Philipps-Universität Marburg Fachbereich Chemie
Ausarbeitung des Experimentalvortrages
„Öle und Fette“
Hinweis:
Dieses Protokoll stammt von der Seite www.chids.de (Chemie in der Schule).
Dort können unterschiedliche Materialien für den Schulunterricht heruntergeladen werden, unter anderem hunderte von Experimentalvorträgen so wie der vorliegende:
http://online-media.uni-marburg.de/chemie/chids/veranstaltungen/uebungen_experimentalvortrag.html
Gehalten am 22.06.2006 Referent: Felix von Lehmden Matrikel-Nr. 1460501
Inhalt
1 Einleitung
1.1 Versuch 1: Bestimmung der Iodzahl
1.1.1 Auswertung der indirekten Titration nach Margosches 1.2 Reaktionsmechanismus
1.3 Farbe
2 Struktur
2.1 Versuch 2: Verseifung von Olivenöl 2.2 Geschichte der Seife
2.3 Versuch 3: Wirkung von Seife
3 Anwendungen
3.1 Streichfett
3.1.1 Demonstration 1: Herstellung von Margarine 3.1.2 Versuch 4: Zerstörung einer W/O – Emulsion 3.2 Ricinusöl
3.2.1 Demonstration 2: Chromatographie von Ricinusöl 3.2.2 Versuch 5: Polyester auf Ricinusöl-Basis
3.2.3 Demonstration 3: Reaktivkleben mit Ricinusöl
4 Schulrelevanz
5 Literatur 6 Anhang
1 Einleitung
Fette und fette Öle (auf die Unterscheidung zwischen diesen beiden Kennzeichnungen soll in Kapitel 2 eingegangen werden) werden bereits seit der Steinzeit (ca. 2,5 Mio bis ca. 8000 v.Chr.) vom Menschen gezielt gewonnen und in den Speiseplan integriert. Damit bilden Fette einen wesentlichen Bestandteil der Nahrung. Im Verlauf der letzten Jahrtausende sind die zwei wesentlichen Methoden zur Gewinnung von Fetten bzw. fetten Ölen bis heute prinzipiell unverändert geblieben. Bei Fetten tierischen Ursprungs wird tierisches Gewebe in Wasser gekocht, wobei die Hitze zur Folge hat, dass das Fett aus dem Gewebe herausgeschmolzen wird und schließlich auf der dichteren wässerigen Phase schwimmt. Bei fetten Ölen pflanzlichen Ursprungs werden Pflanzenkerne und -früchte, die reich an fetten Ölen sind (z.B. Sonnenblumenkerne, Raps etc.), maschinell in dafür vorgesehenen Apparaturen ausgepresst (siehe Abb.1 und 2).
Heute kategorisiert man Speiseöle – je nach Herstellungsschritt – in verschiedene Qualitätsstufen. Die "erste Qualität" wird durch die erste, kalte Pressung der frischen Samen gewonnen. Bei der Kaltpressung entsteht eine Wärme von höchstens 35°C, sodass alle natürlichen Inhaltsstoffe erhalten bleiben. Bei der zweiten und dritten Pressung sind die Inhaltsstoffe durch die erhöhte Temperatur zum Teil bereits verändert. Alle weiteren Qualitäten werden durch noch
Abb. 1: Rapsölpresse (Demonstration)
(Quelle: Haus- und Landwirtschaftliche
Schule Öhringen)
stärkeres Pressen, zusätzliches Erhitzen oder durch Extraktion mit chemischen
Lösungsmitteln gewonnen. Dieses fette Öl ist jedoch ungenießbar.
Bei den letzten Pressschritten werden mit hohem Druck, Temperaturen bis zu 270° C und aggressiven Laugen Öle geschaffen, die keinen Eigengeschmack und nur wenige der ursprünglichen Inhaltsstoffe besitzen. Manche Öle müssen aber aus einem anderen Grund raffiniert werden. Sie wären im kaltgepressten Zustand unbrauchbar, weil sie Giftstoffe besitzen, die durch das Raffinieren gelöst werden können. Ein Beispiel dafür ist das Rizinusöl, auf das in Kapitel 3.2 näher eingegangen werden soll) mit seinem Gehalt an giftigem Rizin 1 [1].
1
Bekannt wurde Rizin als Biowaffe im sog. „Regenschirmattentat“ auf den bulgarischen Schriftsteller Georgi Markov in London 1978. Der Täter verletzte das Opfer scheinbar zufällig mit einer präparierten Regenschirmspitze. Dabei wurde ein winziges vor dem Abschuss mit Zucker kandiertes Platinkügelchen von ca. 1 mm Durchmesser in den Abb. 2: Schema einer modernen Rapsölpresse
(Quelle: LTV-Arbeitskreis dezentrale Pflanzenölgewinnung, Weihenstephan)
Die wichtigsten Inhaltsstoffe von kaltgepressten Ölen sind die ungesättigten Fettsäuren, bei den Stoffwechselvorgängen im Körper eine zentrale Rolle spielen, so z.B. zur Biosynthese von Arachidonsäure, die zur Herstellung von Prostaglandinen benötigt wird. Weitere Inhaltsstoffe sind Lecithin, Histidin, Phytosterol, Chlorophyll, Spurenelemente und die Vitamine A, B, D, E, F, H und K, die u.a. als Koenzyme (Vitamin B, H), Radikalfänger (Vitamin E) und Blutgerinnungsfaktor (Vitamin K) im Körper fungieren.
Vergleicht man Fette bzw. fette Öle mit den anderen Hauptbestandteilen der Nahrung, den Kohlenhydraten und Eiweißen, so fällt zunächst der hohe Brennwert gegenüber diesen Bestandteilen auf. Tabelle 1 zeigt die Brennwerte der Nahrungshauptbestandteile in Kilojoule pro Gramm.
Der hohe Brennwert von Fetten und fetten Ölen im Vergleich zu Kohlenhydraten und Eiweiß ergibt sich dadurch, dass die Kohlenstoffatome in den Triacylglyceriden zum größten Teil die Oxidationsstufe –2 aufweisen, wohingegen in Kohlenhydraten und Eiweißen die Kohlenstoffatome durch Bindung an elektronegativere Heteroatome (Sauerstoff, Stickstoff) z.T. eine höhere Oxidationszahl besitzen. Aufgrund der Tatsache, dass der Brennwert den energetischen Unterschied zwischen Ausgangsstoffen und den Verbrennungsproduktion Kohlenstoffdioxid und Wasser kennzeichnet, ist
Oberschenkel des Opfers injiziert. In der Platinkugel fanden sich 2 dünne Röhren, die mit 40 μg Rizin gefüllt waren und die daraufhin kontinuierlich dieses Gift freisetzten. Zunächst als harmloser Zwischenfall abgetan, wurde die Ursache der spät einsetzenden Symptome der Vergiftung erst spät erkannt. Markov starb 3 Tage nach dem Attentat. Das Platinkügelchen wurde bei der Obduktion entdeckt. (Quelle: Die Welt, 31.01.2006)
Brennwert*
Eiweiß 17,1 kJ/g Kohlenhydrat 17,1 kJ/g Fett 38,9 kJ/g
Tabelle 1: Brennwerte der Nahrungshauptbestandteile
(Quelle: Deutsche Gesellschaft für Ernährung e.V.)
ersichtlich, dass zur Verbrennung (Oxidation) von Kohlenhydraten bzw.
Eiweißen weniger Energie aufgewendet werden muss, da im Mittel beim Kohlenstoff bereits eine höhere Oxidationsstufe als –2 (wie bei Fetten) vorliegt.
Im Körper werden Fette im Gegensatz zu Kohlenhydraten nicht durch Glycolyse, bei der aus einem Glucose-Molekül zwei Acetyl-CoA-Moleküle entstehen, sondern via β-Oxidation abgebaut, die z.B. eine C 18 -Fettsäure zu 9 Acetyl-CoA-Molekülen aufspaltet. Die so entstandenen Acetyl-CoA-Moleküle werden zwecks Energiegewinnung in den Citrat-Zyklus eingespeist und unter Gewinnung von Reduktionsäquivalenten (NADH und FADH 2 ) abgebaut. Diese werden in der Atmungskette mit Sauerstoff kontrolliert zu Wasser oxidiert und die dabei frei werdende Energie in Form des Energieäquivalents Adenosintriphosphat (ATP) gespeichert. Um eine Überversorgung mit Energie und die daraus resultierenden Gesundheitsproblee zu vermeiden, sollte daher eine möglichst fettarme Ernährung anvisiert werden. Dies lässt sich auch am Aufbau der sog. „Ernährungspyramide“ erkennen, da fettreiche Nahrungsmittel wie Butter und Kuchen hier an der Spitze stehen, d.h. nur in geringen Mengen aufgenommen werden sollten, wohingegen die Basis der Ernährung aus kohlehydratreichen Lebensmitteln wie Backwaren, Obst und Gemüse bestehen sollte.
Abb. 6: Die Ernährungspyramide
(Quelle: www.wellcon.ch)
1.1 Versuch 1: Bestimmung der Iodzahl
Die Iodzahl ist generell bestimmt als diejenige Masse Iod, die an 100 Gramm einer Probe eines beliebigen Fettes addiert werden kann. Wenn eine Fett-Portion der Masse m(Fett) eine Portion Iod der Masse m(I 2 ) bindet, ergibt sich für die Iodzahl IZ des Fettes:
Ungesättigte Fettsäurereste finden sich in praktisch allen natürlichen Fetten, deren Art (einfach oder mehrfach ungesättigt) und Mengenanteil die Eigenschaften der Fette wesentlich beeinflussen. In der Schule wird die Frage nach dem Gehalt an ungesättigten Fettsäureestern im Rahmen der Diskussion des Schmelzpunktes von Fetten bzw. fetten Ölen (Verknüpfung von Molekülstruktur und Stoffeigenschaft) sowie bei der Frage nach der ernährungsphysiologischen Bedeutung von fetten Ölen relevant. Unter der Prämisse, dass ungesättigte Fettsäuren für den Menschen gesünder sind als gesättigte Fettsäuren, kann die Iodzahl als ein Indikator für die Qualität eines Fettes bzw. fetten Öls angesehen werden. Eine höhere Iodzahl ist damit gleichbedeutend mit einem größeren Anteil an Doppelbindungen in den Fettsäureresten der Triacylglyceride. Eine Unterscheidung zwischen einfach und mehrfach ungesättigten Fettsäureresten ist jedoch aufgrund der Iodzahl nicht möglich. Um den Gehalt an einzelnen Fettsäureresten (z.B. von der Öl- oder Linolsäure) zu bestimmen, ist der Einsatz weiterer analytischer Methoden notwendig (z.B. Ionenchromatographie oder Kapillarelektrophorese).
Um die Iodzahl zu bestimmen, stehen prinzipiell zwei unterschiedliche volumetrische Verfahren zur Verfügung: Die direkte Methode, bei der eine eingewogene Fettmenge mit halogenhaltiger Lösung bis zum Umschlagspunkt titriert wird oder die indirekte Methode, bei der eine abgemessene Menge Halogen zu dem eingewogenen fetten Öl gegeben wird, wobei im Anschluss an
m(Fett)
100
)
IZ m(I
2
die Addition an die Doppelbindungen die verbleibende Menge Halogen mit einem Reduktionsmittel bis zum Umschlag titriert wird. Zu der ersten Klasse der direkten Verfahren gehören dabei das Verfahren nach Wijs (Lösung von Iodtrichlorid), das Verfahren nach Hofmann/Green (Iodtrichlorid mit Quecksilber(II)acetat), das Verfahren nach Hanus (Iodmonobromid in Essigsäure) sowie das Verfahren nach Kaufmann (Brom in Methanol mit Natriumbromid gesättigt). Als Beispiel für die zweite Klasse von Verfahren der indirekten Methoden zur Iodzahlbestimmung kann das Verfahren nach Margosches angeführt werden, bei dem die Fettmenge mit ethanolischer Iod- Lösung im Überschuss umgesetzt wird und man nach fünf Minuten mit Thiosulfat-Lösung zurücktitriert. Prinzipiell ist festzuhalten, dass bei einigen der angewendeten Verfahren nicht Iod, sondern ein anderes Halogen bzw.
Interhalogen zwecks Addition and die Doppelbindungen der Fettsäurereste eingesetzt wird, da die Addition von Iod an Doppelbindungen im Gegensatz zu Brom oder Interhalogenen reversibel ist und so zu Fehlern bei der Umsetzung führen kann. Trotzdem wird augrund der Tradition bei der Berechnung stets die molare Masse von Iod zu Grunde gelegt. In der Schule stehen im Wesentlichen zwei Methoden zur Verfügung, die bei annehmbarem Planungsaufwand mit den Schülerinnen und Schülern durchgeführt werden können. Einerseits kann die direkte Titration nach Winkler in modifizierter Form (vgl. den Beitrag von Kugel, W. in: PdN-C 4 (1984), S. 120ff.) durchgeführt werden. Als Lösungsmittel für die eingewogenen Fettportion wird salzsaures 1-Propanol der Qualität „purum“ verwendet, als Halogen kommt in situ hergestelltes Brom in wässriger Lösung der Konzentration c(Br 2 ) = 0,05 mol/L zum Einsatz („Winkler-Lösung“ aus Kaliumbromat und Kaliumbromid im Überschuss).
Andererseits kann in der Schule auch mit dem modifizierten Verfahren nach
Margosches gearbeitet werden, dass ein Beispiel für die indirekte
Iodzahlbestimmung darstellt (Erläuterung s.o.). Bei diesem Verfahren wird die
eingewogenen Fettportion ebenfalls in 1-Propanol gelöst (ggf. das Fett zuvor
leicht im Wasserbad erhitzen) und im Anschluss mit einer abgemessenen Menge
in 1-Propanol gelöstem Iod versetzt. Die Titration des verbliebenen Iods erfolgt mit Natriumthiosulfat-Lösung der Konzentration c(Na 2 S 2 O 3 ) = 0,1 mol/L.
1.1.1 Auswertung der indirekten Titration nach Margosches
Zunächst sind die im Rahmen der Messung festgesetzten bzw. gemessenen Werte angegeben:
Vorgelegte Iodportion (festgesetzt) V(Iod) vorgelegt = 10,0 mL
Blindverbrauch an Thiosulfat-Lösung V’(Na s S 2 O 3(aq) ) blind = 18,4 mL Eingewogene Fettportion (Palmin) m(Palmin) = 0,629 g Eingewogene Fettportion (Rapsöl) m(Rapsöl) = 0,129 g Verbrauch Thiosulfat-Lösung (Palmin) V’(Na s S 2 O 3(aq) ) Palmin = 12,6 ml Verbrauch Thiosulfat-Lösung (Rapsöl) V’(Na s S 2 O 3(aq) ) Rapsöl = 8,5 ml Um die Iodzahl nach dem Verfahren nach Margosches praktisch zu bestimmen (Versuchsvorschrift siehe Anhang), ist es bei der Rechnung notwendig, die Stöchiometrie der Reaktion zu betrachten, um anschließend daraus die Formel zur Berechnung der Iodzahl herzuleiten.
Da ein Molekül Iod mit zwei Thiosulfat-Anionen reagiert, gilt für den Umsatz an Iod am Umschlagspunkt der Titration:
- 2 6(aq) 2,5
(aq) 4 1 - - -
2 3(aq) 2
2 2(aq)
0
2 6(aq) 2,5
4 2
3(aq) 2
2
1 2(aq)
0
O S I
2 O
S 2 I
) (Oxidation 2e
O S O
S 2
) (Reduktion I
2 2e
I
2
) O S ) n(Na
n(I
2
2 2 3(aq)(1)
(2)
2
) O S V(Na )
O S ) c(Na
n(I
2 2 2 3(aq)
2 2 3(aq)
Dies lässt sich durch Einsetzen der Beziehung n = c ∙ V auch anders ausdrücken:
Da insgesamt vorgelegten 10 ml Iod-Lösung (c = 0,1 mol/L) nicht vollständig verbraucht werden, gilt für die verbrauchte (= addierte) Menge Iod:
Die Stoffmenge n(Iod) vorgelegt wurde im Blindversuch (nur Lösungsmittel mit 10 mL der Iod-Lösung versetzt) ermittelt, die Stoffmenge n(Iod) übrig wird als n(Iod) Probe durch Titration mit Thiosulfat-Lösung als n(Iod) Probe bestimmt. Somit lässt sich (4) auch folgendermaßen ausdrücken:
Da die Iodzahl jedoch nicht über die Stoffmenge n, sondern über Masse m definiert ist, muss mit Hilfe der molaren Masse M umgerechnet werden:
Einsetzen von (6) in (5) sowie Umformung ergibt:
Einsetzen von (3) in (7) [c(Na 2 S 2 O 3 ) = 0,1 mol/L und M(I 2 ) = 126,9 g/mol]:
(3)
übrig vorgelegt
addiert
n(Iod) n(Iod)
n(Iod) (4)
M n m
(6)
Probe blind
addiert
n(Iod) n(Iod)
n(Iod) (5)
] ) n(I )
[n(I ) M(I )
m(I
2
2
2 blind
2 Probe(7)
Nun benötigt man die Definition der Iodzahl:
Einsetzen von (8) in (9) ergibt (wobei V in ml):
Bzw. kürzer:
Setzt man in (11) die eingangs aufgeführten empirisch durch Titration bzw.
Einwaage bestimmten Werte ein, so erhält man folgende Iodzahlen:
IZ (Palmin) = 11,7 IZ (Rapsöl) = 97,4
Die Literaturwerte weisen – je nach Quelle – z.T. große Unterschiede auf. So werden beispielsweise die Werte für Kokosfett (Palmin) mit ca. 9 bzw. für Rapsöl mit ca. 102-112 2 angegeben. Es lassen sich jedoch viele abweichende Angaben finden, da es sich nicht um Reinstoffe handelt. Allerdings liefert das hier durchgeführte Verfahren in der Größenordnung ein korrektes Ergebnis, das auch im Versuch angemessen reproduzierbar (Abweichung < 3 %) ist.
2
Quelle: Widmann, Dr. B. A., „Pflanzenölbetriebene Blockheizkraftwerke Teil I“ Bayerisches Staatsministerium für Landesentwicklung und Umweltfragen (StMLU) München 2002, Tab. 3, S. 11
m(Fett)
ml 1,269 g )
V (V
IZ
blind
Probe
ml 10 g 126,9 ]
) O S V´(Na )
O S [V´(Na )
m(I
2
2 2 3(aq) blind
2 2 3(aq) Probe
4(8)
m(Fett) 100 )
IZ m(I
2 (9)
m(Fett) ml
1,269 g ]
) O S V´(Na )
O S [(V`(Na
IZ
2 2 3(aq) blind
2 2 3(aq) Probe
(10)
(11)
1.2 Reaktionsmechanismus
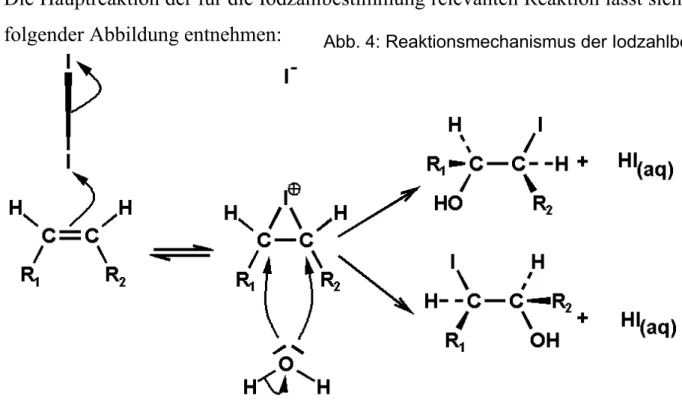
Die Hauptreaktion der für die Iodzahlbestimmung relevanten Reaktion lässt sich folgender Abbildung entnehmen:
Dabei wird die σ-Bindung des Iodmoleküls durch Annäherung an die π- Bindung der Kohlenwasserstoffkette polarisiert, so dass Elektronendichte zum weiter von der Doppelbindung entfernten Iod-Atom verschoben und das näher gelegene Iod-Atom positiv polarisiert wird. Im Anschluss kommt es zum Bindungsbruch, wobei der Iodonium-Komplex und Iodid gebildet wird. Die insgesamt positive Ladung des Iodonium-Komplexes ermöglicht es nun Nukleophilen, an der Rückseite eines der beiden Kohlenstoff-Atome anzugreifen. Nach Abspaltung des Protons bildet sich die abgebildeten Isomere, sowie hydratisierter Iodwasserstoff. Da Iodid ein besseres Nukleophil ist als Wasser oder Hydroxid, könnte man erwarten, dass als Hauptprodukt eine Diiodfettsäure gebildet wird. Dagegen spricht jedoch zunächst, dass Wasser im großen Überschuss zugesetzt wird sowie weiterhin, dass die Addition von Iod an Doppelbindungen reversibel verläuft.
Abb. 4: Reaktionsmechanismus der Iodzahlbestimmung
An der Iodzahlbestimmung lassen sich verschiedene chemische Konzepte illustrieren. Einerseits ist hier natürlich die Chemie der Addition von Halogenen an Doppelbindungen zu nennen. Dieser Inhalt, sowie das Thema
„Eliminierung“, ist nach dem Lehrplan des Landes Hessen für die Jahrgangsstufe 12 im Grundkurs fakultativ, im Leistungskurs obligatorisch und damit zentraler Teil der Oberstufenchemie. Andererseits kann die Thematik der Konkurrenzreaktionen anhand der Iodzahlbestimmung besprochen werden.
Konkret lassen sich an diesem Beispiel Lösungsmitteleinfluss (Wasser ist protisch und polar), Vergleich von Nukleophilen für den Rückseitenangriff (Hydroxid vs. Iodid) und der Konzentrationseinfluss aufgreifen und gemeinsam mit den Schülerinnen und Schülern diskutieren. Durch die Titration, die von den Schülern selbst durchgeführt werden kann, wird zusätzlich eine elementare praktische Fähigkeit des Chemieunterrichts trainiert. Weiterhin kann das Thema
„Redoxreaktionen“ anhand der Reduktion des Überschüssigen Iods durch Thiosulfat wiederholt werden. Somit ermöglicht die Iodzahlbestimmung sowohl die Erarbeitung neuer Aspekte als auch die Wiederholung von bereits gelernten Inhalten.
1.3 Farbe
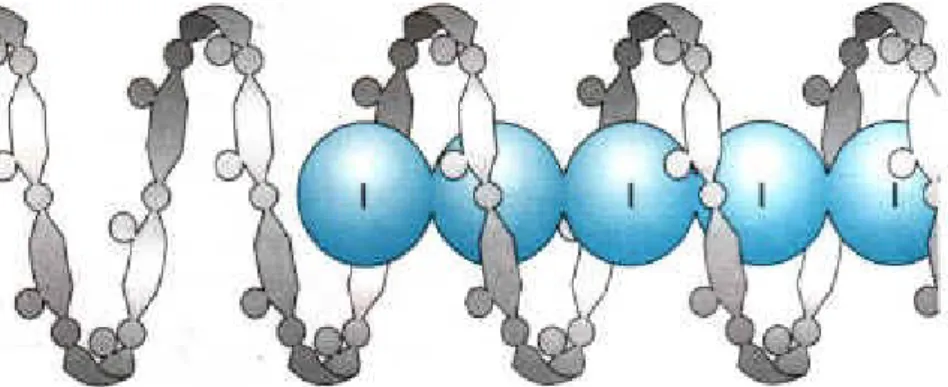
Die dunkelviolette Farbe der Fettlösung nach Zugabe von Stärke-Lösung (w = 0,01) ist durch die Ausbildung eines Charge-Transfer-Komplexes zu erklären.
Strukturell werden dabei Polyiodid-Ionen (I 3 - , I 5 - ) in den „Sesseln“ der Amylose eingelagert (siehe Abb. 5).
Abb. 5: Einschluss von Polyiodid-Ionen in Amylose
(Quelle: www.bio-modelle.de)
C
C
C O O
O C
C
C H
H H
H
H O
O
O
R 1
R 2
R 3
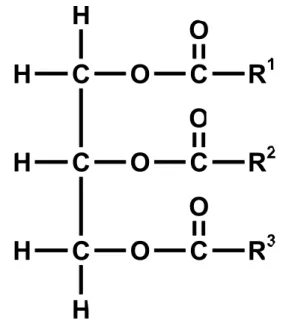
Abb. 7: Allg. Struktur eines Triacylglycerids
Durch Lichtanregung können nun Elektronen vom Donor [(Poly-)Iodid] zum Akzeptor (Iod) verschoben werden. Der Rest des Spektrums des eingestrahlten Lichtes verursacht den für das Auge sichtbaren Farbeindruck (siehe Abb. 6)
2 Struktur
Fette bzw. fette Öle lassen sich chemisch gesehen als 1,2,3-Propantriolester (siehe Abb. 7) von Fettsäuren der Kettenlänge C 8-12 (mittlere Fettsäuren) bzw.
C 12-18 (höhere Fettsäuren) auffassen, wobei auch die Trivialnamen „Triglyceride“
oder „Triacylglyceride“ (IUPAC) gebräuchlich sind.
Abb. 6: Schema der Absorption von Licht zur Anregung von Elektronen
(Zeichnung hergestellt mit Microsoft Paint)
Die Namen leiten sich dabei vom Glycerin, dem Trivialnamen des 1,2,3- Propantriols ab. Als „fette Öle“ werden dabei Triacylglyceride bezeichnet, die bei Raumtemperatur und Atmosphärendruck flüssig sind, als „Fette“
dementsprechend unter diesen Bedingungen feste Triacylglyceride. Die am häufigsten vorkommenden Fettsäuren sind C 16-18 -Körper, d.h. beispielsweise Palmitinsäure, Stearinsäure, Ölsäure, Linolsäure und Linolensäure. Einen deutlich kleineren Anteil am natürlichen Gesamtfettaufkommen haben pflanzliche Fette, die mittelkettige Fettsäuren C 10-14 (Caprinsäure, Laurinsäure und Myristinsäure) enthalten. Im Alltag sind Fettsäuren besonders in Form von Natrium- oder Kaliumsalzen als Seifen gebräuchlich (siehe Kap. 2.1). Bei der Unterteilung der Fettsäuren gibt es verschiedene mögliche Einteilungen. Erstens gibt es die biologisch relevante Unterscheidung von essentiellen und nicht essentiellen Fettsäuren. Zur ersten Klasse zählen dabei primär Linol- und Linolensäure, da sie vom menschlichen Organismus nicht synthetisiert werden können und daher mit der Nahrung aufgenommen werden müssen (also
„essentiell“ sind). Zweitens können Fettsäuren bezüglich ihres Gehalts an C-C- Doppelbindungen charakterisiert werden. Enthält der Fettsäurerest keine solchen Bindungen, spricht man von „gesättigten“ Fettsäuren, da kein Wasserstoff mehr addiert werden kann. Enthält der Fettsäurerest eine C-C-Doppelbindung, spricht man von „einfach ungesättigten“ Fettsäuren und entsprechend von „zweifach“
bzw. „dreifach ungesättigten“ Fettsäuren bei zwei bzw. drei C-C- Doppelbindungen. Dieser Aspekt hat Folgen für die physikalisch-chemischen Eigenschaften von Fetten und fetten Ölen, da die in der Natur vorkommenden ungesättigten Fettsäuren stets cis-Konfiguration besitzen, was strukturell einem
„Knick“ in der Kohlenwasserstoffkette gleichkommt. Die π-Bindung erlaubt
zudem keine Rotation um die C-C-Bindung, was zur erhöhter Starrheit der Kette
führt. Bedingt durch den „Knick“ und die Starrheit der Kohlenwasserstoffkette
in ungesättigten Fetten bzw. fetten Ölen, liegt der Schmelzpunkt wesentlich
niedriger als bei den korrespondierenden gesättigten Fetten, da letztere sich gut
aneinander anlagern können und somit stärkere London-Kräfte wirksam sind,
die beim Erhitzen erst aufgebrochen werden müssen. Daraus resultiert, dass bei erhöhter Kettenlänge eine Zunahme des Schmelzpunktes zu erwarten ist. Diese beiden Sachverhalte illustriert folgende Tabelle, in der die Schmelzpunkte einiger Fettsäuren aufgeführt sind:
SmP. in °C
Caprinsäure (C 10 ) 31,0
Palmitinsäure (C 16 ) 62,8
Stearinsäure (C 18 ) 69,6
Ölsäure (C 18:1 ) 16,0
2.1 Versuch 2: Verseifung von Olivenöl
C O OH
C O OH
C O OH
C O OH
Die alkalische Esterspaltung ist als Sonderfall der Verseifung, die überdies noch die hydrolytische Esterspaltung durch Säuren oder Enzyme (Esterasen) umfasst, ein zentrales Thema in der organischen Chemie der Oberstufe. Bei der hier durchgeführten alkalischen Esterspaltung werden die Triacylglyceride in Ethanol gelöst und mit 30prozentiger Kaliumhydroxid-Lösung umgesetzt.
Mechanistisch greift in einem ersten Schritt das Hydroxid-Anion am positiv polarisierten Kohlenstoffatom der Esterbindung an, wodurch das bindende π- Elektronenpaar der Carbonylgruppe zum Sauerstoffatom verschoben wird. Das Produkt dieses nukleophilen Angriffes stellt die tetraedrische negativ geladene Zwischenstufe dar (vgl. Abb.8). Im nächsten Gleichgewichtsschritt kommt es zum Bindungsbruch der Esterbindung bei gleichzeitiger Rekonfiguration der Carbonylgruppe. Das dabei formal gebildete Alkoholat-Anion spaltet daraufhin sehr schnell ein Proton von der Carbonsäure ab. Dieser Schritt ist irreversibel, da das gebildete Carboxylat-Anion durch Mesomerie sehr gut in wässriger bzw.
ethanolischer Lösung stabilisiert wird.
Durch erneue alkalische Esterspaltung nach gleichem Mechanismus führt schließlich zu einem Glyerin-Molekülen und 3 Fettsäure-Anionen pro Triacylglycerid-Molekül. Als Nachweis des entstandenen Glycerins stehen in der Schule prinzipiell zwei Verfahren zur Verfügung: Der Nachweis von Glycerin als durch Erhitzen gebildetes Acrolein mit Fehling-Reagenz oder der Nachweis als Kupfer(II)-Komplex. Die erste Möglichkeit ist hier jedoch aus zwei Gründen von Nachteil: Erstens ist Acrolein giftig und karzinogen und daher für den Schulversuch wenig geeignet und zweitens liefert der Blindversuch mit dem Lösungsmittel Ethanol ebenfalls einen positive Fehling- Probe beim Erhitzen mit dem Bunsenbrenner. Somit wäre eine positive Reaktion auch bei Abwesenheit von Glycerin bzw. Acrolein zu erwarten. Die zweite Möglichkeit des Glycerinnachweises ist hingegen schneller, wesentlich ungefährlicher und zudem bei Blindprobe negativ (siehe Anhang). Zu dem Reaktionsprodukt der Verseifung werden dabei einige mL Kupfer(II)-sulfat- Lösung (c = 0,1 mol/L) gegeben und gut durchmischt. Es kommt zur Ausbildung des Tris-propantriolato-tricuprat(II)-anions (siehe Abb. 9), das in Lösung tiefblau ist. Wichtig ist dabei ein stark alkalischer pH-Wert, da Protonen von den Hydroxylgruppen des Glycerins abgespalten werden müssen. Dies ist jedoch durch die in der Verseifung eingesetzte 30prozentige Kaliumhydroxid- Lösung gegeben und muss daher nicht separat hinzugegeben werden. Die Blindproben mit Olivenöl und ethanolischem Olivenöl fallen dabei deutlich sichtbar negativ aus.
Abb. 8: Reaktionsmechanismus der Verseifung eines Triacylglycerids
H
2C
HC
H
2C OH OH
OH +
3 3 OH
-Cu
2+3 +
O Cu
O C H
2CH O
Cu O CH
2O O
CH
2CH
CH
2O
Cu O H
2C
CH
H
2C
O
3-
+ 3 H
2O
2.2 Geschichte der Seife
Eine Vorform der heute verwendeten Handseife kannten die Menschen bereits etwa 2500 Jahre v.Chr. Auf einer Tontafel der Sumerer (siehe Abb. 10) ist das erste überlieferte Seifenrezept der Menschheit in Keilschrift verewigt. Die Rezeptur beinhaltet bereits eine Anleitung zum "Kochen" von Seifen aus Pottasche und Ölen. Um an die benötigte Pottasche
zu gelangen, verbrannten die Sumerer wahrscheinlich Tannenzapfen oder Dattelpalmen.
Ein ähnliches Rezept dürften auch die Ägypter des Alten Reiches (ca. 2700-2200 v. Chr.) verwendet haben. Diese Seife der Frühzeit, die wohl eher einer Schmierseife ähnelte, nutzten die Menschen weniger zur Körperreinigung, sondern wohl vorrangig zum Wäsche waschen. Vor allem diente sie ihnen aber als Medizin bei verschiedenen Hautkrankheiten. Die hautreinigende Wirkung von Seife wurde erst relativ
spät von dem römischen Arzt Galenus ca. 200 n.Chr. dokumentiert obwohl die Römer bereits zuvor eine hoch entwickelte Badekultur pflegten. Die Germanen und Gallier waren es dann, die Seife zuerst als Kosmetikum verwendeten, indem sie die aus Ziegen-, Rinder- oder Hirschtalg hergestellte Seife als Bleichmittel für ihre Haare nutzten oder sich mit einer Art Seifen-pomade frisierten. Auch im
Abb. 9: Glycerin-Nachweis mit Kupfer(II)-Sulfat-Lösung
Abb. 10: Tontafel der Sumerer
(Quelle: www.planet-wissen.de)
alten Testament lassen sich zahlreiche Verweis auf die Verwendung von Seife zwecks Reinigung finden, so heißt es beispielsweise in Jeremia 2,22 „Das treulose Volk“ 2,1-37:
„Selbst wenn du dich mit Lauge waschen und noch soviel Seife verwenden wolltest deine Schuld bleibe doch ein Schmutzfleck vor meinen Augen –
Spruch Gottes des Herrn.“
In der weiteren Entwicklung der Seifensiederkunst zeigten sich arabische Stämme als besonders einfallsreich. Sie setzten in ihren Rezepten als erste gebrannten Kalk (CaCO 3 ) ein, um besonders feste Seifen zu erhalten, die sog. „Kalkseife“, die sich heute einfach im Schulversuch durch Zugabe von Calciumchlorid zu dem Produkt der Olivenöl- Verseifung herstellen lässt. Über die Araber kamen diese Erkenntnisse nach Spanien, wo sich bald die ersten Seifenhandelszentren entwickelten. Noch begehrenswerter wurde die Seife durch beigefügte Duftkomponenten und wurde somit zu einem luxuriösen Kosmetikum. In England und in den Niederlanden entwickelte sich sogar eine eigene Seifenindustrie. Im Mittelpunkt der Seifenherstellung standen jedoch weiterhin die Mittelmeerländer mit den Zentren Alicante, Sevilla, Savona, Venedig und Genua. Erstmals finden sich in Seifenstücken eingeprägte Markenzeichen. In Mitteleuropa waren die Zentren der Seifenherstellung Augsburg, Prag, Marseilles und Wien im 14.
Jahrhundert. Im 17. Jahrhundert bildeten sich in den bürgerlichen Häusern die ersten Waschküchen
aus. Zunehmend begannen sich die Naturwissenschaftler auch für die Chemie der Waschmittel zu
interessieren. Entscheidendes für die Entstehung einer Waschmittelindustrie im 19. Jahrhundert hat
der Franzose Michel Eugène Chevreul geleistet. Er prägte den Namen Stearinsäure für eine Fettsäure
talgiger Konsistenz und den Namen Oleinsäure für eine flüssige, ölige Fettsäure. Er entdeckte 1823
zudem, dass sich einfache Fette nicht mit Alkalien verbinden und so Seife bilden, sondern dass sie
zuerst in Fettsäuren und Glycerin zerfallen. Die Seifensiederei profitierte so von den Forschungen
Chevreuls. Ernest Solvay (1838-1922) wurde bei der Suche nach einer Verwendung für das im
väterlichen Gaswerk anfallende Ammoniak fündig. Bald wurde nur noch nach seinem einfacheren und
billigen Verfahren Soda hergestellt. Doch den Seifensiedern fehlten die geeigneten Fette. Die
gepressten Öle und Talge aus dem Inland reichten bei weitem nicht aus, so dass tropische
Pflanzenöle eingeführt werden mussten. Der Durchbruch von der Waschküche mit Kessel zur
Haushaltswaschmaschine gelang erst Anfang der Fünfzigerjahre des 20. Jahrhunderts, als die
Trommelwaschmaschine – die schon einige Jahrzehnte im Gewerbebereich angewendet wurde – als
Haushaltsgerät auf den Markt kam. Weiterhin kam 1907 mit „Persil“ das erste Vollwaschmittel auf den
Markt, das neben Seifenpulver auch Natriumperborat als Bleichmittel und Natriumsilicat als
Stabilisator enthielt. Heute wird eine Vielzahl unterschiedlicher Tenside verwendet:
Tensidklasse Modell Beispiel
Anionische Tenside
Kationische Tenside
Amphotere Tenside
Nichtionische Tenside
2.3 Versuch 3: Wirkung von Seife
Das in der Schule verwendete Tensidmodell erinnert an ein Streichholz, wobei die rote Spitze den hydrophilen Kopf und der lange Holzstiel den hydrophoben Schwanz repräsentiert.
Schematisch lässt sich die im Versuch (siehe Anhang) demonstrierte Waschwirkung von Tensiden in 4 Schritte unterteilen. Zunächst wird die Oberfläche des mit Cereinsrot eingefärbten Speiseöls mit Tensid-Molekülen benetzt. Dadurch, dass die hydrophilen Köpfe an der Oberfläche des Fettes nun zur wässrigen Phase orientiert sind, wird nun die Haftung des Fettes an der Oberfläche des Textils vermindert.
Abb. 12: Streichholz als Tensid-Modell
Abb. 11: Schematische Struktur verschiedener Tenside
(Quelle: www.uni-essen.de)
Im nächsten Schritt wird das Speiseöl weiter von der Oberfläche abgelöst und schließlich durch Micellenbildung gehalten in Lösung gehalten.
In Abb. 16 sind die Kugelmicellen dargestellt, weitere Micellentypen sind in Abb. 17 abgebildet:
Eine weitere wichtige Funktion der Tenside besteht in der Herabsetzung der Oberflächenspannung des Waschwassers, was erst sein Eindringen in das verschmutzte Kleidungsstück ermöglicht und somit zentrale Bedeutung für den Waschvorgang hat. In reinem Wasser kommt es zur Ausbildung von Wasserstoffbrückenbindungen zwischen den Wassermolekülen. Wirken in der Flüssigkeit Wechselwirkungen in alle Raumrichtungen, so ist dies für ein Teilchen an der Oberfläche nicht der Fall. Hier hört die Flüssigkeit auf und die
Abb. 13: Benetzen der Oberfläche Abb. 14: Verminderung der Haftung
Abb. 15: Ablösen des Fettes Abb. 16: Solvatisieren des Fettes
Abb. 17: Verschiedene Micellentypen
(Quelle: www.uni-paderborn.de)
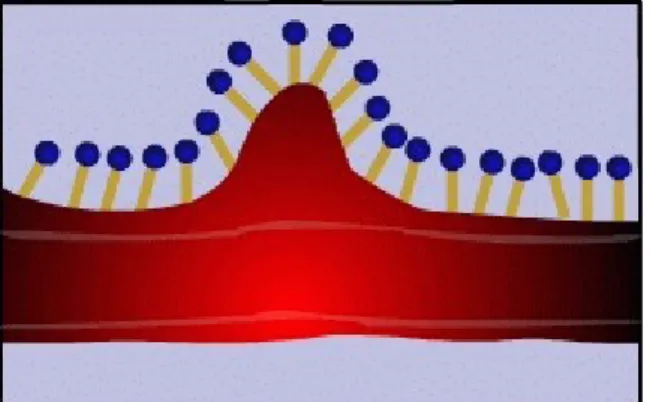
Gasphase beginnt. Ein Teilchen aus der Lösung kann an die Oberfläche gelangen, wenn dafür ein anderes Teilchen von der Oberfläche in die Flüssigkeit wandert. Dieser Vorgang ist energetisch neutral, da genau so viele Bindungen getrennt wie gebildet werden. An der Oberfläche richten sich die Wassermoleküle mit einer Seite des Tetraeders in Richtung der Gasphase aus, um Wechselwirkungen mit benachbarten Molekülen zu maximieren. Dies ist in folgender Abbildung schematisch dargestellt. Dabei stellen die straffierten Linien Wasserstoffbrückenbindungen dar, die dünnen durchgezogenen Linien jeweils die freien Elektronenpaare der Sauerstoffatome.
Prinzipiell ist es auch hier möglich, einzelne Wassermoleküle an der Oberfläche durch Wassermoleküle aus dem inneren der Flüssigkeit auszutauschen. Die Struktur bleibt bei reinem Wasser jedoch erhalten, da sie energetisch durch die maximal mögliche Anzahl an ausgebildeten Wasserstoffbrückenbindungen begünstigt ist. So kommt es schließlich zur Ausbildung einer belastbaren Schicht an der Wasseroberfläche (siehe Wasserläufer). Die Oberflächenspannung von reinem Wasser verhindert somit ein tiefes Eindringen des Wassers in die verschmutzte Kleidung.
Durch den Zusatz von Tensid-Molekülen verändern sich nun die Eigenschaften des Wassers. An der Wasseroberfläche taucht der hydrophile Kopf des Tensid- Moleküls in das Wasser ein, wohingegen der hydrophobe Schwanz zur Gasphase orientiert ist. Die Wassermoleküle in der Umgebung des hydrophilen Kopfes haben nun viele energetisch ähnliche Möglichkeiten, Wechselwirkungen (Wasserstoffbrückenbindungen) mit diesem Teil des Tensid-Moleküls
O H H
O H
H
O H H
O H
H
Gasphase
einzugehen. Die Beweglichkeit der Wasser-Moleküle an der Oberfläche wird erhöht, was zu einer Absenkung der Oberflächenspannung führt.
3 Anwendungen
Im Folgenden sollen zwei zentrale Anwendungen von Fetten bzw. Fettsäuren vorgestellt werden, wobei der Fokus auf der Durchführbarkeit in der Schule liegt, d.h. die Versuche sollen auch als Schülerversuch geeignet sein. Zum Einen soll die Herstellung von Streichfett (Margarine) und zum Anderen die chemischen Einsatzmöglichkeiten von Ricinusöl diskutiert werden.
3.1 Streichfett
Seit etwa Christi Geburt wurden feste Streichfette für die menschliche
Ernährung durch Pressen fettreicher Pflanzensamen oder aus tierischen Fetten
gewonnen. Dabei war die Fettversorgung aufgrund ihres hohen Energiegehalts
(vgl. Kapitel 1) stets eine Säule der menschlichen Ernährung. Aufgrund der
Veränderung der Lebensbedingungen in Europa während der vergangenen 200
Jahre kam es zu einem rapiden Ansteigen der Bevölkerung, was eine Anpassung
der Streichfettversorgung erforderlich machte. Neben weiteren Ursachen sind
hier primär die Industrialisierung und die daraus resultierenden verbesserten
Lebensverhältnisse (Ernährung, Hygiene, medizinische Versorgung etc.) zu
nennen. Allein in der Zeit von 1800 bis 1850 verdoppelte sich die
Bevölkerungszahl in Deutschland. Zunächst konnte die kommerzielle
Fettversorgung, die sich hauptsächlich im landwirtschaftlichen Sektor vollzog,
nicht mit der rasch steigenden Nachfrage mithalten, so dass der Preis von
Streichfetten drastisch anstieg. Dieses Problem erkannte auch Napoleon III., der
1866 den Auftrag erteilte, eine neue Methode der Speisefettgewinnung zu
entwickeln, um die Fettversorgung der französischen Armee zu sichern. Als
C C R 1 R 2
+ H 2 C C
R 1 R 2
H H
H H
H H
<Ni-Kat.>
Reaktion auf diese, mit einer großen Belohung dotierten Ausschreibung, setzten sich viele Wissenschaftler mit dem Problem auseinander, fanden jedoch zunächst keine zufriedenstellende Lösung. Allerdings erkannte Hippolyte Mouriés im Jahr 1869 durch Experimente, dass Kühe auch dann Milchfett abgeben, wenn sie über einen längeren Zeitraum hungern. Dieses Wissen nutzte der Franzose zu dem Schluss, dass Milchfett von Kühen offenbar aus eigenem Körperfett erzeugt werden konnte. Mouriés erhitzte Rindertalk, ließ das reine Fett ausschmelzen und erkalten. Durch Auspressen gewann er die weichen, öligen Bestandteile, das sogenannte „Oleomargarin“. Dieses verbutterte er mit Magermilch. Er erhielt ein haltbares, streichbares Speisefett, das er wegen seines
„perligen“ Schimmerns - in Anlehnung an das Wort "Margaron" (gr.) - Margarine nannte. 1869 ließ sich Mouriés die Erfindung patentieren. Ab 1874 wurde nach seinem Verfahren in Europa und Amerika Margarine produziert.
Diese Alternative bot jedoch keine finale Lösung, da tierische Fette wie bereits erwähnt nicht in ausreichender Menge anfielen. Man wusste zwar, dass fette Öle in Form von Pflanzenfetten billig und in ausreichender Menge verfügbar waren, es mangelte jedoch an einem Verfahren, aus diesem ein festes und streichfähiges Fett herzustellen.
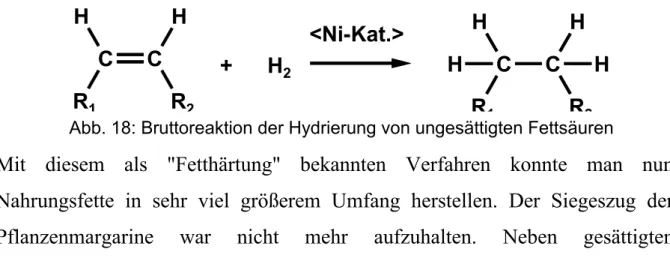
Im Jahre 1902 entdeckte der deutsche Chemiker Wilhelm Normann, dass sich flüssige Pflanzenöle unter Einsatz eines Katalysators aus elementarem Nickel durch Anreicherung der Fettmoleküle mit reinem Wasserstoff (Hydrierung) in feste Fette wandeln lassen.
Mit diesem als "Fetthärtung" bekannten Verfahren konnte man nun Nahrungsfette in sehr viel größerem Umfang herstellen. Der Siegeszug der Pflanzenmargarine war nicht mehr aufzuhalten. Neben gesättigten
Abb. 18: Bruttoreaktion der Hydrierung von ungesättigten Fettsäuren
Triacylglyceriden enthält Margarine 18 bis 20 % entrahmte Milch bzw. Wasser, Emulgatoren wie Lecithine (siehe 3.1.2), die Vitamine A und D und schließlich Aroma- und Farbstoffe natürlicher Herkunft, wie z.B. das Provitamin A, (Karotin). Ferner werden geringe Mengen an Salz sowie ein gesetzlich vorgeschriebenen Zusatz von Stärke als chemisch nachweisbare Substanz zur Unterscheidung von Butter zugesetzt. Der Fettanteil der Margarine muss des Weiteren bei mindestens 80 % liegen. Die wichtigsten Rohstoffe zur Herstellung von Margarine sind Pflanzenöle und Pflanzenfette aus der Sojabohne, Sonnenblumenkernen, Erdnüssen, Baumwollsaat, Kokosnüssen und Palmen.
Raps bzw. Rüben sind die einzigen bedeutenden Ölpflanzen, die im gemäßigten Klima Europas und Ostasiens in größerem Maßstab angebaut werden. Der überwiegende Teil der Ölfrüchte kommt jedoch aufgrund der günstigeren klimatischen Bedingungen aus südlicheren Gefilden (ungefähr zwischen dem 30. Breitengrad oberhalb und unterhalb des Äquators).
Fette und fette Öle werden zur Margarine-Herstellung in einem genau festgelegten Verhältnis zu einer "Fettkombination" zusammengestellt und bereits auf die Bedürfnisse des späteren Einsatzgebietes (z.B. Backen, Brotaufstrich, Braten) abgestimmt. Der heutige Pro-Kopf-Verbrauch an Margarine liegt mit 9,0 kg/a 3 in Deutschland wesentlich höher als der Pro-Kopf- Verbrauch an Butter (6,5 kg/a), was die Bedeutung von Streichfetten auf Pflanzenöl-Basis unterstreicht.
3.1.1 Demonstration 1: Herstellung von Margarine
Wie im vorherigen Kapitel bereits angedeutet, ist die Kennzeichnung
„Margarine“ heute an die Einhaltung bestimmter Kriterien gebunden. Diese werden in der EG-Streichfettverordnung von 1.1.1996 fixiert. So ist Margarine
„Ein bei einer Temperatur von 20°C fest bleibendes, streich- fähiges Erzeugnis in Form einer festen, plastischen Emulsion,
3
Quelle: www.milchindustrie.de
überwiegend nach dem Typ Wasser in Öl [...]“
(EG-Streichfettverordnung vom 1.1.1996)
Im Schulunterricht kann Margarine relativ leicht selbst hergestellt und in weiteren Versuchen verwendet werden und ist daher im experimentellen Chemieunterricht von Interesse. Wird bei der Herstellung auf den Gebrauch von Laborgerät verzichtet, kann die hergestellte Margarine sogar verköstigt werden.
Dazu sind neben einem Küchenmixer nur noch einige Plastikschüsseln erforderlich. Die Zutaten sind in jedem Supermarkt erhältlich. Zunächst werden 100 g Kokosfett (Palmin) erwärmt und in flüssigem Zustand mit 50 g Rapsöl in einer Plastikschüssel vermischt. Anschließend gibt man unter ständigem Rühren nacheinander 15 mL Vollmilch, 15 mL Wasser, 1 g Kochsalz und 1 Eigelb hinzu. Ist alles gut vermengt, stellt man die Plastikschüssel in ein Eisbad und rührt weiter bis das Gemisch sicht verfestigt.
3.1.2 Versuch 4: Zerstörung einer W/O – Emulsion
Der vorliegende Versuch (Vorschrift siehe Anhang) dient im Wesentlichen der
Einführung des Begriffes der Emulsion und des Emulgators (von lat. emulgere =
ausmelken). Der einfache Versuch zeigt den Unterschied zwischen einem
Reinstoff, dem praktisch wasserfreien Kokosfett und dem kolloiddispersen
Stoffgemenge Margarine in Form einer Emulsion. Dabei ist die Micellengröße
in der Regel nicht einheitlich und schwankt in einem Bereich zwischen 10 -2 und
10 -6 m. Als Emulgator fungieren die in der selbst hergestellten Margarine
enthaltenen Phosphatidylcholine (früher Lecithine) aus dem Eigelb, deren
allgemeine Struktur in Abb. 19 dargestellt ist (Reste R 1 und R 2 :
Kohlenwasserstoffketten der Kettenlänge C 15 oder C 17 und bis zu 4
Dopelbindungen). Wie bei Tensiden ist die grenzflächenaktive Wirkung des
Emulgators auf die intramolekular-räumliche Separation von hydrophilen und
hydrophoben Bereichen zurückzuführen.
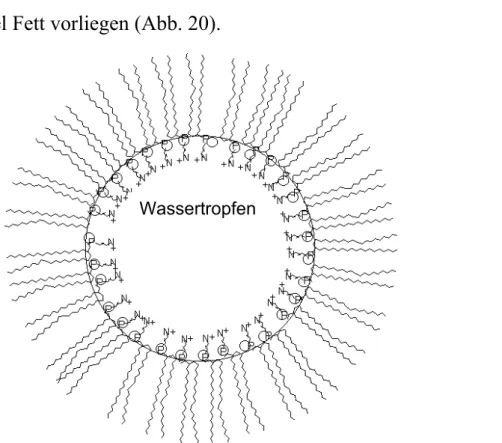
Da Margarine eine Emulsion vom Typ „Wasser in Öl“ (W/O) mit einem Wassergehalt von ca. 20% darstellt, kann man sich die Struktur so vorstellen, dass kleine Wassertröpfchen vom Emulgator eingeschlossen im Dispersionsmittel Fett vorliegen (Abb. 20).
Durch das Erhitzen kommt es nun zunächst zu einem Verdampfen des in der Margarine emulgierten Wassers. Dabei werden kleine Tröpfchen flüssigen Fettes mitgerissen, was beim Braten im Haushalt zu Verbrennungen führen kann. Das spontane Aufspritzen von Fetttröpfchen ist während des Versuchs sowie beim Braten am heimischen Herd in Form eines „Brutzelns“ deutlich zu vernehmen. Nach dem Abkühlen im Eisbad bleibt den in der Margarine
Abb. 19: Allg. Struktur eines Phosphatidylcholins (X = Cholinrest)
(Quelle: CD Römpp Chemie Lexikon, 9. Auflage Georg Thieme Verlag Stuttgart)
Abb. 20: Struktur einer Wasser-Phosphatidylcholin-Micelle (Quelle: Chemie Studienseiten www.ch-4.de)
Wassertropfen
enthaltenen Wasser nicht genug Zeit, um wieder vollständig zu emulgieren, so dass es zu einem sichtbaren Zwei-Phasen-System kommt. Zum Vergleich ist bei dem fast wasserfreien Kokosfett sowohl keine Phasentrennung als auch kein
„Brutzeln“ zu beobachten.
3.2 Ricinusöl
In dem nun folgenden Kapitel soll das Ricinusöl als ein fettes Öl vorgestellt werden, dass sich sowohl in seinem chemischen Aufbau als auch in seinen Eigenschaften wesentlich von den bisher diskutierten Fetten und fetten Ölen unterscheidet. Nicht nur hat das Ricinusöl von allen fetten Ölen mit 0,961-0,963 g/cm 3 die größte Dichte, es weist auch ein für fette Öle atypisches Chromatographieverhalten auf (siehe 3.2.1). Gewonnen wird das Ricinusöl durch Kaltpressen aus den Samen der Ricinusstaude (ricinus communis, Abb.21 und 22), die zu diesem Zweck vor allem in Indien, Thailand und Brasilien angebaut wird.
In Deutschland kann Ricinusöl problemlos günstig in der Apotheke erworben werden. Da es zudem ungefährlich ist, eignet es sich hervorragend für den Einsatz im Chemieunterricht. Wie alle anderen hier besprochenen Fette und fetten Öle ist auch das Ricinusöl ein Triacylglycerid, wobei das Glyceringrundgerüst in diesem Fall zu 80 – 85% mit
Abb. 21: Ricinusstaude (ricinus communis) (Quelle : www.enature.com)
Abb. 22: Ricinussamen
(Quelle: www.ruhr-uni-bochum.de)
COOH
HO
Ricinol- (siehe Abb. 23), zu 7% mit Öl-, zu 3% mit Linol-, zu 2% mit Palmin- und zu ca. 1% mit Stearinsäure verestert ist.
Als Speiseöl ist das Ricinusöl aufgrund seines unangenehmen Geschmacks nicht geeignet. Es riecht schwach, ist brennbar, zähflüssig und vor allem unverdaulich, was seinen Einsatz als Abführmittel erklärt. Es erreicht unverdaut den Dünndarm, wo es durch Lipasen zu Glyerin und Ricinolsäure gespalten wird. Die freie Ricinolsäure hat eine antiabsorptive (nicht aufsaugende/verschluckende) und hydragoge (stark abführende) Wirkung. Die Synthese von Prostaglandin E2, das eine Vermehrung der Sekretion von Elektrolyten und Wasser in das Darmlumen bewirkt, wird möglicherweise verstärkt. Der Teil des Rizinusöls, der nicht verseift ist, erhöht die Gleitfähigkeit des Darminhalts und damit die abführende Wirkung. Dabei bleibt die Wirkung aufgrund der Resorption weitgehend auf den Dünndarm beschränkt. Nach der Einnahme tritt die Wirkung bei niedriger Dosierung nach etwa 8 Stunden ein, bei hoher Dosierung schon nach etwa 2-4 Stunden. Der abführende Effekt von Ricinusöl ist bereits seit über 3500 Jahren bekannt und wird auch heute noch zum Teil ausgenutzt. Insbesondere unter Naturheilkundlern erfreut sich Ricinusöl als Abführmittel großer Beliebtheit.
3.2.1 Demonstration 2: Chromatographie von Ricinusöl
Um Aufschluss über die Bestandteile eines Fettes zu erlangen, bietet sich im Schulversuch neben der Papierchromatographie die Dünnschichtchromato- graphie (DC) mit Silicagelplatten an. Als Kontrastmittel werden dabei einige Iod-Kristalle verwendet, die in der luftdichten DC-Kammer liegen und von
Abb. 23: Ricinolsäure
Außen mit Hilfe eines Föns erhitzt werden. In der Literatur 4 findet sich dabei folgende Einteilung:
Betrachtet man das Chromatogramm von Ricinusöl, so fällt auf, dass nahezu keine Färbung an der Stelle auftritt, an der man die Triacylglyceride erwarten würde. Bei allen anderen untersuchten Fetten traten an dieser Stelle hingegen deutliche Orangefärbungen auf. Zu erklären ist dieser Befund durch den Hinweis, dass der Hauptbestandteil des Ricinusöls im Gegensatz zu den anderen bisher diskutierten Fettsäuren Hydroxylgruppen enthält (siehe Abb. 23), die die Polarität deutlich erhöhen und dazu führen, dass der R f -Wert der Ricinolsäure in dem unpolaren Fließmittel Trichlormethan deutlich geringer ist als beispielsweise der R f -Wert von Palmitinsäure. Insofern kann Ricinusöl nicht als
„typisches Triacylglycerid“ bezeichnet werden. Die zusätzliche Hydroxylgruppe der Ricinolsäure ermöglicht so auch chemische Reaktionen, die über die für Fette und fette Öle ansonsten typischen Reaktionen hinaus gehen. Einige davon sollen nun angeführt werden.
4