AUS DEM LEHRSTUHL FÜR INNERE MEDIZIN II PROF. DR. LARS MAIER DER FAKULTÄT FÜR MEDIZIN DER UNIVERSITÄT REGENSBURG
142
0
0
Volltext
(2)(3)
(4)
(5)(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
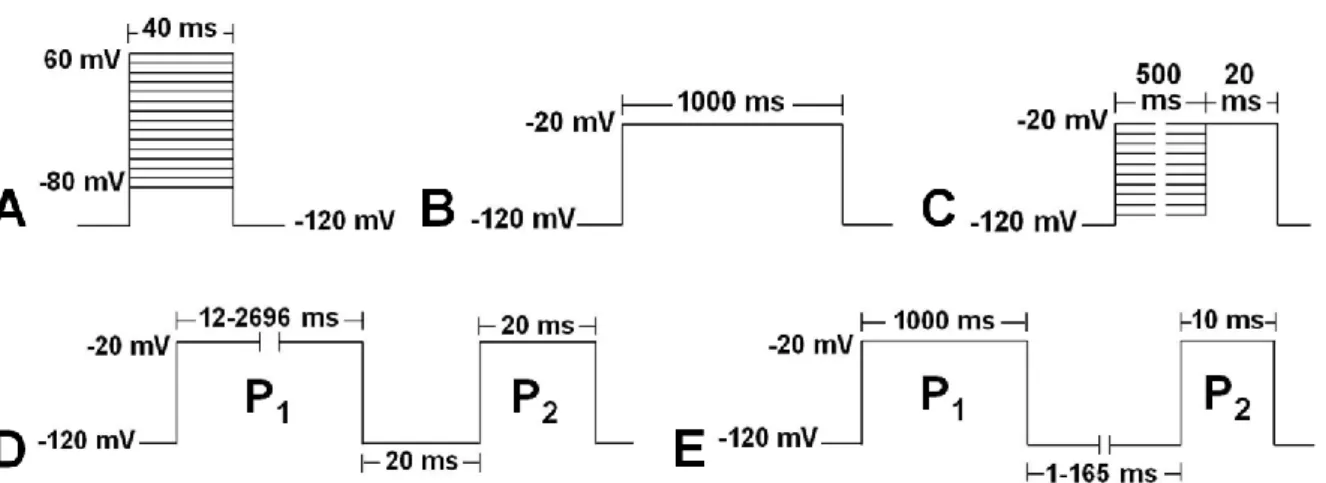
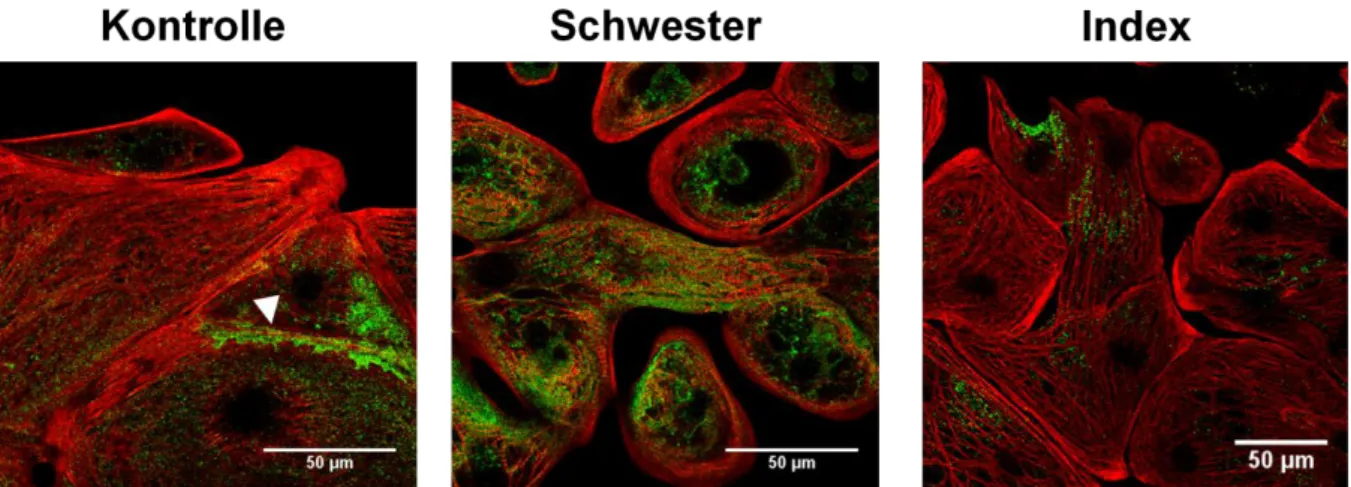
Abbildung
+7
ÄHNLICHE DOKUMENTE
After excluding several patients who were found to have AML at baseline, the overall response rate (complete response and partial response) in patients who were treated with
EFfects of continuous positive airway pressure treatment and withdrawal in patients with obstructive sleep apnea on arterial stiffness and central bp*. Buchner NJ,
Die Gruppe der ER-positiven adipösen Probanden in unserem Studienkollektiv lässt sich durch einen insgesamt gesünderen Phänotyp im Vergleich zu den ER-negativen
Eine chronische GvHD nach allogener SZT führte im Vergleich zum Gesamtkollektiv zwar kaum zu einem längeren progressionsfreien Überleben und Gesamtüberleben, jedoch
Im Gegensatz zum allosterischen CaMKII-Inhibitor KN-93 (grün) ist AS100105 (rot) aufgrund seiner kompetitiven Wirkung an der ATP-Bindungsstelle in der Lage sowohl die
In vorliegender Untersuchung wurde entsprechend den Empfehlungen der Society for Cardiovascular Magnetic Resonance (SCMR) eine Kombination von verschiedenen
Bei den Ergebnissen für NGAL zeigte sich bei der mit Omapatrilat behandelten herzinsuffizienten Gruppe eine leicht, aber nicht signifikant höhere NGAL-Expression als bei
Dabei konnte man die geschlechtsspezifischen Unterschiede in Bezug auf Nüchternglukose auch bei schlanken Männern feststellen, was insbesondere nach Adjus- tierung für Alter,
