20 Triplett-Zustände berechnet und der CI-Referenzraum auf 10 Elektronen in 10 Orbitalen mit maximal Doppelanregungen beschränkt. Bei den Berechnungen des Moleküls ohne explizites Lösemittel wurden die ersten 48 Orbitale ein- gefroren, beim Molekül mit explizitem Lösemittel die ersten 51. Für beide Durchläufe der DFT/MRCI-Rechnung wurde der Selektionsschwellwert auf 0,8 Eh gesetzt und der
18 4 Technische Details der Rechnungen Parameter short/tight verwendet. Der Einfluss des Lösemittels wurde über exportierte Punktladungen berücksichtigt.
Franck-Condon-Emissionsspektren wurden mithilfe des Programms VIBES33bei 77 Kelvin und Raumtemperatur (298,15 K) erstellt. Die Berechnung erfolgte mit einem Zeitintervall von 300 fs, 16384 Gitterpunkten und einer Breiten der Dämpfungsfunktion von 200,0cm−1. Für die Darstellung der Geometrien und Molekülorbitale wurde Jmol34 verwendet, für die Erstellung der Absorptions- und Emissionsspektren Gnuplot.35 Experimentelle Spektren der Literatur wurden mittels Insidias, ein von Fabian Dinkelbach geschriebenes Programm, exportiert.36
19
5 Ergebnisse und Auswertung
5.1 Grundzustand
5.1.1 Geometrie
Bei der optimierten Grundzustandsgeometrie im Vakuum handelt es sich beim Zink(dtsq)- (batho)-Komplex um ein Cs-symmetrisches Molekül. Dabei liegt die Spiegelebene in der Ebene des Dithioquadratsäure-Liganden. Eine Drehachse liegt nicht vor, da die Ringstruk- tur mit den Stickstoffatomen des Bathocuproin-Liganden nicht vollständig planar ist und auch die Phenylringe nur durch eine Spiegelung ineinander überführt werden können.
Im Lösemittel ändert sich die Symmetrie zu C1. Es könnte zwar immer noch eine Spie- gelebene durch den Bathocuproinliganden gelegt werden, jedoch geht das Symmetrieele- ment um das Zink-Atom verloren, da sich die Bindungslängen sowie -winkel der jeweiligen Stickstoff- und Schwefel-Atome zum Zink unterscheiden. Innerhalb der Dithioquadratsäu- re ist ebenfalls keine Symmertrie mehr vorhanden, da auch hier die gegenüberliegenden Bindungslängen und -winkel unterschiedlich sind.
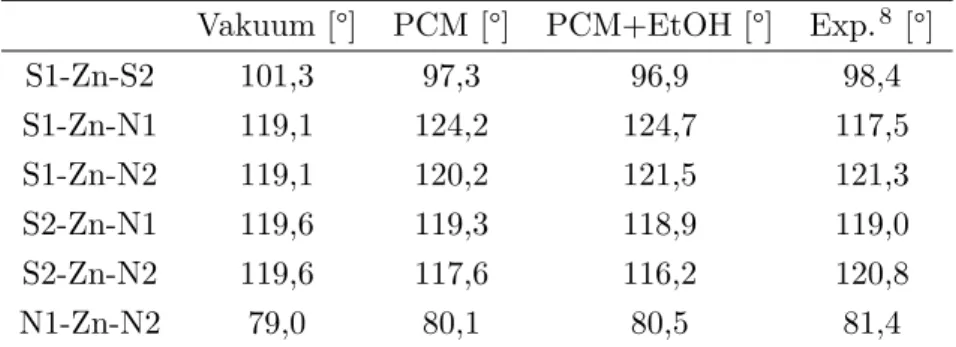
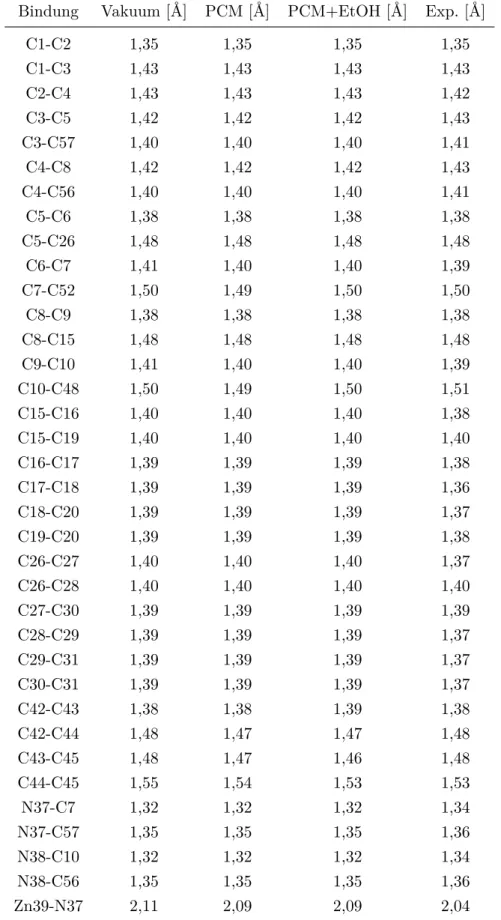
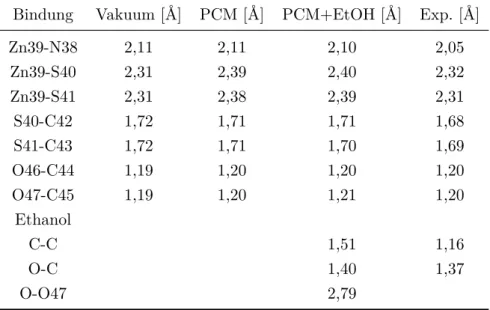
Von besonderem Interesse sind die Bindungslängen und -winkel, in denen das Zink-Atom involviert ist. Dazugehörige Werte sind in Tabelle 5.1 und 5.2 aufgelistet. Die Zuordnung der Atome erfolgt exemplarisch an der Grundzustandsgeometrie für die Berechnung mit PCM und explizitem Ethanol-Molekül und ist Abbildung 5.1 zu entnehmen. Eine voll- ständige Auflistung der Daten befindet sich im Anhang.
Abb. 5.1: Optimierte Grundzustandsgeometrie des Zn(dtsq)(batho)-Komplexes der Be- rechnung mit PCM-Umgebung und explizitem Ethanol-Molekül.
Dabei fällt auf, dass bei der Berechnung im Vakuum die Zink-Schwefel-Bindungen sehr gut beschrieben werden. Sie zeigen kaum eine Abweichung zum Experiment. Diese Über- einstimmung geht jedoch bei den Berechnungen mit Lösemitteleffekten verloren und es kommt zu Abweichungen von bis zu 0,083 Angstroem.
20 5 Ergebnisse und Auswertung Die Zink-Stickstoff-Bindungen hingegen zeigen bei der Berechnung im Vakuum eine er- höhte Abweichung im Vergleich zum Experiment. Sie liegt bei ca. 0,6 Ångstroem. Eine Verbesserung der Zn-N1-Bindungs zeigt sich dieses Mal bei Berücksichtigung der Lösemit- teleffekte, jedoch bleibt eine Abweichung von 0,048 und 0,041 Ångstroem bestehen. Für die Zn-N2-Bindung ergibt sich kein Unterschied bei den verschiedenen Berechnungen.
Tabelle 5.1: Zusammenfassung der wichtigsten Bindungslängen des optimerten Grundzu- standes des Zn(dtsq)(batho)-Komplexes bei unterschiedlichen Umgebungsein- flüssen und des Experiments.
Vakuum [Å] PCM [Å] PCM+EtOH [Å] Exp.8 [Å]
Zn-S1 2,313 2,388 2,398 2,315
Zn-S2 2,312 2,383 2,395 2,313
Zn-N1 2,106 2,093 2,086 2,045
Zn-N2 2,106 2,107 2,105 2,047
Bei den Bindungswinkeln des Zink-Atoms für die Berechnung im Vakuum werden alle ähnlich gut beschrieben. Die mittlere Abweichung ist im Vergleich zu den anderen Berech- nungen am geringsten und liegt bei 1,8 °. Der kleinste Wert der Differenz zum Experiment beträgt 0,6 °, der größte 2,8 °. Die Streuung um den Mittelwert ist somit gering.
Das Verhalten der Streuung ändert sich bei der Berechnung mit PCM und explizitem Ethanol-Molekül. Hier werden vier der sechs Winkel besonders gut beschrieben. Dies gilt vor allem für die S1-Zn-N2- und S2-Zn-N1-Winkel, die lediglich eine Abweichung von 0,2 bzw. 0,1 ° aufweisen. Die mittlere Abweichung liegt jedoch für die drei Berechungen mit 2,4 ° trotzdem am höchsten, da die übrigen beiden Winkel eine größere Differenz zum Experiment besitzen. Es handelt sich dabei um die gegenüberliegenden Winkel S1-Zn-N1 und S2-Zn-N2, die eine Abweichung von 7,2 bzw. 4,6 ° aufweisen.
Für die Berechnung mit PCM ohne explizitem Ethanol zeigt sich ein ähnliches Verhalten.
Die mittlere Abweichung ist mit 2,3 ° etwas niedriger und auch die Streuung der Werte nimmt ab. Die Winkel verhalten sich in Bezug auf die Differenz zum Literaturwert, außer bei wenigen Ausnahmen gleich.
Für den Vergleich der Bindungslängen und -winkel ist zu beachten, dass die Geometrie des Zink-Komplexes für das Experiment im Kristall bestimmt wurden. Dies ist für die hier durchgeführten Rechnungen nicht der Fall. Kristallpackungseffekte wurden nicht berück- sichtigt, wodurch Abweichungen der Längen und Winkel zustande kommen können.
5.1 Grundzustand 21
Tabelle 5.2: Zusammenfassung der wichtigsten Bindungswinkel des optimerten Grundzu- standes des Zn(dtsq)(batho)-Komplexes bei unterschiedlichen Umgebungsein- flüssen und des Experiments.
Vakuum [°] PCM [°] PCM+EtOH [°] Exp.8 [°]
S1-Zn-S2 101,3 97,3 96,9 98,4
S1-Zn-N1 119,1 124,2 124,7 117,5
S1-Zn-N2 119,1 120,2 121,5 121,3
S2-Zn-N1 119,6 119,3 118,9 119,0
S2-Zn-N2 119,6 117,6 116,2 120,8
N1-Zn-N2 79,0 80,1 80,5 81,4
5.1.2 Anregung und Absorption
Der Zink-Komplex besitzt im Grundzustand ein hohes Dipolmoment. Bei der Berechnung im Vakuum liegt es bei 22,69 Debye. Bei Berücksichtigung des Lösemittels wird es noch höher und liegt ohne explizites Ethanol-Molekül bei 29 Debye, mit bei 32,44 Debye.
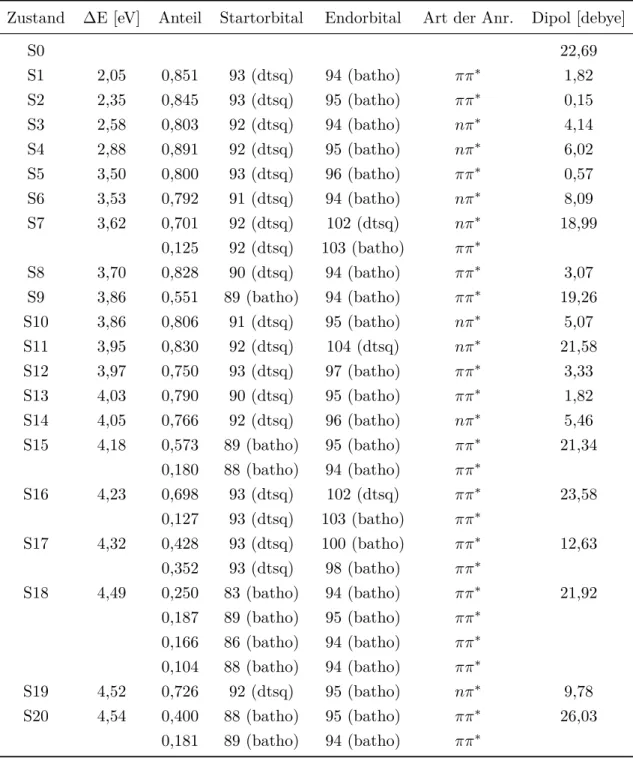
Bei der Berechnung im Vakuum sind 13 der ersten 20 Singlett-Anregungen Charge-Transfer- Übergänge. Dabei findet in jedem Fall eine Ladungsübertragung von der Dithiosäure zum Bathocuproin-Liganden statt, wobei das Dipolmoment stark abnimmt. Die erste lokale An- regung tritt erst beim Übergang in den S7-Zustand auf. Auch bei der Triplett-Anregung sind 13 der ersten 20 Anregungen Charge-Transfer-Übergänge.
Wird die Lösemittelumgebung hinzugefügt, treten unter den ersten 20 angeregten Zu- ständen deutlich mehr lokal angeregte Zustände auf. Die Anzahl der Charge-Transfer- Übergänge verringert sich mit und ohne explizitem Ethanol-Molekül für die Singulett- Zustände auf 6. Auch bei den Triplett-Anregungen zeigt sich ein ähnliches Verhalten.
Die Anzahl der Charge-Transfer-Übergänge reduziert sich ohne explizites Ethanol auf 5 und mit auf 6. Die zunehmende Anzahl lokaler Anregungen und abnehmende Anzahl von Charge-Transfer-Übergängen bei den niedrigsten Anregungen ist auf den Einfluss des Lö- semittels auf die energetische Lage der Zustände zurückzuführen. Dabei werden in der Regel am meisten solche Zustände stabilisiert, die ein hohes Dipolmoment aufweisen. Da das Dipolmoment der lokal angeregten Zustände deutlich höher liegt als das der Charge- Transfer-Übergänge, erfolgt hier eine stärkere Absenkung der energetischen Lage. Somit kann es zu einer Umverteilung der Zustände kommen.
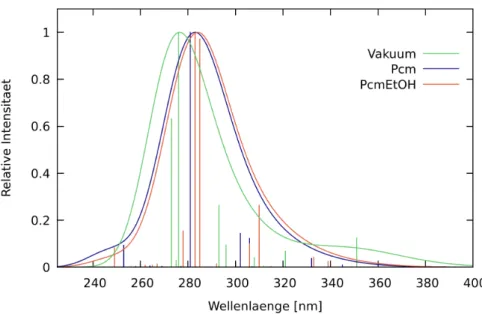
Auch im Absorptionsspektrum ist der stabilisierende Effekt des Lösemittels sichtbar. Die- ses wurde mithilfe der Energieabstände der angeregten Zustände und den entsprechenden Oszilatorstärken erstellt. Ein Absorbtions- oder Anregungsspektrum zum Vergleich konnte in der Literatur nicht gefunden werden.
22 5 Ergebnisse und Auswertung
Abb. 5.2: Absorptionsspektren des Zn(dtsq)(batho)-Komplexes im Grundzustand für die Berechnungen im Vakuum, PCM und PCM mit explizitem Ethanol-Molekül.
Alle Spektren wurden mit einer Halbwertsbreite von 0,471 eV, ausgehend von den Linienspektren, berechnet.
Die Anregung in den S1-Zustand erfolgt im Vakuum bei Bestrahlung mit einer Wellenlän- ge von 605 nm, mit PCM bei 383 nm und mit PCM + explizitem Ethanol bei 375 nm.
In allen Fällen ist die zugehörige Oszillatorstärke so gering, dass sie im Linienspektrum keinen signifikanten Peak verursacht. In dem von Gronlund durchgeführten Experiment erfolgte die Anregung mit einer Wellenlänge von 364 nm. Den Berechnungen mit PCM entsprechend könnte hier nur eine Anregung in den S1-Zustand erfolgen.
Deutlich im Spektrum zu erkennen ist eine Rotverschiebung des intensivsten Peaks bei Berücksichtigung des Lösemitteleinflusses. Dieser Effekt verstärkt sich minimal bei zu- sätzlichem Hinzufügen eines expliziten Ethanol-Moleküls. Der Peak wird in allen Fällen durch lokale Anregungen, die mit den höchsten Oszillatorstärken verknüpft sind, hervorge- rufen. Die Rotverschiebung hängt mit den leicht erhöhten Dipolmomenten der angeregten Zustände gegenüber dem Grundzustand zusammen. In geeignetem Lösemittel erfolgt eine bessere Stabilisierung der Zustände mit höherem Dipolmoment. Dadurch nehmen die lokal angeregten Zustände energetisch stärker ab als der Grundzustand und es wird eine we- niger energiereiche Strahlung, also solche mit höherer Wellenlänge, zur Anregung benötigt.
Im Absorptionsspektrum des Moleküls im Vakuum ruft nur ein Charge-Transfer-Übergang eine signifikante Intensität hervor und sorgt für einen zweiten, schwach ausgeprägten Peak.
Dabei handelt es sich um die Anregung in den S6-Zustand bei einer Wellenlänge von 351 nm. Dieser Übergang ist in den anderen Spektren nicht mehr zu erkennen, da er dort energetisch deutlich weiter oben liegt. Im Spektrum der Berechnung mit PCM findet sich
5.1 Grundzustand 23 hingegen ein anderer Charge-Transfer-Peak. Er liegt bei 302 nm, erzeugt jedoch keinen zweiten Peak mehr, da er durch den ersten überlagert wird. Beim Spektrum der Berech- nung mit PCM und explizitem Ethanol-Molekül ist kein Charge-Transfer-Übergang mit einer nennenswerten Intensität mehr vorhanden.
24 5 Ergebnisse und Auswertung
5.2 Geometrien der angeregten Zustände
Im folgenden werden die Geometrien der angeregten Zustände und die Änderungen gegen- über dem Grundzustand für die Berechnung mit PCM und explizitem Ethanol-Molekül betrachtet. Für die Änderungen der Bindungslängen ist dabei eine Betrachtung der Dich- tedifferenzen besonders anschaulich, da über die Verschiebung der Elektronendichte eine Aussage über die Änderung der Geometrie ermöglicht wird. Dabei ist in den folgenden Bildern die Abnahme der Elektronendichte in rot gekennzeichnet, die Zunahme in gelb.
Die Betrachtung der Änderungen der Bindungslängen der Liganden erfolgt nur qualitativ.
Eine genaue Auflistung der Werte befindet sich im Anhang. Die Abstände zum Zink-Atom werden am Ende des Kapitels jedoch noch einmal gesondert aufgeführt.
Unter anderem wurde die Geometrie des tiefliegendsten Singulett- und Triplett-Charge- Transfer-Zustands bestimmt. Dabei handelt es sich um die Anregungen in den S1- bzw.
T4-Zustand. Eine Abweichung der Geometrien untereinander ist kaum vorhanden. Anhand der Dichtedifferenzen in Abbildung 5.3 ist eindeutig zu erkennen, dass ein Ladungstrans- fer von der Dithioquadratsäure zum Bathocuproin erfolgt. Erwartungsgemäß sollten somit die Bindungslängen innerhalb des Dtsq-Liganden zunehmen und in vielen Bereichen des Batho-Liganden abnehmen. Dieses Verhalten spiegelt sich auch in den Werten wider. In- nerhalb des Kohlenstoffgerüsts der Dithioquadratsäure werden alle Kohlenstoffbindungen länger. Beim Batho-Liganden ist die Änderung der Bingungslängen nicht komplett einheit- lich. Bindungen, bei denen eine Zunahme der Elektronendichte eingezeichnet ist, werden kürzer, die anderen werden länger oder bleiben unverändert. Die größten Änderungen tre- ten jedoch bei den Bindungen zum Zink-Atom auf. Dabei zeigt sich der eindeutige Trend, dass die Bindungslängen in Richtung des Bathocuproin-Ligandes abnehmen und zu den Schwefel-Atomen der Dithioquadratsäure zunehmen. Hierbei treten kleine Änderungen zwischen der Geometrie desS1- und T4-Zustandes auf.
Abb. 5.3: Änderung der Elektronendichte bei der Anregung in denS1/T4-Zustand an der Grundzustandsgeometrie des Zn(dtsq)(batho)-Komplexes mit einem wasserstoff- brückengebundenem Ethanol-Molekül.
5.2 Geometrien der angeregten Zustände 25 Bei der Anregung in denT3-Zustand handelt es sich um eine lokale Anregung auf dem Ba- thocuproin. Dabei nimmt die Bindungslänge in den rot makierten Bereichen aus Abbildung 5.4 zu und in den gelb markierten Bereichen ab. Auf dem Dithioquadratsäure-Liganden gibt es, mit einer Ausnahme bei einer Bindung, überhaupt keine Änderungen. Dadurch, dass die Ladung nur auf dem Batho-Liganden umverteilt wird, ändern sich die Bindungs- längen zum Zink-Atom nicht so stark wie beim Charge-Transfer-Zustand. Die Bindungen des Zinks zum Schwefel werden minimal kürzer, die zum Stickstoff-Atom länger.
Abb. 5.4: Änderung der Elektronendichte bei der Anregung in den T3-Zustand an der Grundzustandsgeometrie des Zn(dtsq)(batho)-Komplexes mit einem wasserstoff- brückengebundenem Ethanol-Molekül.
Ebenfalls wurde die Geometrie des tiefliegendsten Triplett-Zustandes berechnet. Dieser kennzeichnet sich durch eine lokale Anregung auf der Dithioquadratsäure. Dabei werden zwei Bindungen des Kohlenstoffgerüst kürzer und die anderen beiden, sowie die Bindungen zum Sauerstoff-Atom länger. Wie beim lokal angeregten Batho-Zustand sind die Ände- rungen zum Zink-Atom nicht so ausgeprägt wie beim Charge-Transfer-Übergang. Es tritt lediglich eine mäßige Verlängerung der Bindungslänge zwischen dem Zink- und Schwefel- Atom auf, sowie eine Verkürzung zum Stickstoff.
Abb. 5.5: Änderung der Elektronendichte bei der Anregung in den T1-Zustand an der Grundzustandsgeometrie des Zn(dtsq)(batho)-Komplexes.
26 5 Ergebnisse und Auswertung
Tabelle 5.3: Zusammenfassung der Bindungslängen zum Zink-Atom für die unterschiedli- chen Geometrien der angeregten Zustände. Die Nummerierung der Atome ist Abbildung 5.1 zu entnehmen.
Bindung S0 [Å] SCT [Å] TCT [Å] TLE(batho) [Å] TLE(dtsq) [Å]
Zn-S1 2,398 2,469 2,463 2,394 2,412
Zn-S2 2,395 2,511 2,504 2,392 2,417
Zn-N1 2,086 1,993 1,994 2,095 2,078
Zn-N2 2,105 2,009 2,010 2,117 2,100
Anhand der Abbildungen der Dichtedifferenzen ist ebenfalls zu sehen, dass durch die An- regungen kein Ladungstransfer zum Zink-Atom stattfindet. Auch metallzentrierte Über- gänge sind bei den niedrigen Anregungen nicht vorhanden. Durch die vollständig besetzte d-Schale ist das Bestreben zur Elektronenaufnahme des Zink-Atoms gering. Erst bei hö- heren Anregungen findet ein Ladungstransfer zu den unbesetzten p-Orbitalen statt.
5.3 Adiabatische Anregungsenergien 27
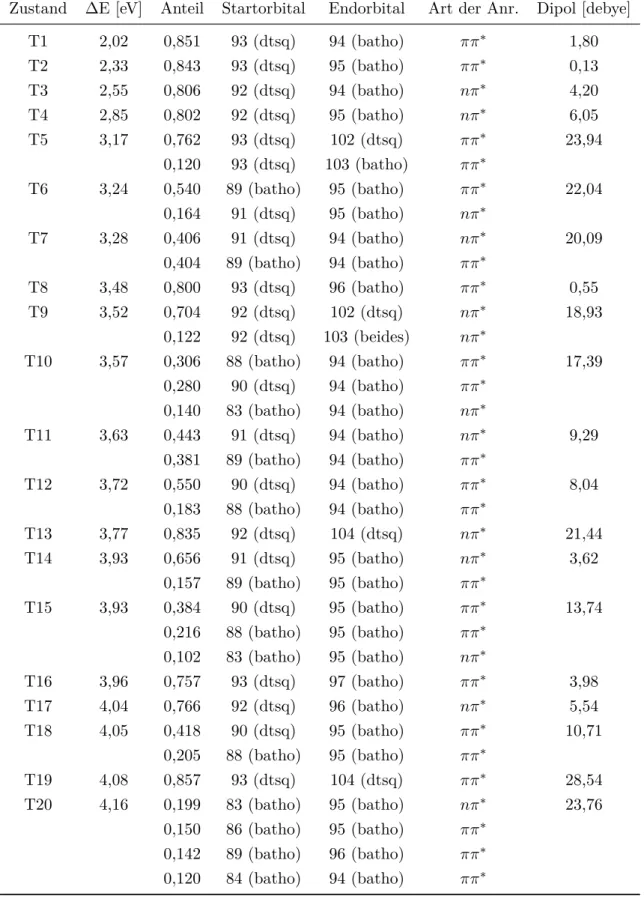
5.3 Adiabatische Anregungsenergien
Ausgehend von den, durch TDDFT ermittelten, Geometrien der angeregten Zustände wur- den MRCI-Rechnungen durchgeführt. Bei der Auswertung dieser liegt der Schwerpunkt bei den niedristen angeregten Zuständen der verschiedenen Übergänge (Charge-Transfer, lokal angeregte Dtsq- und Batho-Übergänge). Dafür erfolgt eine schematische Zusammen- fassung und ein Vergleich der energetischen Lage der adiabatischen Anregungsenergien.
Diese werden berechnet, indem die Differenz zwischen dem angeregten Zustand und der Grundzustandsenergie an der S0-Geometrie gebildet wird. Eine vollständige Zusammen- fassung aller Werte befindet sich im Anhang. Zudem werden wieder alle Rechnungen mit verschiedenen Umgebungseinflüssen berücksichtigt und nicht, wie im vorangegangen Ab- schnitt, nur die mit explizitem Ethanol-Molekül.
5.3.1 Vakuum
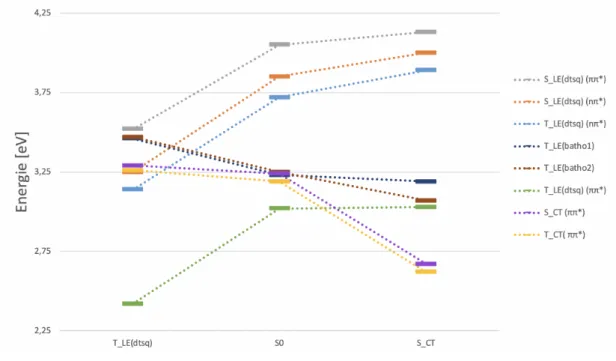
Für die Berechnungen im Vakuum konnten lediglich die Geometrien der angeregten Zu- stände der Charge-Transfer-Übergänge für Singuletts und Tripletts gefunden werden. Eine Zusammenfassung der energetischen Lage der niegristen Singulett- und Triplett-Charge- Transfer-Übergänge, der Singulett und Triplett lokal angeregten Zustände der Dithioqua- dratsäure und der Triplett lokal angereten Zustände des Bathocuproins, anhand der drei Geometrien, befinden sich in Abbildung 5.6.
Abb. 5.6: Zusammenfassung der adiabatischen Anregungsenergien der niedrigsten ange- regten Zustände anhand derS0-,SCT- undTCT-Geometrien für die Berechnung im Vakuum.
28 5 Ergebnisse und Auswertung Dabei zeigt sich, dass bei allen Geometrien die Charge-Transfer-Zustände mit Abstand am niedrigsten liegen. Der Triplett-Zustand ist minimal günstiger als der des Singuletts.
Durch den Übergang von der Geometrie des Grundzustandes zurSCT bzw-TCT-Geometrie sinkt die Energie dieser, wie erwartet, weiter ab. Im Gegensatz dazu erfolgt für alle lokal angeregten Zustände eine Erhöhung der Energie. Dabei sind die Ergebnisse der Zustände für dieSCT und TCT-Geometrie bis auf sehr wenige Ausnahmen identisch.
Bei den lokal angeregten Zuständen ist zu erkennen, dass die Triplett-Zustände niedriger als vergleichbare des Singuletts liegen. Der tiefliegendste lokal angeregte Zustand ist für alle Geometrien ein auf der Dithioquadratsäure lokalisierter und kommt durch einenππ∗- Übergang zustande. Darauf folgen zwei sehr nah beieinander liegende Batho-Zustände.
Aufgrund des hohen Dipolmoments des Moleküls im Grundzustand ist zu erwarten, dass das Lösemittel einen großen Einfluss auf die energetische Lage der lokal angeregten Zu- stände hat. Da sie das hohe Dipolmoment beibehalten, kann hier eine Stabilisierung durch das Lösemittel besonders gut erfolgen. Durch das sinkende Dipolmoment bei den Charge- Transfer-Zuständen ist dieser Effekt im Lösemittel geringer ausgeprägt.
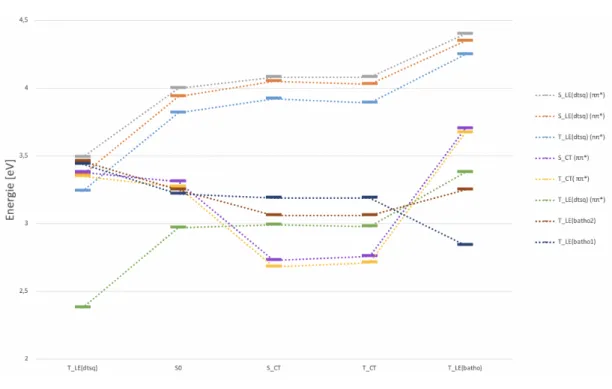
5.3.2 PCM
Bei den Berechnungen mit PCM konnten die Geometrien desSCT- undTLE(batho)-Zustan- des berechnet werden. Dabei erfolgt im Vergleich zur Berechnung im Vakuum eine gerin- gere Aufspaltung der betrachteten Zustände untereinander. Die tiefliegendsten Zustände der Berechnung mit PCM liegen höher als vergleichbare im Vakuum und die höchsten Zu- stände der Berechnung mit PCM liegen tiefer als vergleichbare der Berechnung im Vakuum.
Die Annahme zum Einfluss des Lösemittels auf die energetische Lage der Charge-Transfer- und lokal angeregten Zustände lässt sich bestätigen. Bei der Grundzustandsgeometrie liegt ein lokal angeregter Zustand nun niedriger als die beiden CTs. In Bezug auf die Reihenfol- ge der übrigen Zustände ändert sich anhand derS0-Geometrie im Vergleich zur Rechnung im Vakuum nichts.
Bei der Geometrie desTLE(dtsq)s werden alle auf der Dithioquadratsäure lokal angeregten Zustände energetisch günstiger. Vor allem der in grün eingezeichnete, niedrigste, lokal an- geregteππ∗. Er besitzt an dieser Geometrie den energetisch günstigsten Zustand in Bezug auf alle Geometrien. Die Batho-Zustände hingegen nehmen in ihrer Energie zu, ebenfalls, sehr gering ausgeprägt, auch die des Charge-Transfers.
5.3 Adiabatische Anregungsenergien 29
Abb. 5.7: Zusammenfassung der niedrigsten angeregten Zustände der MRCI-Rechnungen mit PCM anhand der ermittelten Geometrien.
Anders verhält es sich für die SCT-Geometrie. Hierbei sind die entsprechende Charge- Transfer-Zustände energetisch deutlich günstiger. Sie sinken soweit ab, dass sie für diese Geometrie am tiefsten liegen. Auch die Energie der Batho-Zustände nimmt ab, lediglich die der lokal angeregten Dithioquadratsäure-Zustände nimmt zu.
5.3.3 PCM+EtOH
Für die Berechnung mit einem expliziten Ethanol-Molekül konnten die im vorherigen Ab- schnitt beschriebenen vier Geometrien der angeregten Zustände und der Grundzustand ermittelt werden. Wie bereits im Abschnitt "Technische Details zur Rechung" erwähnt, wurden dabei zwei der Geometrien (TCT,TLE(batho)) mit einem leicht veränderten Pseu- dopotential des Zink-Atoms berechnet. Durch die zusätzliche f-Funktion im ECP sind die Bedingungen der Berechnungen untereinander nicht mehr vollständig identisch. Die da- durch auftretenden Abweichungen sind jedoch so gering (unterhalb von 0,01 eV), dass diese nicht weiter berücksichtigt wurden.
Anhand derTLE(dtsq)-, S0-, undSCT-Geometrie ist zu erkennen, dass die Ergebnisse sich kaum von der Rechnung mit PCM ohne explizitem Ethanol-Molekül unterscheiden. Die einzig nennenswerte Abweichung zeigt sich beim Vergleich der S0-Zustände. Dabei lie- gen die Charge-Transfer-Zustände bei der Rechnung mit dem expliziten Ethanol-Molekül um 0,08 bzw. 0,07 eV höher, wodurch sie sich nun energetisch über den beiden Batho-
30 5 Ergebnisse und Auswertung Zuständen befinden.
Abb. 5.8: Zusammenfassung der niedrigsten angeregten Zustände der MRCI-Rechnungen mit PCM und explizitem Ethanol-Molekül anhand der ermittelten Geometrien.
Da die SCT- und TCT-Geometrien, wie bereits in Abschnitt 5.2 erwähnt, kaum vonein- ander abweichen und auch die Abbildungen der Dichtedifferenzen gleich aussehen, ist zu erwarten, dass sich auch ein ähnliches Verhalten in der MRCI-Rechnung widerspiegelt.
Wie Abbildung 5.8 zu entnehmen ist, ist dies auch der Fall.
Die TLE(batho)-Geometrie ist hingegen wieder mit einer Umverteilung der Zustände ver- knüpft. Diesmal findet im Vergleich zum Grundzustand eine energetische Absenkung nur für denTLE(batho1)-Zustand statt. Dieser wird dabei zum niedrigst liegendsten dieser Geo- metrie. Der andere lokal angeregte Batho-Zustand verändert sich energetisch gar nicht.
Ansonsten nehmen alle anderen Zustände energetisch mit bis zu 0,5 eV zu.
In Bezug auf alle Geometrien ist, wie auch bei der Berechnung mit PCM ohne explizites Ethanol-Molekül, wieder derTLE(dtsq)-Zustand der energetisch niedrigste.
Wie bereits erwähnt, reicht die Energie der Strahlung des Experiments nur zur Anregung in denS1-Zustand aus. Das ist in diesem Fall der violett eingezeichnete Singulett-Charge- Transfer-Übergang. Von diesem Standpunkt aus sind nun mehrere Konkurrenzprozesse möglich.
Eine Möglichkeit ist die strahlungsreiche Energieabgabe aus selbigem Zustand in Form von Fluoreszenz. Dies ist hier jedoch eher unwahrscheinlich, da mehrere energetisch tiefer- liegende Triplett-Zustände vorhanden sind. Bei niedrigen ISC-Raten kann ein Übergang
5.3 Adiabatische Anregungsenergien 31 in diese erfolgen. Anbieten würden sich beispielsweise das ISC in denTLE(batho)- oderTCT- Zustande. Von hier aus kann nun Phosphoreszenz aus einem der beiden Zustände ablau- fen, aber auch die experimentell ermittelte duale Lumineszenz, postuliert für die beiden Zustände, ist anhand der berechneten energetischen Lagen durchaus möglich. Duale Lu- mineszenz tritt vor allem bei energetisch nah beieinander liegenden Zuständen auf. Dies passt für die beiden Triplett-Zustände, die einen Abstand von 0,13 eV an den entsprechen- den Geometrien besitzen. Ein weiteres Kriterium für die duale Lumineszenz sind ähnliche Lebenszeiten der infrage kommenden Übergänge.
Ebenfalls möglich ist ein ISC in den tiefliegensten TLE(dtsq)-Zustand mit anschließender Phosphoreszenz. Aber auch strahlungslose Prozesse können aus allen Zuständen erfolgen.
Von den Experimentatoren ermittelte Lumineszenzlebenszeiten weisen darauf hin, dass ein Großteil der Energieabgabe aus den angeregten Zuständen strahlungslos erfolgt. Die Lebenszeiten liegen in der Größenordnung von 4-7 Nanosekunden und sind somit deutlich geringer als typische Phosphoreszenzlebensdauern. Da auch emittiertes Licht beobachtet worden ist, scheinen mehrere Prozesse im Lauf der Relaxion parallel stattzufinden.
Um genaue Aussagen über das photophysikalische Verhalten anhand der energetischen La- ge der Zustände treffen zu können, sind Berechnungen der ISC-, Strahlungs- und Fluores- zenz- bzw. Phosphoreszenz-Raten notwenig.
32 5 Ergebnisse und Auswertung
5.4 Emissionsspektren
5.4.1 Raumtemperatur
Da die Experimentatoren vermuten, dass die Emission bei Raumtemperatur durch einen Triplett-Charge-Transfer-Übergang verursacht wird, wurde dieser Zustand zur Erstellung der in Abbildung 5.9 gezeigten Emissionsspektren gewählt. Dabei ist in violett das Spek- trum des Experiments dargestellt, in grün das der Berechnung mit PCM und explizitem Ethanol-Molekül und in blau das der Berechnung im Vakuum.
Abb. 5.9: Vergleich der Emissionsspektren bei Raumtemperatur.
Dabei zeigt sich, dass alle Spektren aus nur einem Peak bestehen und die strukturlosen Banden eine ähnliche Form besitzen. Die Emission des Spektrums der Rechnung mit ex- plizitem Ethanol-Molekül zeigt eine deutlich bessere Übereinstimmung von der Lage der Wellenzahl im Vergleich zur Berechnung im Vakuum. Die Peaks des blauen und violetten Spektrums weichen um ca. 12500cm−1 ab, die des grünen und violetten nur noch um ca.
1800cm−1.
Dabei ist die emititerte Strahlung der Berechnung im Vakuum deutlich weniger energie- reich. Das hängt damit zusammen, dass die Energieäbstände zwischen den angeregten Zuständen und dem Grundzustand deutlich geringer sind (siehe MRCI-Auswertung), als bei den Berechnungen mit berücksichtigtem Lösemitteleinfluss.
5.4 Emissionsspektren 33 5.4.2 77 Kelvin
Abbildung 5.10 enthält verschiedene Emissionspektren bei 77 Kelvin. Dabei ist in vio- lett das experimentelle Spektrum dargestellt, in gelb und orange die der Triplett-Charge- Transfer-Übergänge für die Berechnungen im Vakuum und bei explizitem Ethanol-Molekül und in blau und grün die lokal angeregten Zustände auf der Dithioquadratsäure für die Berechnungen mit PCM und explizitem Ethanol-Molekül.
Abb. 5.10: Vergleich der Emissionspektren bei 77 K für die Berechnungen bei unterschied- lichen Lösemitteleffekten.
Bei den Charge-Transfer-Übergängen ist das Spektrum der Berechnung im Vakuum wie- der, wie bei der vorherigen Abbildung, deutlich weiter in Richtung niedrigere Wellenzahl verschoben, als das Spektrum der Berechnung mit explizitem Ethanol-Molekül. Auch die Lage der Maxima ist ähnlich wie bei den Emissionsspektren bei Raumtemperatur. Eine deutliche Veränderung zeigt lediglich die Form des Spektrums der Berechnung mit expli- zitem Ethanol-Molekül. Es besitzt nun zwei schwach ausgeprägt Peaks in der Mitte und leichte Schultern an der linken und rechten Seite. Die Struktur des Spektrums im Vakuum ist hingegen unverändert und zeigt nach wie vor eine strukturlose Bande mit nur einem klaren Peak.
Die lokal angeregten Zustände unterscheiden sich kaum von einander. Die Form ist nahezu identisch und auch in Bezug auf die Wellenzahlen ist das Spektrum der Berechung mit PCM nur um wenigecm−1 in Richtung energiereichere Strahlung verschoben. Diese Ver- schiebung kommt zustande, da der lokal angeregten Triplett-Zustand auf der Dithioqua- dratsäure bei der Berechnung mit explizitem Ethanol-Molekül einen energetisch kleineren
34 5 Ergebnisse und Auswertung
Abstand (0,04eV) zum Grundzustand besitzt.
Abbildung 5.11 zeigt ebenfalls Emissionsspektren bei 77 K. Abgesehen vom experimen- tellen Spektrum sind diesmal jedoch nur die Spektren für die Berechnung mit explizitem Ethanol-Molekül aufgetragen. Dabei sind die Daten des Experiments wieder in violett dargestellt, in grün die des lokal angeregten Triplett-Zustands des Bathocuproins, in blau die des lokal angeregte Zustands der Dithioquadratsäure und in orange die des Triplett- Charge-Transfer-Übergangs.
Wie in der Einleitung erwähnt, vermuten die Experimentatoren, dass das Emissions- spektrum bei 77 K durch einen lokal angeregten Triplett-Batho-Zustand hervorgerufen wird, der die Emission eines Triplett-Charge-Transfer-Übergangs überlagert. Da aus den Ergebnissen der MRCI-Rechnungen hervorgeht, dass ebenfalls der lokal angeregte Triplett- Dtsq-Zustand aufgrund der niedrigen energetischen Lage für eine Emission in Frage kommt, wurde das Spektrum dieses Zustandes ebenfalls in die Abbildung mit aufgenommen.
Abb. 5.11: Vergleich der Emissionsspektren bei 77 K für die Berechnung mit explizitem Ethanol-Molekül.
Dabei ist in der Abbildung zu sehen, dass das Maximum des Spektrums des lokal ange- regten Dtsq-Zustandes weit (mehr als 5000 cm−1) vom experimentellen abweicht. Eine Übereinstimmung mit dem Spektrum des Experiments könnte bei einer Blauverschiebung des Dtsq-Zustandes und anschließende Überlagerung desTCTs erhalten werden. Da für die anderen Übergänge die enregetische Lage jedoch deutlich besser passt, ist es unwahrschein- lich, dass durch die Berechnung nur der TLE(dtsq)-Zustand bei zu niedrigerer Wellenzahl liegt.
5.4 Emissionsspektren 35
Die Zustände des lokal angeregten Bathos und des Charge-Transfers zeigen eine geringere Abweichung zum Experiment. Auch die Form könnte durch Überlagerung der Zustände dem eperimentellen entsprechen. Unterschiede in Bezug zu den Wellenzahlen sind zwar auch hier noch vorhanden, liegen jedoch im Rahmen der Messgenauigkeiten. Eine leichte Verschiebung kann bereits durch die Unterschiede in den Bindungslängen und -winkel her- vorgerufen werden. Ebenfalls ist die DFT/MRCI-Rechnung für organische Übergangsme- tallkomplexe bei Benutzung des Hamilton-Operators R2018 mit einem durchschnittlichen Fehler von 0,19 eV behaftet, das sind ca. 1500cm−1.20
Ein weiteres Indiz dafür, dass es sich bei der Emission um die Überlagerung des TCT- und TLE(batho)-Zustandes handelt, liefert der Vergleich mit Spektren ähnlicher Moleküle.
In der Arbeit von A. M. Galinet al.wurde ein (P hS)2ZnP hen-Molekül untersucht. Das Emissionspektren zeigt Ähnlichkeiten zu dem des Zn(dtsq)(batho)-Komplexes und wird von den Experimentatoren dem ILCT-Übergang auf dem Phenantrolin zugeordnet.37
37
6 Fazit
Mithilfe der ausgewählten Rechenmethode konnten gute Ergebnisse für die Grundzu- standsgeometrien des Zink(dtsq)(batho)-Komplexes erzielt werden. Vor allem die Berech- nung im Vakuum zeigt bis auf die Zink-Stickstoff-Abstände kaum eine Differenz gegen- über dem Experiment. Für die Berechnungen mit Berücksichtigung der Lösemitteleffekte tauchen vermehrt Abweichungen der Geometrie um das Zink-Atom auf. Hierbei ist zu be- rücksichtigen, dass die experimentelle Geometrie des Moleküls im Kristall ermittelt wurde und Kristallpackungseffekte bei der Modellierung nicht berücksichtigt wurden.
Anhand der Absorptionsspektren und der energetischen Lage der niedrigsten Anregungen lässt sich festellen, dass sich der Einfluss des Lösemittels auf das Molekül stark bemerkbar macht. Es kommt unter anderem zur Umverteilung der Zustände. Dafür verantwortlich ist die deutlich geringere Polarität der LLCT-Zustände gegenüber dem Molekül im Grundzu- stand und den ILCT-Übergängen. Zudem wird ersichtlich, dass in einer polaren Umgebung wegen der verwendete Anregungswellenlänge im Experiment, nur eine Absorption in den S1-Zustand erfolgen kann.
Die Abbildung der adiabatischen Anregungsenergien zeigt, dass der Prozess der Ener- gieabgabe aus dem niedrigsten Singulett-Zustand auf verschiedene Weisen ablaufen kann, da drei Triplett-Zustände unterhalb dieses Zustandes liegen. In Konkurrenz stehen die Fluoreszenz aus dem SCT, die Phosphoreszenz aus dem TCT, dem TLE(batho) bzw. dem TLE(dtsq) und strahlungslose Prozesse aus allen genannten Zuständen. Die von den Ex- perimentatoren vermutete duale Lumineszenz aus dem lokal angeregten Batho- und dem Charge-Transfer-Zustand ist dabei aufgrund der Nähe der energetischen Lagen ein mögli- cher Prozess, aber auch die Emission aus dem niedrigsten Triplett-Zustand, demTLE(dtsq) muss in betracht gezogen werden. Kurze Lebenszeiten, die deutlich geringer sind als typi- sche Phosphoreszenzlebensdauern, weisen jedoch darauf hin, dass ein Großteil der Ener- gieabgabe aus dem angeregten Triplett-Zustand strahlungslos erfolgt.
Emissionen können dennoch in entsprechenden Spektren bei Raumtemperatur und 77 Kelvin detektiert werden. Das experimentelle Spektrum bei Raumtemperatur zeigt im Rahmen der Genauigkeit der DFT/MRCI eine gute Übereinstimmung mit der vermu- teten Emission des Triplett-Charge-Transfer-Übergangs. Auch bei 77 Kelvin lässt sich die Vermutung der Experimentatoren bestätigen. Das experimentelle Spektrum stimmt am besten mit der Überlagerung des TCT- und TLE(batho)-Zustandes überein. Die Emis- sion der Überlagerung des Triplett-Charge-Transfer-Übergangs mit dem lokal angeregten Triplett-Zustand der Dithioquadratsäure könnte zwar auch der Form des experimentellen Spektrums entsprechen, jedoch ist derTLE(dtsq) deutlich rotverschoben.
Um die genannten Ergebnisse und Vermutungen zu den photophysikalischen Eigenschaften des Zink(dtsq)(batho)-Komplexes weiter zu untermauern, bieten sich vor allem Berechun-
38 6 Fazit
gen der ISC-, Strahlungs-, Fluoreszenz- und Phosphoreszenz-Raten an.
Trotz möglicher geeigneter photophysikalischer Eigenschaften des Zink(dtsq)(batho)-Kom- plexes ist der Einsatz als phosphoreszenter Emitter schwierig. Die Experimentatoren ver- muten eine Zersetzung des Moleküls bei Bestrahlung mit UV-Licht. Diese könnte innerhalb des Dithioquadratsäure-Liganden stattfinden, da vor allem die enthaltenen Keto-Gruppen empfindlich gegenüber Photoreduktion sind. Auch die kurzen Lebenszeiten, die vermutlich durch strahlungslose Prozesse hervorgerufen werden und eine Verringerung der internen Quantenausbeute zur Folge haben, schmälern die Attraktivität des Einsatzes in OLEDs, da dies die Effizienz erheblich einschränkt.
Literaturverzeichnis 39
Literaturverzeichnis
[1] https://cynora.com/de/technologie-produkte/oled-displays/, (18.08.2020).
[2] Leo, K.; Lüssem, B.; Polte, A.; Reineke, S.Optik & Photonik 2010,5, 32–35.
[3] Li, W.; Pan, Y.; Xiao, R.Advanced Functional Materials 2014,24, 1609–1614.
[4] Hartmann, F.-F. Neuartige Organische Funktionsmaterialien zum Einsatz in flüssig- phasenprozessierten OLEDs. Ph.D. thesis, 2019.
[5] Fu, H.; Cheng, Y.-M.; Chou, P.-T.; Chi, Y.Materials Today 2011,14, 472 – 479.
[6] Gernert, M.; Müller, U.; Haehnel, M.; Pflaum, J.; Steffen, A.Chemistry – A European Journal 2017,23, 2206–2216.
[7] Barbieri, A.; Accorsi, G.; Armaroli, N. Chemical Communications 2008, 19, 2185–
2193.
[8] Gronlund, P.; Burt, J.; Wacholtz, W.Inorganica Chimica Acta 1995,234, 13–18.
[9] http://www.chemie.uni-jena.de/institute/oc/weiss/lumineszenz.htm, (20.08.2020).
[10] Stohrer, W.-D.Photochemie; Wiley-VCH, 2005; Chapter 2, pp 5–81.
[11] Mustroph, H.; Ernst, S.Chemie in unserer Zeit 2011,45, 256–269.
[12] Püschner, D.Quantitative Rechenverfahren der Theoretischen Chemie; Springer Fach- medien Wiesbaden, 2017.
[13] Becke, A. D.The Journal of Chemical Physics 1993,98, 1372–1377.
[14] Lee, C.; Yang, W.; Parr, R. G.Physical Review B 1988,37, 785–789.
[15] Goerigk, L.; Grimme, S.WIREs Computational Molecular Science2014,4, 576–600.
[16] Marian, C. M.Jahrbuch der Heinrich Heine Universität 2003 2004, 175–194.
[17] Grimme, S.; Waletzke, M.The Journal of Chemical Physics 1999,111, 5645–5655.
[18] Marian, C. M. Praktikumsskript zur Vorlesung Angewandete Quantenchemie und Computerchemie, Heinrich Heine Universität 2020, 67–69.
[19] Kleinschmidt, M.; Marian, C.; Waletzke, M.; Grimme, S. The Journal of chemical physics 2009,130, 044708.
[20] Heil, A.; Kleinschmidt, M.; Marian, C. M. The Journal of Chemical Physics 2018, 149, 164106.
[21] Dreuw, A.; Head-Gordon, M.Chemical Reviews 2005,105, 4009–4037.