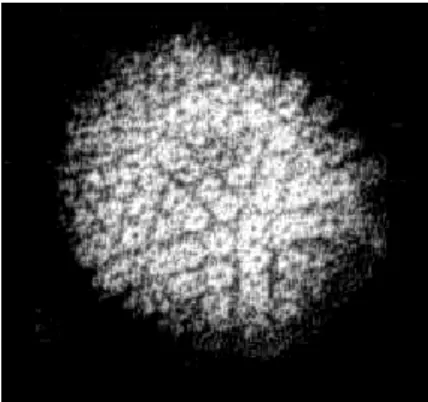
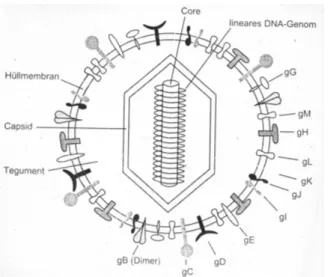
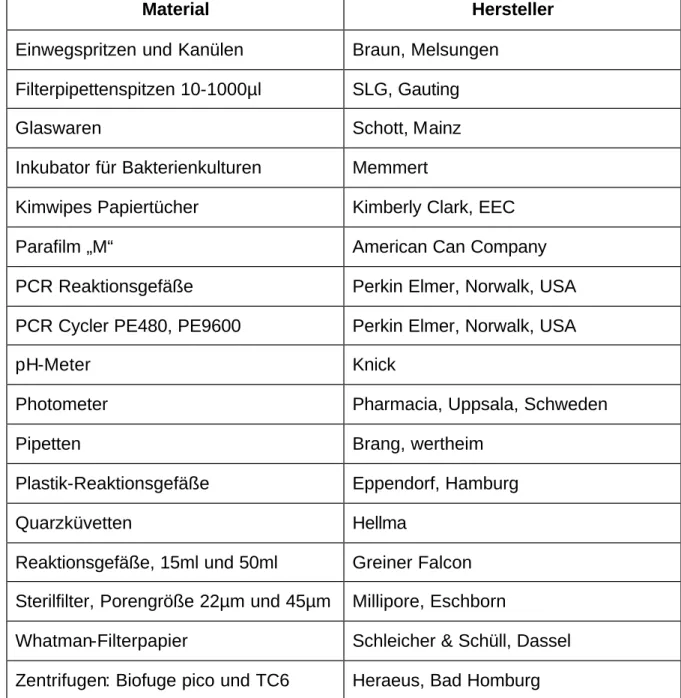
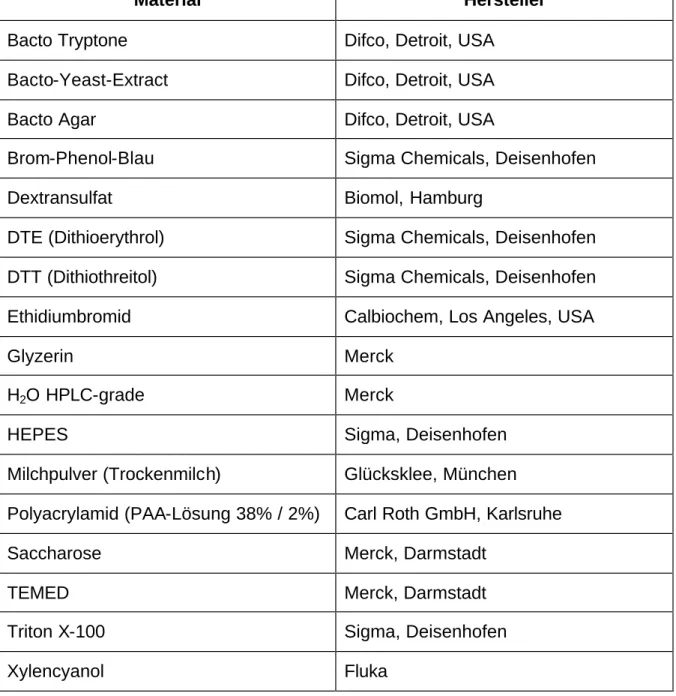
Zur DNA Sequenz eines klinischen Isolaten ähnlichen HCMV (Humanes Cytomegalievirus) Stammes (Toledo):
91
0
0
Volltext
(2)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
(39)
(40)
(41)
Abbildung

+7

ÄHNLICHE DOKUMENTE
Bei den Untersuchungen iiber die Pedimente der Montes de Toledo ergab sich eine Frage, zu der hier wegen der regionalen Beschrankung auf ein relativ kleines Gebiet
Theorem 5.1 characterizes the final payoff allocation in the protocol with non-uniform price for 2-lessor land rental problems.... The disagreement point in this (out of
Um den Baum höchster Parsimonität zu finden, müssen wir berechnen können, wie viele Zustandsänderungen für einen gegebenen Baum nötig sind. Dieser Baum stelle die Phylogenie
Es ist viel sinnvoller, die Sequenzen in die entsprechenden Proteinsequenzen zu übersetzen, diese zu alignieren und dann in den DNS-Sequenzen
Um den Baum höchster Parsimonität zu finden müssen wir berechnen können, wie viele Zustandsänderungen für einen gegebenen Baum nötig sind. Dieser Baum stelle die Phylogenie des
Um den Baum höchster Parsimonität zu finden müssen wir berechnen können, wie viele Zustandsänderungen für einen gegebenen Baum nötig sind. Dieser Baum stelle die Phylogenie des
K U R Z I N F O R M I E R T Fahrverbot – Würde ein Fahrverbot (hier: für einen Monat) einen Berufstätigen (hier einen Koch) zwangsläu- fig den Arbeitsplatz kosten, da wegen
Wenn ein Spiel zum Beispiel für Kontext c 1 eine stabile Strategie erreicht hat, dies aber noch nicht für einen weiteren Kontext gilt, so wird das Spiel weiter laufen. Und zwar
