Philipps-Universität Marburg Fachbereich 15: Chemie Sommersemester 2010
Übung „Übungen im Experimentalvortrag für Studierende des Lehramts“
Übungsleitung: Prof. Dr. Bernhard Neumüller, Dr. Philipp Reiß
Verfasser: Andreas Trabert Matrikel-Nr.: 2144140 Datum: 30.09.2010
e-Mail: AndreasTrabert@web.de
Inhaltsverzeichnis
1. Einführung in die Thematik...4
2. Definitionen und Terminologie...6
3. Historische Entwicklung...9
4. Systematik und Synthese...11
4.1. Direkte Synthese aus nachwachsenden Rohstoffen...14
4.1.1. Einführung...14
4.1.2. Versuch 1: Synthese thermoplastischer Stärke...14
4.1.2.1. Versuchsdurchführung...14
4.1.2.2. Wissenschaftliche Analyse...17
4.1.2.3. Technische Analyse...20
4.2. Indirekte biologisch-chemische Synthese aus nachwachsenden Rohstoffen...20
4.2.1. Einführung...20
4.2.2. Versuch 2: Synthese von Polymilchsäure...21
4.2.2.1. Versuchsdurchführung...21
4.2.2.2. Wissenschaftliche Analyse...23
4.2.2.3. Technische Analyse...26
4.3. Chemische Synthese aus fossilen Rohstoffen...28
4.3.1.Einführung...28
4.3.2. Versuch 3: Synthese von Polyesteramid...29
4.3.2.1. Versuchsdurchführung...29
4.3.2.2. Wissenschaftliche Analyse...33
4.4. Chemische Synthese von Copolymeren...36
4.5. Chemische Synthese verzweigter Polymere...38
5. Verarbeitung...40
5.1. Industrielle Verarbeitung...40
5.1.1. Einführung...40
5.1.2. Versuch 4: Silanierung thermoplastischer Stärke...44
5.1.2.1. Versuchsdurchführung...44
5.1.2.2. Wissenschaftliche Analyse...46
5.2. Produkteigenschaften und Verwendung...48
5.2.1. Einführung...48
5.2.1. Versuch 5: Nachweis von Stärke in Verpackungsmaterialien...52
5.2.1.2. Versuchsdurchführung...52
5.2.1.3. Wissenschaftliche Analyse...54
5.3. Ökonomische Analyse...54
6. Biologischer Abbau...59
6.1. Ablauf des biologischen Abbaus...59
6.1.1. Einführung...59
6.1.2. Versuch 6: Abiotische Hydrolyse von Polymilchsäure...61
6.1.2.2. Versuchsdurchführung...61
6.1.2.3. Wissenschaftliche Analyse...63
6.1.3. Demonstration 1: Kompostierung...64
6.1.3.1. Versuchsdurchführung...64
6.1.3.2. Wissenschaftliche Analyse...68
6.2. Ökotoxikologische Analyse...70
7. Lebenszyklusanalyse und Ökobilanzierung...71
7.1. Lebenszyklus und Ökobilanz biologisch abbaubarer Kunststoffe...71
7.2. Zusammenfassende Bewertung von Vor- und Nachteilen...76
8. Didaktische Analyse...78
8.1. Schulische Relevanz...78
8.2. Ansätze für fächerübergreifende Projekte...80
Literaturverzeichnis...82
Abbildungsverzeichnis...84
1. Einführung in die Thematik
Die Verschmutzung terrestrischer und mariner Ökosysteme kann als bedeutendes globales Problem der heutigen Zeit angesehen werden. Neben der als „Littern“
bezeichneten, im Allgemeinen bekannten Landschaftsverschmutzung durch das „achtlose Wegwerfen von Produkten“ (vgl. ENDRES et al. 2009, S. 266) existieren weitere, in Gesellschaft und Medien wenig beachtete, jedoch mit weitreichenden Folgen verbundene Probleme im Zusammenhang mit der Belastung von Ökosystemen durch anthropogene Abfälle.
Als bedeutendes Beispiel kann die Belastung der Weltmeere mit langlebigen Kunststoffabfällen aufgeführt werden, die dazu geführt hat, dass global heute mit „bis zu 18.000 Plastikteile[n] in jedem Quadratkilometer der Weltozeane“ gerechnet werden muss (vgl. UNEP 2006, zitiert in GREENPEACE e. V. 2006, S. 1). In pazifischen Zirkulationsströmungen ist mittlerweile „ein nahezu geschlossener Müllteppich“
akkumuliert worden, der die „Größe Zentraleuropas“ erreicht hat (vgl. GREENPEACE e. V.
2006, S. 1). Derartige marine Kunststoffmüllteppiche stellen eine bedeutende Bedrohung für Mensch und Tierwelt dar. Insbesondere Seevögel verwechseln Kunststoffteile mit Nahrung und eröffnen so Kunststoffpartikeln einen Eintritt in die Nahrungskette. Durch die nachgewiesene bindende Wirkung, die die Kunststoffpartikel auf „[w]asserunlösliche, giftige Substanzen wie DDT oder PCB“ ausüben, werden diese ebenfalls in die Nahrungskette eingetragen (vgl. GREENPEACE e. V. 2006, S. 1 – 2).
Vor diesem Hintergrund erscheinen Erzeugnisse wünschenswert, die auch bei nicht fachgerechter Entsorgung keine dauerhafte Belastung für Mensch und Umwelt darstellen.
Als möglicher Lösungsansatz treten immer wieder Kunststofferzeugnisse in Erscheinung, die als „biologisch abbaubar“ deklariert werden. Dabei handelt es sich in chemischer Hinsicht bis auf wenige Ausnahmen um Polymere, die in ihrer Gestalt den herkömmlichen Standardkunststoffen durchaus ähnlich sind, jedoch mit „Sollbruchstellen“ innerhalb der Polymerstruktur ausgestattet wurden, welche sie einem Angriff durch Mikroorganismen zugänglich machen. Solche „Sollbruchstellen“ liegen dann vor, wenn sich Heteroatome,
„insbesondere Sauerstoff und Stickstoff“, zwischen den Kohlenstoffatomen der Polymerkette befinden (vgl. ENDRES et al. 2009, S. 6).
Abb. 1: Aus dem Meer geborgene Kunststoffabfälle.
Quelle: GREENPEACE e. V. o. J. Abb. 2: Animation der Akkumulation von nicht abbaubaren Abfällen im nördlichen Pazifiks. Quelle:
GREENPEACE e. V. o. J.
Die vorliegende Arbeit soll einen objektiven Zugang zu dieser aktuellen und innovativen Thematik schaffen, um das technische, ökonomische und ökologische Potential derartiger Verbindungen im Kontext relevanter Umwelt- und Rohstoffprobleme nach dem heutigen Stand der Wissenschaft beurteilen zu können. Besondere Beachtung soll dabei der naturwissenschaftlichen und ökologischen Bildung im schulischen Bereich zukommen. Die strukturelle Konzeption der Arbeit sieht vor, nach der Schaffung der notwendigen Voraussetzungen den Lebenszyklus biologisch abbaubarer Kunststoffe von der Synthese über die Verarbeitung bis hin zur Entsorgung anhand theoretischer Betrachtungen und praktischer Experimente nachzuzeichnen, um abschließend sowohl auf fachwissenschaftlicher, als auch auf fachdidaktischer Ebene mit einem fundierten Fazit zu schließen.
Einer einleitenden Festlegung einer verbindlichen terminologischen Grundlage in Anlehnung an die heute in Deutschland und zahlreichen anderen Staaten gültigen Normen und Regelungen und kurzen Betrachtung der historischen Entwicklung des Themenfeldes folgt daher die Darstellung der grundlegenden Systematik der Biokunststoffe im Allgemeinen und der biologisch abbaubaren Kunststoffe im Speziellen, welche die Basis für eine theoretische und experimentelle Auseinandersetzung mit den Synthesewegen der heute technisch relevanten Polymere bildet. Den Lebensweg weiter beschreitend, erfolgt ein Überblick über Verarbeitung und Verwendung, dem sich die Analyse des biologischen Abbaus als für diese Kunststoffe charakteristische Verwertungsoption anschließt. Analog zur Vorgehensweise im Falle der Synthesewege werden auch die nachfolgenden Schritte durch Experimente unterstützt, wobei stets beachtet wird, dass eine Umsetzung selbiger innerhalb eines unterrichtlichen Rahmens grundsätzlich möglich ist.
Den Abschluss dieser Arbeit bilden die fachwissenschaftliche und fachdidaktische Bewertung der Thematik. Die Analyse des vorangehend nachvollzogenen Lebenszyklus biologisch abbaubarer Kunststoffe inklusive möglicher Alternativen dient ebenso wie die Ermittlung des heutigen Standes der Ökobilanzierung entsprechender Produkte auf Basis biologisch abbaubarer Kunststoffe der Schaffung einer möglichst objektiven Faktengrundlage, die für die im nachfolgenden Kapitel näher beschriebene Umsetzung der Thematik im Rahmen schulischer und außerschulischer Bildungsvorhaben von hoher Bedeutung ist.
Abb. 3: Strukturausschnitte aus Polymerketten mit Heteroatomen.
Quelle: Eigene Darstellung nach VAN DER ZEE 2005, S. 21.
Abb. 4:
Strukturausschnitt aus einer Polymerkette ohne Heteroatome. Quelle:
Eigene Darstellung nach VAN DER ZEE 2005, S.
21.
2. Definitionen und Terminologie
Als charakteristische Problematik des Fachgebiets der Polymerchemie, das allgemein häufig unter dem Begriff „Biokunststoffe“ zusammengefasst wird, können die Verwechselungen, Irrtümer und Idealisierungen angesehen werden, die in terminologischer Hinsicht oftmals im Zusammenhang mit dieser Thematik auftreten.
Erklärt werden kann dieses Phänomen primär mit einer bis heute uneinheitlichen Definition entsprechender Begriffe, die durch die aktuell stattfindende Entwicklung und Einführung einer einheitlichen Terminologie „durch nationale und internationale Normungsgremien“
endgültig standardisiert werden soll (vgl. UMWELTBUNDESAMT 2009, S. 3). Neben den bisher noch uneinheitlichen Definitionen tritt als weiteres problematisches Phänomen die irreführende Verwendung von Termini, die ursprünglich dem Bereich der Biokunststoffe entstammen oder solchen sehr ähnlich sind, dabei jedoch jeglicher normierenden Grundlage entbehren, als Marketingbegriffe zur Vortäuschung bzw. Idealisierung bestimmter Eigenschaften von Produkten, die in wissenschaftlicher Hinsicht keine Analogien mit den heute anerkannten Biokunststoffen aufweisen, auf (vgl. EUROPEAN BIOPLASTICS e. V. 2009, S. 3 – 4).
Die beschriebenen Gründe machen eine Erfassung des aktuellen Standes der terminologischen Normierung im Bereich der Biokunststoffe ebenso unabdingbar wie eine Festlegung verbindlicher Definitionen und Termini für die weiteren Inhalte dieser Arbeit.
Eine Orientierung soll dabei weitestgehend an der Systematik und den Festlegungen erfolgen, die das deutsche Umweltbundesamt aktuell vertritt (vgl. UMWELTBUNDESAMT 2009, S. 3 – 4):
• Biokunststoff: Der Terminus „Biokunststoff“ dient als Sammelbegriff für Kunststoffe, die entweder über die Möglichkeit der biologischen Abbaubarkeit oder über eine nachwachsende Rohstoffbasis verfügen. Auch Kunststoffe, die beide Eigenschaften in sich vereinen, werden als Biokunststoffe bezeichnet. Somit umfasst der Begriff „Biokunststoff“ zwei grundlegende, nicht voneinander abhängige Konzepte, die eine weitere terminologische Differenzierung ermöglichen (vgl.
UMWELTBUNDESAMT 2009, S. 3 – 4)
• Biobasiert: Als „biobasiert“ werden unter dem übergeordneten Sammelbegriff der „Biokunststoffe“ Erzeugnisse bezeichnet, „die teilweise oder vollständig aus nachwachsenden Rohstoffen“
hergestellt werden (vgl. UMWELTBUNDESAMT 2009, S. 3). Das ausschließende Kriterium stellt in diesem Fall die Rohstoffbasis dar. Eine Biobasierung kann mit der biologischen Abbaubarkeit eines Erzeugnisses einhergehen, impliziert diese jedoch nicht gezwungenermaßen, sodass auch Erzeugnisse existent sind, die zwar „biobasiert“, jedoch eindeutig nicht biologisch abbaubar und kompostierbar sind. Der Nachweis einer Biobasierung kann auf analytischem Wege („ASTM D-6866“) erfolgen (vgl. EUROPEAN BIOPLASTICS e. V. 2008, S. 3).
• Biologisch abbaubar / kompostierbar: Als „biologisch abbaubar / kompostierbar“
werden unter dem übergeordneten Sammelbegriff der „Biokunststoffe“ Erzeugnisse bezeichnet, die innerhalb einer definierten Zeitspanne unter vorgegebenen Bedingungen zu einem bestimmten Grad „zu Wasser, Kohlen[stoff]dioxid und
Abb. 5: Zertifikat für biobasierte
Produkte. Quelle:
DIN CERTCO 2010.
Biomasse“ zersetzbar sind (vgl. UMWELTBUNDESAMT 2009, S. 3). Das ausschließende Kriterium stellt in diesem Fall die chemische Struktur der einem Erzeugnis zu Grunde liegenden Verbindung dar, die einen biologische Abbau ermöglicht. Eine biologische Abbaubarkeit / Kompostierbarkeit kann mit der Biobasierung eines Erzeugnisses einhergehen, impliziert diese jedoch nicht gezwungenermaßen, sodass auch Erzeugnisse aus nicht nachwachsenden Rohstoffen existent sind, die biologisch abbaubar und kompostierbar sind (vgl. ebd., S. 3). Eine biologische Abbaubarkeit / Kompostierbarkeit muss ebenfalls auf analytischem Wege („EN 13432“, „EN 14995“, „ASTM D-6400“) nachgewiesen werden (vgl. EUROPEAN BIOPLASTICS e. V. 2008, S. 3). Die im europäischen Raum gültigen Normen EN 13432 und EN 14995 definieren dabei folgende Kriterien, die für eine Anerkennung der biologischen Abbaubarkeit / Kompostierbarkeit erfüllt sein müssen:
• Information über alle Inhaltsstoffe, Einhaltung der Grenzwerte für Schwermetalle
• im wässrigen Medium: Nachweis einer Umsetzung von mindestens 90 %des „organischen Materials“ zu Kohlenstoffdioxid in einem Zeitraum von 6 Monaten
• im Kompost: Nachweis von weniger als 10 % der ursprünglichen Stoffmasse an Rückständen bei einer Aussiebung (Maschenweite: 2 mm) nach einem Kompostierungszeitraum von 3 Monaten
• keine negative Beeiflussung des Kompostierungsprozesses
• Bestehen eines agronomischen Tests und eines Ökotoxizitätstests
(vgl. EUROPEAN BIOPLASTICS e. V. o. J.)
• Abbaubar, biologisch abbaubar, oxo-abbaubar, oxo-biologisch abbaubar:
Neben den vorangehend definierten Termini existieren diverse weitere Begriffe, die normgemäße Eigenschaften suggerieren sollen, dabei jedoch keiner der aufgeführten Normen entsprechen und in vielen Fällen einer wissenschaftlich fundierten Nachweisbarkeit entsprechender Eigenschaften entbehren. Dazu zählt auch die Bezeichnung „biologisch abbaubar“ ohne den Zusatz „kompostierbar“. Der Branchenverband der europäischen Biokunststoffindustrie bezeichnet derartige, hauptsächlich zu Marketingzwecken eingesetzte Termini zum Teil als „irreführend“
und „ohne jede Substanz“ (vgl. EUROPEAN BIOPLASTICS e. V. 2009, S. 3).
Besonders eindringlich wird in diesem Zusammenhang vor sog. „oxo-abbaubaren“
bzw. „oxo-biologisch abbaubaren“ Erzeugnissen gewarnt. Im Falle dieser Produkte erfolgt die Zugabe übergangsmetallbasierter Katalysatoren zu „herkömmlichen Kunststoffen wie Polyethylen (PE), Polypropylen (PP), Polystyrol (PS), Polyethylenterephtalat (PET) und manchmal auch Polyvinylchlorid (PVC)“, die, in Kombination mit dem Einsatz von Stabilisatoren zur zeitlichen Steuerung der
Abb. 6: Zertifikat für biologisch abbaubare / kompostierbare Produkte.
Quelle: DIN CERTCO 2010a.
ablaufenden Prozesse, unter Energiezufuhr (Wärme, ultraviolette Strahlung etc.) eine oxidative Spaltung der Kunststoffpolymerketten ermöglicht. Daraus resultiert die Fragmentierung des sichtbaren Kunststofferzeugnisses in „weniger leicht identifizierbare[...]“ Kunststoffteile, die nicht mit einer biologischen Abbaubarkeit / Kompostierbarkeit gleichgesetzt werden kann, aber die Gefahr einer entsprechenden Fehlinterpretation potentiell mit sich bringt (vgl. ebd., S. 3 – 4). Die im Zuge dieser Abbauvariante entstehenden Spaltprodukte weisen in den meisten Fällen Kettenlängen auf, die den Voraussetzungen für einen biologischen Abbau nicht entsprechen (vgl. ENDRES et al. 2009, S. 29). Ein an die oxidative Fragmentierung anschließender biologischer Abbau der entstandenen Kunststoffteile kann somit aktuell nicht endgültig nachgewiesen werden, sodass nicht nur mit einer Akkumulation von Kunststofffragmenten im Boden, sondern auch mit der weitreichenden äolischen und fluvialen Verbreitung in terrestrischen und marinen Ökosystemen inklusive der biologischen Anreicherung in Organismen gerechnet werden muss (vgl. EUROPEAN BIOPLASTICS e. V. 2009, S. 5).
Verbindungen, deren Abbau „durch biologisch Aktivität“ zwar prinzipiell möglich ist, aber nicht innerhalb der normgemäß definierten Zeitspanne erfolgt, werden ausschließlich als
„biologisch abbaubar“ bezeichnet (vgl. ENDRES et al. 2009, S. 27).
3. Historische Entwicklung
Die historische Entwicklung der Biokunststoffe reicht bis zu den Anfängen der modernen Polymerchemie zurück und ist somit deutlich älter, als es die erst in den letzten Jahrzehnten intensivierten Forschungs- und Entwicklungsaktivitäten zunächst vermuten lassen. Die ersten in technischem Maßstab für den Massenmarkt produzierten Kunststoffe waren sowohl biobasiert, als auch biologisch abbaubar / kompostierbar. Der Beginn der technischen Produktion von Celluloid aus Cellulose und Campher ab 1869 stellt somit gleichzeitig den Ausgangspunkt der Entwicklung von
Biokunststoffen dar. Ursprünglich zur Substitution von Elfenbein in Billardkugeln entwickelt, stellte das thermoplastische Celluoid bald eine essentielle Ausgangsverbindung zur Produktion von „Filme[n], dekorative[r] Manufakturware, Brillengestelle[n], Kämme[n], Tischtennisbälle[n]“ und zahlreichen weiteren Erzeugnissen dar (vgl. FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V. 2005, S. 6). Ab 1923 folgte mit Cellophan („Zellglas“) ein weiterer, hauptsächlich zur Herstellung von Verpackungsmaterialien eingesetzter, biobasierter und biologisch abbaubarer / kompostierbarer Kunststoff, der im technischen Maßstab produziert wurde (vgl. ebd., S. 6). Die 1924 erstmals auf biologisch-chemischem Wege zur
technischen Verwendung synthetisierte
Polyhydroxybuttersäure konnte dagegen zu dieser Zeit nicht den Status der kommerziellen Produktion erreichen (vgl.
FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V.
o. J.). Mit der aufkommenden Entwicklung von auf fossilen Rohstoffen basierenden, nicht biologisch abbaubaren
Kunststoffen, die mit Bakelite (1907) einsetzte und über Acrylglas („Plexiglas“, 1930), Nylon, Perlon, Polystyrol und Teflon (1930 – 1950) zu den „heutigen Standardkunststoffen“
Polyethylen und Polypropylen (ab 1956) führte, folgte die fast vollständige Verdrängung der bezüglich der Materialeigenschaften (Celluloid) und in ökonomischer Hinsicht (Cellophan) nicht mehr wettbewerbsfähigen Biokunststoffe aus der Anfangszeit der technischen Kunststoffproduktion (vgl. FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V, 2005, S. 6 – 7). Auf der Basis fossiler Rohstoffe konnten schließlich auch erste biologisch abbaubare / kompostierbare Polymere „wie z. B. Polyvinylalkohole und Polycaprolacton“, zunächst für spezielle „Nischenanwendungen“ realisiert werden (vgl. ENDRES et al. 2009, S. 21).
Die technische Entwicklung biobasierter, biologisch abbaubarer / kompostierbarer Kunststoffe wurde in Forschung und Entwicklung ab 1980 vor dem Hintergrund einer stärkeren argumentativen Gewichtung von Nachhaltigkeitsaspekten wie einer erneuerbaren Rohstoffbasis und „geschlossene[n] Stoffkreisläufe[n]“ eingeläutet (vgl.
FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V. 2005, S. 6). Als eine 1. Generation dieser Werkstoffklasse können nach ENDRES et al. die Erzeugnisse bezeichnet werden, die „gegen Ende der 80-er, Anfang der 90er-Jahre“ eine kommerzielle Verfügbarkeit erreichten (vgl. ENDRES et al. 2009, S. 22). Hierzu zählen neben der 1988 entwickelten thermoplastischen Stärke (vgl. FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V. 2005, S. 12) vor allem „fermentativ hergestellte Polyhydroxyalkanoate“
(vgl. ENDRES et al. 2009, S. 22). Die marktwirtschaftliche Durchsetzung biologisch
Abb. 7: Cellophan. Quelle:
FACHAGENTUR NACHWACHSENDE
ROHSTOFFE e. V. 2005, S. 7.
abbaubarer / kompostierbarer Kunststoffe konnte jedoch erst mit der 2. Generation dieser Werkstoffklasse erreicht werden, die im Kontext verbesserter Materialeigenschaften und günstigerer „politische[r] und wirtschaftliche[r] Rahmenbedingungen“ für „bestimmte Anwendungen […] [eine] zunehmend konkurrenzfähig[e]“ Verwendung ermöglichte (vgl.
ebd., S. 22). Entsprechende Kunststoffe haben in den letzten Jahren sowohl bezüglich der Patentaktivitäten, als auch der Produktionskapazitäten eine rasante Entwicklung erfahren (vgl. FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V. 2005, S. 7 – 8) und im Falle von Stärke- und Polymilchsäurepolymeren sowie verschiedenen, nicht oder nur teilweise biobasierten, biologisch abbaubaren / kompostierbaren Kunststoffen das Niveau großtechnischer Produktion erreicht (vgl. ENDRES et al. 2009, S. 22 – 23), sodass mittlerweile zahlreiche technisch ausgereifte Kunststoffe für verschiedenste Anwendungen zur Verfügung stehen (vgl. UMWELTBUNDESAMT 2007, S. 8).
Jüngste Entwicklungen zeigen eine Tendenz hin zu beständigen Kunststoffen auf Basis nachwachsender Rohstoffe. Vor dem Hintergrund einer „Limitierung […] petrochemische[r]
Rohstoffe“ sollen diese Biokunststoffe der 3. Generation Anwendung in Bereichen finden, in denen eine biologische Abbaubarkeit / Kompostierbarkeit nicht erwünscht ist (vgl.
ENDRES et al. 2009, S. 23). Hierbei erfolgen auch Aktivitäten zur Entwicklung biobasierter Synthesen ursprünglich fossiler Rohstoffe, mittels derer eine Herstellung klassischer Standardkunststoffe wie beispielsweise Polyethylen auf der Basis nachwachsender Rohstoffe ermöglicht werden soll (vgl. ebd., S. 23 – 24).
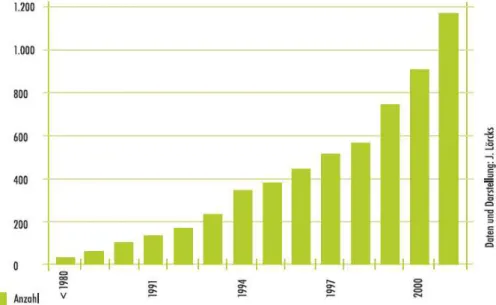
Abb. 8: Entwicklung der Patentaktivitäten im Bereich der modernen
Biokunststoffe. Quelle: FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V., S. 8.
4. Systematik und Synthese
Die heute existente Vielfalt an Produkten und Möglichkeiten im Bereich der Biokunststoffe macht eine nachvollziehbare Struktur für eine adäquate Erfassung der relevanten Inhalte unabdingbar. Daher soll an dieser Stelle eine entsprechende Strukturierung erfolgen, die zugleich die Grundlage für eine nähere Betrachtung relevanter Verbindungen und Synthesen darstellen soll. Eine angemessene Orientierung für diese Zwecke bietet die durch das deutsche Umweltbundesamt publizierte Systematik, die das Themengebiet der Biokunststoffe in 3 elementare Bereiche untergliedert (vgl. UMWELTBUNDESAMT 2009, S. 4 – 5):
Abb. 9: Systematik der Biokunststoffe. Quelle: UMWELTBUNDESAMT 2009, S. 4.
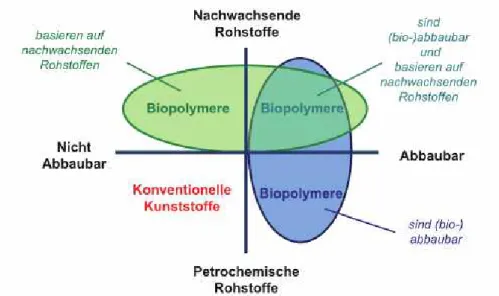
Abb. 10: Differenzierung zwischen Biokunststoffen und konventionellen Kunststoffen. Quelle: ENDRES et al. 2009, S. 6.
• Biologisch nicht abbaubare Biokunststoffe: Die Kategorie „Biologisch nicht abbaubare Biokunststoffe“ umfasst Werkstoffe, die gemäß der in Kapitel 2 definierten Terminologie normgerecht biobasiert, aber nicht biologisch abbaubar / kompostierbar sind (vgl. UMWELTBUNDESAMT 2009, S. 4).
Eine bedeutende Werkstoffklasse innerhalb dieser Kategorie stellen die
„naturfaserverstärkte[n] Kunststoffe“ dar (vgl. UMWELTBUNDESAMT 2009, S. 4).
Hierbei handelt es sich primär um „Standardkunststoffe“, die, analog zur technisch etablierten Verstärkung von Werkstoffen mit Glasfasern, zur Optimierung ihrer Materialeigenschaften mit Naturfasern verstärkt werden, sodass z. B. eine Erhöhung von Steifigkeit und Festigkeit bei gleichzeitig niedriger Dichte erreicht wird. Zur Gewinnung geeigneter Fasern werden hauptsächlich Flachs, Hanf, Jute, Kenaf (sog. „Bastfaserpflanzen“), Sisal, Abaca und Baumwolle eingesetzt (vgl.
FACHAGENTUR NACHWACHSENE ROHSTOFFE e. V. 2010, S. 43). Verwendung finden die ab den 1980er Jahren entwickelten Erzeugnisse aus naturfaserverstärkten Kunststoffen seit Mitte der 1990er Jahre in der Automobilindustrie sowie bei der Fertigung von Strukturbauteilen für Innen- und Außenanwendungen (vgl. ebd., S. 43 – 45).
Eine zweite relevante Werkstoffklasse bilden die „Holz-Kunststoff- Verbundwerkstoffe“. Bei der Herstellung dieser Werkstoffe erfolgt die Kombination von Holz und Kunststoffen mit dem Ziel einer Vereinigung der Materialeigenschaften beider Werkstoffe. Anwendungsbereiche stellen wiederum die Automobilindustrie sowie die Fertigung von Strukturbauteilen, Konsumgütern und Möbelteilen dar (vgl. FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V.
2010, S. 46).
Da diese Arbeit, entsprechend ihrem Titel, ausschließlich biologisch abbaubare / kompostierbare Kunststoffen behandeln soll, findet die Kategorie der biologisch nicht abbaubaren Biokunststoffe in den nachfolgenden Ausführungen keine weitere Berücksichtigung.
• Biologisch abbaubare Biokunststoffe aus nachwachsenden Rohstoffen: Die Kategorie „Biologisch abbaubare Biokunststoffe aus nachwachsenden Rohstoffen“
umfasst Werkstoffe, die gemäß der in Kapitel 2 definierten Terminologie in normgerechter Weise sowohl biobasiert, als auch biologisch abbaubar / kompostierbar sind und stellt ein bedeutendes Gebiet innerhalb der Biokunststoffe dar (vgl. UMWELTBUNDESAMT 2009, S. 4). Resultierend aus der Vielfalt der nutzbaren Rohstoffe und Synthesewege erfolgt innerhalb dieser Kategorie eine weitere Differenzierung in 3 Unterkategorien (vgl. ebd., S. 43 – 45):
• Biologisch abbaubare Biokunststoffe aus nachwachsenden Rohstoffen pflanzlichen Ursprungs: „Biologisch abbaubare Biokunststoffe aus nachwachsenden Rohstoffen pflanzlichen Ursprungs“ sind Kunststoffe, die durch die Modifikation pflanzlicher Rohstoffe gewonnen werden (vgl.
FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V. 2010, S. 38). Eine dominierende Rolle als Rohstoff spielt in dieser Unterkategorie die Stärke, die durch Modifikation in thermoplastische Stärke und anderweitige Stärkederivate überführt werden kann. Weitere relevante Ausgangsverbindungen stellen die Cellulose und ihre Derivate sowie Lignin dar (vgl. UMWELTBUNDESAMT 2009, S. 4 - 5). Eine nähere Betrachtung dieser Kategorie von Biokunststoffen erfolgt in Kapitel 4.1. vor dem Hintergrund der industriell relevanten Synthese von thermoplastischer
Stärke.
• Biologisch abbaubare Biokunststoffe mikroorganismischer Synthese:
Eine zweite Unterkategorie der biologisch abbaubaren Biokunststoffe aus nachwachsenden Rohstoffen bilden die auf mikroorganismischem Wege synthetisierbaren Biokunststoffe (vgl. UMWELTBUNDESAMT 2009, S. 4 – 5).
Den technisch bedeutendsten Vertreter dieser Unterkategorie stellt die Polymilchsäure dar, die durch eine Kombination von biologischen und chemischen Verfahrensschritten indirekt aus Zucker oder Stärke synthetisiert wird. Dabei erfolgt in einem ersten, biologischen Verfahrensschritt die Gewinnung von Milchsäure durch Fermentation der Rohstoffe mit Hilfe von Mikroorganismen (z. B. Milchsäurebakterien), der in einem zweiten, chemischen Schritt die Polymerisation zu Polymilchsäure angeschlossen ist (vgl. FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V. 2005, S. 15).
Als Beispiel für ein technisch relevantes, rein biologisches Verfahren kann die Gewinnung von Polyhydroxyfettsäuren aus Zucker oder Stärke „durch die Einwirkung von Bakterien oder Pilzen“ aufgeführt werden, aus deren Zellen gespeicherte Polyhydroxyfettsäuren direkt extrahiert werden können (vgl.
UMWELTBUNDESAMT 2009, S. 4). Eine weitere Vertiefung dieser Unterkategorie der Biokunststoffe erfolgt in Kapitel 4.2. am Beispiel der Gewinnung von Polymilchsäure.
• Biologisch abbaubare Biokunststoffe aus nachwachsenden Rohstoffen tierischen Ursprungs: Neben pflanzlichen Rohstoffen können auch Rohstoffe tierischen Ursprungs zur Gewinnung von Biokunststoffen verwendet werden (vgl. UMWELTBUNDESAMT 2009, S. 4 – 5). So ist z. B.
die Verarbeitung von „Chitin und Chitosan aus Krabbenschalen“ und tierischen Proteinen wie Gelatine, Casein etc. als Verfahren zur Gewinnung von Biokunststoffen möglich (vgl. FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V. 2005, S. 10). Auf Grund der „untergeordneten Bedeutung“ dieser Kategorie von Biokunststoffen (vgl.
UMWELTBUNDESAMT 2009, S. 4) erfolgt keine nähere Betrachtung im Rahmen dieser Arbeit.
• Biologisch abbaubare Biokunststoffe aus fossilen Rohstoffen: Da die biologische Abbaubarkeit / Kompostierbarkeit von Kunststoffen, wie eingangs erwähnt, nicht zwangsweise mit einer biobasierten Rohstoffquelle einhergehen muss, sondern lediglich von der chemischen Struktur der jeweiligen Verbindung abhängig ist, ist eine Synthese von biologisch abbaubaren / kompostierbaren Kunststoffen auch aus nicht nachwachsenden, fossilen Rohstoffen möglich.
Technische Relevanz besitzen in dieser Kategorie verschiedene Polyester, Polyamide und Copolymere aus unterschiedlichen Monomereineiten, denen sich Kapitel 4.3. widmet.
Die Anzahl an heute verfügbaren Biokunststoffen übertrifft die Dimensionen dieser Arbeit bei weitem, sodass eine Beschränkung auf die relevantesten Polymere der jeweiligen Kategorie erfolgen muss. Eine ausführliche Übersicht über das aktuelle Angebot an Biopolymeren bieten ENDRES et al. (vgl. ENDRES et al. 2009, S. 91 – 175). Eine zentrale Erfassung von Biokunststoffen und ihren Materialeigenschaften erfolgt zudem seit 2006 durch die Fachhochschule Hannover in einer Biopolymerdatenbank (vgl. FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V. o. J.).
4.1. Direkte Synthese aus nachwachsenden Rohstoffen
4.1.1. Einführung
Die Betrachtung der Synthese eines biologisch abbaubaren Biokunststoffs aus nachwachsenden Rohstoffen pflanzlichen Ursprungs erfolgt auf Grund der hohen industriellen Relevanz am Beispiel der Herstellung thermoplastischer Stärke aus Stärke.
Stärke tritt in Form mikroskopischer Körner in einer Vielzahl von Pflanzen als Energiespeicher auf und ist daher beinahe ubiquitär verfügbar. Als gebräuchliche Stärkequellen dienen in „Europa, Amerika und Südafrika“ hauptsächlich „Mais, Weizen und Kartoffeln“, wohingegen in Asien primär „Tapioka“ verwendet wird (vgl.
FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V. 2005, S. 9). Industrielle Verfahren zur Extraktion hochreiner Stärke aus den entsprechenden Rohstoffquellen sind etabliert und liefern aktuell pro Jahr ca. 45 Mio. t Stärke weltweit, wovon ca. 10 Mio. t auf Europa und ca. 2 Mio. t auf Deutschland entfallen. Ungefähr die Hälfte der industriell gewonnenen Stärke findet Verwendung in technischen Verfahren, hierbei hauptsächlich zur Gewinnung von Glucose. Neben der in diesem Kapitel thematisierten direkten Synthese eines Biokunststoffes aus Stärke kann diese, ebenso wie die aus Stärke gewonnene Glucose, auch als Rohstoff für die in Kapitel 4.2. näher beschriebene biologisch-chemische Synthese von Biokunststoffen, beispielsweise durch fermentative Verfahren, genutzt werden (vgl. ebd., S. 9). Der Anteil der zur „Fermentation und [für]
andere Chemieapplikationen“ genutzten Stärke liegt in Deutschland aktuell bei 20 % (vgl.
NOVA-INSTITUT FÜR POLITISCHE UND ÖKOLOGISCHE INNOVATION GmbH 2010, S. 62).
4.1.2. Versuch 1: Synthese thermoplastischer Stärke
4.1.2.1. Versuchsdurchführung (verändert nach KÜHN 1998, S. 55 – 56) Chemikalien:
Substanz Summen-
formel
Konzentration Menge R-Sätze S-Sätze Gefahren- symbol
HessGISS Stärke (aus
Kartoffeln) (C6H10O5)n(s) - 10,4 g - - - Keine
Beschränkungen
Glycerin (wasserfrei) C3H8O3(l) w = 1,00 12 mL 26 36 Xi Keine
Beschränkungen
Wasser (entionisiert) H2O(l) w = 1,00 100 mL - - - Keine
Beschränkungen
Geräte:
• Magnetrührer mit Rührfisch
• Laborwaage
• Becherglas 400 mL
• Becherglas 250 mL
• Messzylinder 100 mL
• Petrischale, Glasscheibe oder Kunststoffplatte
• Glasstab
• Spatel
• Tiegelzange
Aufbau:
Zeitbedarf:
• Vorbereitung: 5 Minuten
• Durchführung: 4 Minuten (Reaktionsablauf) 1 Tag (Trocknung)
• Nachbereitung: 5 Minuten
Abb. 11: Realisierter Versuchsaufbau. Quelle: Eigene Aufnahme.
Durchführung:
10,4 g Kartoffelstärke werden in ein Becherglas 250 mL gegeben, mit 12 mL Glycerin und 100 mL entionisiertem Wasser versetzt und unter Rühren im kochenden Wasserbad erhitzt. Für eine verbesserte Sichtbarkeit des eintretenden Effekts kann das Reaktionsgemisch zusätzlich mit herkömmlichem Lebensmittelfarbstoff angefärbt werden.
Die nach ca. 3 Minuten entstehende Masse wird anschließend auf eine Petrischale, Glasscheibe oder Kunststoffplatte aufgetragen und mit einem Glasstab oder einem anderen geeigneten Werkzeug glatt verstrichen. Nach 1-tägiger Lufttrocknung wird die entstandene Folie von der Trägerfläche durch Abziehen entfernt.
Entsorgung:
Das entstandene Reaktionsprodukt wird als unbelasteter Feststoffabfall im Hausmüll oder Kompost entsorgt.
Beobachtung:
Kartoffelstärke ist ein feines, weißes Pulver, Glycerin eine viskose, klare Flüssigkeit. Durch das Aufschlämmen der Edukte in erhitztem, entionisiertem Wasser entsteht zunächst eine trübe, weiße Suspension, die nach ca. 3 Minuten in ein trübes, weißes Gel übergeht. Nach dem Auftragen auf eine Trägerfläche und 1-tägiger Trocknung entsteht eine flexible, gummiartige Folie.
4.1.2.2. Wissenschaftliche Analyse
Für eine adäquate wissenschaftliche Analyse der Synthese von thermoplastischer Stärke sind die Zusammensetzung und Struktur des einzelnen Stärkekorns von essentieller Relevanz, sodass eine Betrachtung selbiger weiteren Ausführungen vorangestellt wird.
Stärke besteht aus zwei grundlegenden Makromolekülen, Amylose und Amylopectin, die jeweils aus D-Glucose-Monomereinheiten aufgebaut sind. Amylose setzt sich dabei aus fast ausschließlich linear angeordneten, 1,4-glycosidisch verknüpften D-Glucose-Einheiten zusammen, Verzweigungen treten lediglich in einem sehr geringen Maße auf. Als Sekundärstrukturen sind sowohl Einzel-, als auch Doppelhelices existent, das Molekulargewicht von Amylosemolekülen liegt zwischen 105 und 106. Amylopectin besteht, analog zur Amylose, ebenfalls aus D-Glucose-Einheiten, die jedoch neben 1,4- glycosidischen Verknüpfungen zusätzlich 1,6-glycosidische Verknüpfungen aufweisen, welche für eine hochgradige Verzweigung der Moleküle verantwortlich sind.
Entsprechende Verzweigungspunkte treten alle 22 bis 70 D-Glucose-Einheiten auf. Als übergeordnete Sekundärstrukturen liegen einzel- und doppelhelixförimge Bereiche in den Amylopectinmolekülen vor, deren Molekulargewicht zwischen 107 und 109 liegt (vgl.
ZOBEL 1988, zitiert in AVEROUS 2007).
Abb. 12: Reaktionsgemisch nach erfolgter Gelbildung.
Quelle: Eigene Aufnahme. Abb. 13: Auf eine Petrischale aufgetragener Gelfilm.
Quelle: Eigene Aufnahme.
Das Verhältnis zwischen Amylose und Amylopectin variiert je nach pflanzlichem Ursprung der Stärke. Gewöhnlich liegt der Anteil an Amylose zwischen 20 und 30 % und der Anteil an Amylopectin zwischen 70 und 80 %, wobei bestimmte Pflanzensorten durchaus Stärken mit einem Amyloseanteil von lediglich 3 % bis hin zu mehr als 50 % produzieren können (vgl. LI et al. 2001, zitiert in CHAPLIN 2010).
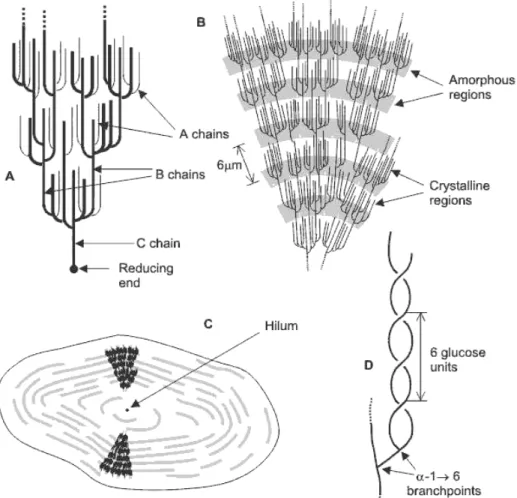
Der strukturelle Aufbau des Stärkekorns wird dominiert durch die radiale Anordnung der Amylopectinmoleküle um eine zentrale Ansatzstelle, das sog. „Hilum“. Zwischen den
Hydroxygruppen der einzelnen Amylopectinmoleküle bestehen
Wasserstoffbrückenbindungen, teilweise unter Beteiligung von Wassermolekülen. Die Verzweigungsbereiche der Amylopectinmoleküle bilden amorphe Zonen innerhalb des Stärkekorns, in denen der maßgebliche Anteil der Amylose eingelagert ist. Die unverzweigten Bereiche der Amylopectinmoleküle bilden dagegen kristalline Zonen, die eine orthogonal zum Radius des Stärkekorns ausgerichtete Lamellenstruktur aufweisen.
Neben der dominierenden doppelhelicalen Sekundärstruktur können in diesem Bereich zudem einzelhelicale Stränge und Einlagerungen von Amylose, freien Fettsäuren und Fetten auftreten (vgl. AVEROUS 2007).
Abb. 14: Ausschnitt aus der Amylose-Struktur.
Quelle: Eigene Darstellung nach AVEROUS 2007.
Abb. 15: Ausschnitt aus der Amylopectin-Struktur.
Quelle: Eigene Darstellung nach AVEROUS 2007.
Abb. 16: Stärkekörner. Quelle: AVEROUS 2007. Abb. 17: Querschnitt eines
Stärkekorns. Quelle: AVEROUS 2007.
Unter der Einwirkung von Wasser und Wärmeenergie erfolgt im Zuge der Synthese thermoplastischer Stärke eine weitestgehende Zerstörung der beschriebenen Strukturen.
Durch das Aufbrechen der Wasserstoffbrückenbindungen und die Einlagerung von Wasser zwischen den einzelnen Makromolekülen tritt einer Quellung der Stärkekörner auf, die schließlich zur Auflösung der semikristallinen Kornstruktur und der Entstehung einer weitestgehend amorphen Masse aus Stärkebestandteilen und zugegebenem Wasser führt. Durch Abkühlung und Trocknung erfolgt die Knüpfung neuer Wasserstoffbrückenbindungen (vgl. LACOURSE et al. 1989, zitiert in AVEROUS 2007).
Neben dem Erhalt einiger kristalliner Strukturen aus den ehemaligen Stärkekörnern können im Laufe der Plastifizierung von Stärke neue kristalline Bereiche entstehen.
Insgesamt bleibt die Kristallinität thermoplastischer Stärke jedoch deutlich hinter der nativer Stärke zurück (vgl. VAN SOEST et al. 1996, zitiert in AVEROUS 2007).
Die Wirkung von Weichmachern wie Glycerin auf thermoplastische Stärke beruht auf der Einlagerung entsprechender Moleküle in die Wasserstoffbrückenbindungen der Polymermatrix. Dadurch treten ab einem bestimmten Wasser- (vgl. MATHEW et al. 2002, zitiert in AVEROUS 2007) und Weichmachergehalt sowohl eine gesteigerte Feuchthaltung, als auch eine durch „Weichmacher-Weichmacher-Interaktionen“ induzierte Quellung des Polymers auf, die zu einem Erweichungseffekt führen (vgl. LOURDIN et al. 1997, zitiert in AVEROUS 2007).
Abb. 18: Struktureller Aufbau eines Stärkekorns. Quelle: CHAPLIN 2010.
4.1.2.3. Technische Analyse
Die technische Darstellung thermoplastischer Stärke erfolgt durch den Einsatz von Extrudern. Hierbei handelt es sich um temperaturregulierte, zylinderförmige Reaktoren, in denen das zuvor aus den Edukten zusammengestellte Reaktionsgemisch durch eine Schnecke bis zu einer Auslassdüse transportiert wird. Der Syntheseprozess gleicht der in Kapitel 4.1.1.3. beschriebenen Synthese im Labormaßstab: Durch das Erhitzen und Umwälzen des Reaktionsgemisches aus nativer Stärke, Wasser und weiteren Additiven erfolgt der Übergang zu thermoplastischer Stärke, die nach anschließender Abkühlung und Trocknung die Möglichkeit einer technischen Weiterverarbeitung bietet (vgl.
FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V. 2005, S. 13).
4.2. Indirekte biologisch-chemische Synthese aus nachwachsenden Rohstoffen
4.2.1. Einführung
Neben der in Kapitel 4.1. beschriebenen direkten Synthese von Biokunststoffen aus nachwachsenden Rohstoffen besteht die Möglichkeit, biobasierte und biologisch abbaubare / kompostierbare Kunststoffe durch biologische und ggf. daran angeschlossene chemische Verfahren zu gewinnen. Im Allgemeinen erfolgt dies durch die fermentative Umsetzung von Stärke oder Saccharose als Rohstoff, die, wie im Falle der Polyhydroxybuttersäuren, zu direkt verwertbaren Biokunststoffen führen oder, wie im Falle der Polymilchsäure, Zwischenprodukte für weitere chemische Verfahrensschritte liefern kann (vgl. UMWELTBUNDESAMT 2009, S. 4 – 5).
Eine nähere Betrachtung dieser Kategorie soll, bedingt durch die hohe technische Relevanz, die Einbindung sowohl biologischer, als auch chemischer Verfahrensschritte und die im Labormaßstab leicht umsetzbare Polymerisationsreaktion am Beispiel der Polymilchsäure erfolgen. Die fermentative Gewinnung der 1780 durch SCHEELE erstmals isolierten Milchsäure aus Stärke, Saccharose und weiteren Verbindungen wurde 1839
Abb. 19: Schematischer Aufbau eines Extruders.
Quelle: FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V. 2005, S. 13.
durch FREMY im Labormaßstab realisiert und erfolgt bereits seit 1881 in industriellen Dimensionen. Heute werden über 90 % der technisch verendeten Milchsäure durch Fermentation gewonnen, als Rohstoffe können mittlerweile eine Vielfalt agrarischer Produkte und Abfälle verwendet werden. Seit den 1960er bestehen zudem Möglichkeiten zur ausschließlich synthetischen Darstellung, die jedoch aus ökonomischen Gründen von lediglich geringer Bedeutung sind. Wirtschaftliche Anwendungsfelder der Milchsäure stellen hauptsächlich die „Lebensmittel-, Pharma- und Kosmetikindustrie“ dar (vgl.
JACOBSEN 2000, S. 54). Die Produktion von Biokunststoffen auf Milchsäurebasis, die auf der 1932 erstmals durch CAROTHERS et al. beschriebenen Synthese „höhermolekularer Milchsäure“ beruht (vgl. CAROTHERS et al. 1932, zitiert in MECKING 2004, S. 1099) und seit den 1970er Jahren technische Verwendung findet (vgl. VERT 2002, zitiert in MECKING 2004, S. 1099), zeigt seit 1996 günstige Zukunftsprognosen für eine großtechnische Verwendung (vgl. BECHTOLD 2003, S. 14). Entsprechende Produktionsanlagen bestehen seit Beginn der 2000er Jahre in mehreren Staaten (vgl.
FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V. 2005, S. 16).
4.2.2. Versuch 2: Synthese von Polymilchsäure
4.2.2.1. Versuchsdurchführung (verändert nach HUNTEMANN et al. 2000, S. 17) Chemikalien:
Substanz Summen-
formel Konzentration Menge R-Sätze S-Sätze Gefahren-
symbol HessGISS
L(+)-Milchsäure C3H6O3(aq) w ≈ 0,9 3 mL 36/38 - Xi Keine
Beschränkungen Zinn(II)-chlorid SnCl2(s) w = 1,00 0,1 g 22-34-37 26-
36/37/39- 45
C Keine
Beschränkungen
Geräte:
• Bunsenbrenner
• Reagenzglas
• Reagenzglasklammer
• Messpipette mit Peleusball
• Petrischale
• Spatel
• Eisbad
Aufbau:
Zeitbedarf:
• Vorbereitung: 3 Minuten
• Durchführung: 4 Minuten
• Nachbereitung: 4 Minuten
Durchführung:
Alle Versuchsschritte werden unter einem laufenden Abzug durchgeführt. 3 mL L(+)- Milchsäure werden in einem Reagenzglas mit 0,1 g Zinn(II)-chlorid versetzt und unter Schütteln ca. 2 Minuten in der leicht rauschenden Brennerflamme erhitzt. Nach erfolgter Reaktion wird das noch flüssige Produkt in eine im Eisbad gekühlte Petrischale überführt.
Entsorgung:
Das entstandene Reaktionsprodukt wird aus der Petrischale entfernt und als chemisch belasteter Feststoffabfall in einem dafür vorgesehenen Behälter entsorgt.
Beobachtung:
L(+)-Milchsäure ist eine viskose, klare Flüssigkeit, Zinn(II)-chlorid ein feines, weißes Pulver. Durch das Erhitzen der Edukte in der leicht rauschenden Brennerflamme tritt im Laufe der 2-minütigen Reaktionszeit unter leichter Rauchentwicklung eine deutliche braune Verfärbung des Reaktionsgemisches auf. Nach dem Überführen in eine im Eisbad
Abb. 20: Realisierter Versuchsaufbau. Quelle: Eigene Aufnahme.
gekühlte Petrischale erstarrt das zuvor flüssige Produkt zu einem harten, braunen Feststoff.
4.2.2.2. Wissenschaftliche Analyse
Milchsäure (2-Hydroxypropansäure) besitzt ein chirales C-Atom und kann somit in Form von 2 Isomeren, der L(+)- Milchsäure und der D(-)-Milchsäure, auftreten. Durch Fermentation wird überwiegend L(+)-Milchsäure gewonnen. Racemische Gemische und reine D(-)-Milchsäure werden durch den
Einsatz „homofermentative[r] Lactobakterien“ oder synthetischer Verfahren erhalten (vgl.
JACOBSEN 2000, S. 54). Für die wissenschaftliche Analyse dieses Versuches wird lediglich die eingesetzte L(+)-Milchsäure berücksichtigt. Jedoch spielt die Stereoisomerie der Milchsäure für eine in Kapitel 4.2.2.3. analysierte Synthese von Milchsäurepolymeren im technischen Maßstab bezüglich verfahrenstechnischer Aspekte und der Anpassung von Produkteigenschaften eine essentielle Rolle.
Die in diesem Versuch durchgeführte Polymerisation von L(+)-Milchsäure zu Polymilchsäure stellt chemisch gesehen eine Polykondensationsreaktion dar (vgl.
HUNTEMANN et al. 2000, S. 17), im Zuge derer über den Mechanismus einer säurekatalysierten Additions-Eliminierungs-Reaktion ein Polyester ausschließlich aus L(+)- Milchsäure-Monomereinheiten gebildet wird. Eine entsprechende Reaktion ist möglich, da die Milchsäure die für eine Veresterung notwendigen Carboxy- und Hydroxygruppen in einem Molekül vereint (vgl. JACOBSEN 2000, S. 54). Dabei ist ein autokatalytischer Verlauf prinzipiell möglich (vgl. HUNTEMANN et al. 2000, S. 17).
Abb. 21: Reaktionsgemisch vor Ablauf der Reaktion. Quelle:
Eigene Aufnahme.
Abb. 22: Reaktionsgemisch nach Ablauf der Reaktion. Quelle:
Eigene Aufnahme.
Abb. 23: Erstarrtes
Reaktionsprodukt. Quelle: Eigene Aufnahme.
Abb. 24: Isomere der Milchsäure. Quelle: Eigene Darstellung nach JACOBSEN 2000, S. 54.
Den 1. Schritt des Reaktionsmechanismus stellt die Protonierung der Carboxygruppe dar, die zu einem delokalisierten Dihydroxycarbenium-Ion als Zwischenprodukt führt, für das 3 mesomere Grenzstrukturen darstellbar sind.
Durch den daraus resultierenden Elektronenmangel wird ein nucleophiler Angriff einer Hydroxygruppe eines zweiten L(+)-Milchsäuremoleküls auf die Carboxygruppe des ersten ermöglicht, der im 2. Schritt des Reaktionsmechanismus erfolgt und zu einem tetraedischen Zwischenprodukt führt.
An dieser Stelle angelangt, sind zwei potentielle Reaktionswege möglich. Einerseits kann durch die Protonierung des der Hydroxygruppe des zweiten L(+)-Milchsäuremoleküls entstammenden Sauerstoffatoms die Rückreaktion zu 2 L(+)-Milchsäure-Monomeren eingeleitet werden. Durch die Protonierung eines der beiden der Carboxygruppe des ersten L(+)-Milchsäuremoleküls entstammenden Sauerstoffatoms kann andererseits eine Weiterreaktion ermöglicht werden, die in einem 3. Schritt unter Wasserabspaltung zu einem Dimilchsäureester führt.
Die Reaktion ist vollständig reversibel und kann durch die Entfernung von Wasser aus dem Reaktionsgemisch in ihrer Gleichgewichtslage zu Gunsten der Produkte beeinflusst werden (abgeleitet nach VOLLHARDT et al. 2007, S. 991 - 992).
Die Weiterreaktion des gebildeten Dimilchsäureesters kann auf 2 Wegen erfolgen, die beide analog zum vorangehend erläuterten Mechanismus zu einer weiteren Esterbildung führen. Zum einen besteht die Möglichkeit einer Reaktion zwischen der endständigen Carboxy- und der endständigen Hydroxygruppe des Dimilchsäureesters, die zum ringförmigen Dilactid führt. Zum anderen ist eine Reaktion der endständigen Carboxy- oder Hydroxygruppe des Dimilchsäureesters mit einer funktionellen Gruppe eines weiteren L(+)-Milchsäuremoleküls möglich, die durch ein analoges Fortschreiten zu einer Polykondesation zahlreicher Moleküle zu Oligomeren mit „molare[n] Massen von 2000 bis 5000 g/mol“ führen kann (vgl. ENTENMANN 1994, zitiert in HUNTEMANN 2000, S. 17).
Wie zu Beginn dieses Kapitels bereits angeführt, verläuft die dargestellte Reaktion durch die in der Milchsäure vorhandenen Protonen „prinzipiell autokatalystisch“ (vgl.
HUNTEMANN 2000, S. 17). In wässriger Lösung neigt Milchsäure zur
„Selbstkondensation“ unter der Ausbildung niedermolekularer Oligomere (vgl. BECHTOLD S. 19). Eine beschleunigte Gleichgewichtseinstellung kann jedoch durch die gezielte Zugabe weiterer Säure, beispielsweise in Form konzentrierter Schwefelsäure, die zusätzliche Protonen zur Verfügung stellt, erreicht werden. Durch die Zugabe von Zinn(II)- chlorid kann zudem eine deutliche Verbesserung der Materialeigenschaften erreicht werden (vgl. HUNTEMANN 2000, S. 17). Dies kann vermutlich mit der Initiation einer Ringöffnungspolymerisation des Dilactids durch das als LEWIS-Säure wirkende Zinn(II)- chlorid erklärt werden, die das Erreichen von Polymerketten mit deutlich höheren Molmassen und daraus resultierend gesteigerten Festigkeitswerten des Polymerfestkörpers ermöglicht (vgl. BECHTOLD 2003, S. 20 – 23). Die im Verlauf der Reaktion auftretende bräunliche Färbung des eigentlich farblosen Produktes lässt auf das Ablaufen von Nebenreaktionen schließen.
4.2.2.3. Technische Analyse
Die Gewinnung von Polymilchsäure in einer technischen Größenordnung nutzt heute weitestgehend Verfahren, die von der in Kapitel 4.1.1.1. analysierten Vorgehensweise im Labormaßstab abweichen. Zwar ist eine Polykondensation von Milchsäuremonomeren
grundsätzlich auch in industriellen Dimensionen möglich, bleibt dabei jedoch auf eine Polykondensation in einem Wasser abführenden Lösungsmittel beschränkt, da lösungsmittelfreie Verfahren keine zufriedenstellenden Ergebnisse im Bezug auf Reaktionszeiten und Molekulargewicht der Polymerketten liefern. Trotz der hohen Reinheit, welche die durch Polykondensation in einem Lösungmittel gewonnenen Polymere aufweisen, bringt die Integration eines solchen Lösungsmittels in verfahrenstechnische Prozesse diverse Probleme mit sich (vgl. JACOBSEN 2000, S. 54 – 55). Als einen bedeutenden Nachteil dieser Vorgehensweise führt BECHTOLD die relativ niedrigen Molekularmassen der entstehenden Polymerketten auf, die mit Werten unterhalb von 104 g/mol außerhalb des Molekularmassenbereichs von 4.104 g/mol bis 3.105 g/mol liegen, der eine ausreichende Festigkeit aliphatischer Polyester für eine technische Verwendung garantiert (vgl. BECHTOLD 2003, S. 23).
Als Alternative zur Polykondensation im Lösungsmittel gilt mittlerweile die ringöffnende Polymerisation von Dilactid als etabliert. Das hierzu notwendige Dilactid wird dabei durch
„Polykondensation [von Milchsäure] und anschließende zyklisierende Depolymerisation“
gewonnen (vgl. BECHTOLD 2003, S. 20). Anschließend erfolgt eine katalysierte Ringöffnungpolymerisation.
Als Katalysatoren eignen sich für dieses Verfahren sowohl LEWIS-Säuren („Metall-Halide, -Oxide und Carboxylate“, in Kombination mit einer eine Hydroxygruppe enthaltenden Verbindung), als auch Metall-Alkoxide (vgl. JACOBSEN 2000, S. 55 – 56), wobei das „für den Kontakt mit Nahrungsmitteln zulässige[...]“ Zinn-Oktoat (Zinn(II)di(ethyl-2-hexanoat) heute eine Schlüsselrolle einnimmt (vgl. ebd., S. 56). Die entscheidenden Vorteile der Ringöffnungspolymerisation von Dilactid stellen im Vergleich zur Polykondensation von Milchsäure-Monomeren die fehlende Notwendigkeit zur Entfernung von Wasser aus dem Reaktionsgemisch mit Hilfe eines Lösungsmittels und die Möglichkeit einer Substitution
„diskontinuierliche[r] Batch-Verfahren“ durch unproblematischere, effektivere
„kontinuierliche Reaktorverahren“ dar, die zur Durchsetzung dieses Synthesewegs in der Industrie geführt haben (vgl. ebd., S. 55). Die durch den bei diesem Verfahren im Kunststoff verbleibenden Katalysator induzierte, nachteilige erhöhte Neigung der Polymermatrix zu thermischen Abbaureaktionen wird durch die Zugabe verschiedener Stabilisatoren kompensiert. Zur Unterscheidung von Milchsäurepolymeren nach dem jeweiligen Ausgangsstoff und Syntheseverfahren erfolgt eine Differenzierung der Begriffe
„Polymilchsäure“ (Polykondensation aus Milchsäure-Monomeren) und „Polylactid“
(Ringöffnungspolymerisation aus Dilactiden), die jedoch beide „das chemisch gleiche Makromolekül“ bezeichnen (vgl. ebd., S. 54 – 55).
Eine bedeutende Rolle für die Beeinflussung der Materialeigenschaften von Milchsäure- Polymeren spielt, unabhängig vom jeweiligen Syntheseweg, die Stereoisomerie von Milchsäure und Dilactid. Aus den 2 existenten Stereoisomeren der monomeren Milchsäure ergeben sich 3 mögliche Stereoisomere des dimeren Dilactids: L,L-Lactid, D,D-Lactid und
meso-Lactid. L,L-Lactid und D,D-Lactid weisen teilkristalline Strukturen auf und verlieren ihre Kristallisationsfähigkeit mit einem steigenden Anteil des jeweils anderen Isomers in der Polymerkette, wohingegen meso-Lactid eine vollständig amorphe Verbindung darstellt (vgl.
JACOBSEN 2000, S. 55). Auf dieser Grundlage basiert die Möglichkeit der Beeinflussung der Materialeigenschaften
wie z. B. der Zersetzungsgeschwindigkeit eines Kunststoffes durch gezielte Kombination von Stereoisomeren. Weitere technische und wirtschaftliche Vorteile der Milchsäurepolymere stellen eine „hohe Festigkeit, […] Transparenz, […], Thermoplastizität“ und die Möglichkeit einer Verarbeitung auf „vorhandenen Anlagen der kunststoffverarbeitenden Industrie“ dar, nachteilig wirkt sich eine niedrige Erweichungstemperatur von ca. 60 °C aus (vgl. FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V. 2005, S. 16).
4.3. Chemische Synthese aus fossilen Rohstoffen
4.3.1.Einführung
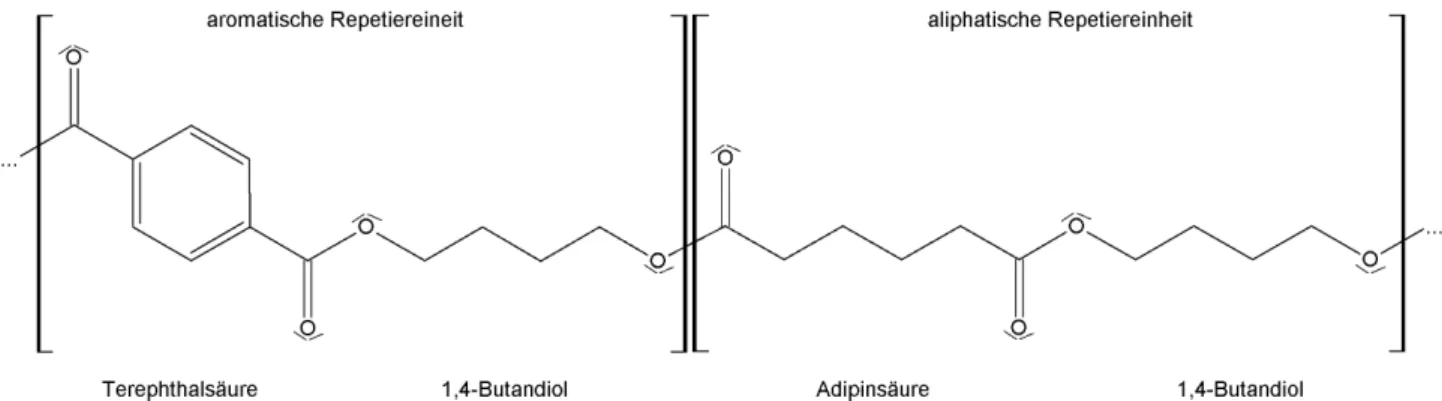
Wie eingangs beschrieben, setzt die biologische Abbaubarkeit / Kompostierbarkeit von Polymeren nicht in zwingender Weise eine Biobasierung der Rohstsoffquelle voraus, sondern resultiert ausschließlich aus der chemischen Struktur der Polymerkette. Neben sterischen Aspekten, die durch die Anteile aliphatischer und aromatischer Monomereinheiten innerhalb des Polymers definiert sind (vgl. MARTEN 2000, S. 11) kommt hierbei der Existenz von Heteroatomen in der Polymerkette eine entscheidende Schlüsselrolle zu (vgl. KLEEBERG 2000, S. 2). Beide Parameter können auch durch die Synthese aus ausschließlich fossilen Rohstoffen derart gestaltet werden, dass eine biologische Abbaubarkeit / Kompostierbarkeit des Produktes vorliegt. Im technischen Maßstab wird diese Möglichkeit in Form der Synthese von Polyester-, Polyamid- und Polyesteramidcopolymeren aus aliphatischen und aromatischen Monomeren genutzt, um die Materialeigenschaften leicht biologisch abbaubarer / kompostierbarer, aber in thermischer und mechanischer Hinsicht oftmals nur in geringem Maße beanspruchbarer, aliphatischer Homopolyester und nicht biologisch abbaubarer / kompostierbarer, aber hinsichtlich ihrer Materialeigenschaften höher beanspruchbarer Polyamide, Polyurethane und aromatischer Homopolyester in einem Polymer zu vereinen (vgl. MARTEN 2000, S. 11). Da auch biobasierte Monomere für entsprechende Synthesen geeignet sind (vgl.
FACHAGENTUR NACHWACHSENDE ROHSTOFFE e. V. 2005, S. 15 – 16), erfolgt eine weiterführende theoretische Betrachtung von Copolymeren in Kapitel 4.4., während dieses Kapitel ausschließlich auf eine fossile Rohstoffbasis beschränkt bleibt. Technisch relevante biologisch abbaubare / kompostierbare Polymere auf Basis fossiler Rohstsoffe stellen Poly(ε-Caprolacton), Polytrimethylencarboant (vgl. BEHNKEN 2008, S. 5 – 6), Poly(butylen-succinat), Poly(butylensuccinat-co-butylenadipat), aliphatische Polyesteramide (vgl. MECKING 2004, S. 1100) und verschiedene aliphatisch-aromatische
Abb. 25: Isomere des Lactids. Quelle: Eigene Darstellung nach JACOBSEN 200, S. 54.
Copolyester auf Basis eines Diols, einer aliphatischen und einer aromatischen Disäure dar (vgl. MARTEN 2000, S. 12 - 13).
Die praktische Umsetzung der Synthese eines biologisch abbaubaren / kompostierbaren Kunststoffs aus fossilen Rohstoffen erfolgt nachfolgend am Beispiel von Polyesteramid.
Obwohl dieses Polymer seit der Einstellung der Produktion durch BAYER aus ökonomischen und rechtlichen Gründen seine technische Relevanz weitestgehend verloren hat (vgl. BAYER AG 2001, zitiert in MECKING 2004, S. 1100), erscheint eine Betrachtung durch die Möglichkeit der kombinierten Analyse zweier unterschiedlicher, in diesem Zusammenhang relevanter Bindungsknüpfungen und die im Vergleich zu anderen Polymeren einfache und unproblematische Umsetzbarkeit im Labormaßstab angemessen (vgl. HUNTEMANN 2000, S. 19 – 20).
4.3.2. Versuch 3: Synthese von Polyesteramid
4.3.2.1. Versuchsdurchführung (verändert nach HUNTEMANN et al. 2000, S. 19 – 20) Chemikalien:
Substanz Summen-
formel
Konzentration Menge R-Sätze S-Sätze Gefahren- symbol
HessGISS Adipinsäure C6H10O4(s) w = 1,00 1,08 g +
8,6 g
36 2 Xi Keine
Beschränkungen 1,6-Diaminohexan C6H16N2(s) w = 1,00 6,84 g 21/22-
34-37
22-26- 36/37/39- 45
C Keine
Beschränkungen
Methanol CH4O(l) w = 1,00 80 mL 11-
23/24/25- 39/23/24/
25
7- 16/36/37- 45
F, T Schülerexperimen te möglich, Exposition für Schwangere und Stillende ausschließen, Ersatzstoffprüfung besonders wichtig Hexamethylendiamin
adipat (AH-Salz)
C12H26N2O4(s )
w = 1,00 1,13 g - - - -
1,4-Butandiol C4H10O2(l) w = 1,00 0,65 g 22 - Xn Keine
Beschränkungen
Geräte:
Abschnitt 1:
• Magnetrührer mit Rührfisch
• Laborwaage
• Saugflasche 500 mL
• Becherglas 250 mL
• Messzylinder 100 mL
• Thermometer
• Büchnertrichter
• Vakuumpumpe
• Trockenschrank
• Spatel Abschnitt 2:
• Bunsenbrenner
• Laborwaage
• Reagenzglas
• Reagenzglasklammer
• Petrischale
• Spatel
• Eisbad
Aufbau:
Zeitbedarf:
Abschnitt 1:
• Vorbereitung: 5 Minuten
• Durchführung: 8 Minuten (Reaktionsablauf und Filtration) 2 Stunden (Trocknung)
• Nachbereitung: 5 Minuten
Abschnitt 2:
• Vorbereitung: 3 Minuten
• Durchführung: 5 Minuten
• Nachbereitung: 4 Minuten
Abb. 27: Realisierter Versuchsaufbau zu Abschnitt 2.
Quelle: Eigene Aufnahme.
Abb. 26: Realisierter Versuchsaufbau zu Abschnitt 1.
Quelle: Eigene Aufnahme.
Durchführung:
Alle Versuchsschritte werden unter einem laufenden Abzug durchgeführt.
Abschnitt 1:
In ein Becherglas 250 mL werden 80 mL Methanol gegeben und 8,6 g Adipinsäure unter Rühren auf dem Magnetrührer darin gelöst. Anschließend werden unter fortgesetztem Rühren 6,84 g 1,6-Diaminohexan zugegeben. Das im Zuge der ablaufenden Reaktion ausfallende Hexamethylendiaminadipat wird durch Filtration abgetrennt und 2 Stunden bei 60°C in einem Trockenschrank getrocknet.
Abschnitt 2:
1,08 g Adipinsäure, 0,65 g 1,4-Butandiol und 1,13 g des in Abschnitt 1 synthetisierten Hexamethylendiaminadipat werden in ein Reagenzglas gegeben und in der leicht rauschenden Brennerflamme zur Reaktion gebracht. Nach 3-minütiger Reaktionszeit wird das noch flüssige Produkt in eine im Eisbad gekühlte Petrischale überführt.
Entsorgung:
Überschüssiges Methanol wird als organischer Lösungsmittelabfall entsorgt. Das entstandene Reaktionsprodukt wird aus der Petrischale entfernt und ebenso wie überschüssiges Hexamethylendiaminadipat als chemisch belasteter Feststoffabfall in einem dafür vorgesehenen Behälter entsorgt.
Beobachtung:
Abschnitt 1:
Adipinsäure und 1,6-Diaminohexan sind weiße Feststoffe, die sich in Methanol vollständig lösen. Ungefähr 10 s nach der Zugabe des 1,6-Diaminohexans fällt unter starker, weißer Trübung und deutlicher Erwärmung der methanolischen Lösung ein Feststoff aus, der nach erfolgter Filtration und Trocknung in Pulverform vorliegt.
Abschnitt 2:
Adipinsäure und das in Abschnitt 1 synthetisierte Hexamethylendiaminadipat sind weiße, pulverförmige Feststoffe. Durch die Vermischung mit farblosem, viskosem 1,4-Butandiol entsteht ein weißes, festes Reaktionsgemisch, das in der leicht rauschenden Brennerflamme zuerst in eine klare Flüssigkeit übergeht, die sich im Laufe der 3-minütigen Reaktionszeit über gelb zu braun verfärbt. Während der Reaktion kann die Kondensation einer klaren Flüssigkeit an der Austrittsöffnung des Reagenzglases beobachtet werden.
Nach dem Überführen in eine im Eisbad gekühlte Petrischale erstarrt das zuvor flüssige Produkt zu einem wachsartigen, braunen Feststoff.
Abb. 28: Reaktionsgemisch vor Ablauf der Reaktion. Quelle:
Eigene Aufnahme.
Abb. 29: Ausfallen des Reaktionsproduktes. Quelle:
Eigene Aufnahme.
Abb. 30: Getrocknetes
Reaktionsprodukt. Quelle: Eigene Aufnahme.
Abb. 31: Reaktionsgemisch vor Ablauf der Reaktion.
Quelle: Eigene Aufnahme. Abb. 32: Erstarrtes Reaktionsprodukt. Quelle: Eigene Aufnahme.
4.3.2.2. Wissenschaftliche Analyse
Im 1. Abschnitt des Versuchs erfolgt die Darstellung von Hexamethylendiaminadipat aus Adipinsäure und 1,6-Diaminohexan. Die ablaufende Reaktion stellt eine Brønsted-Säure- Base-Reaktion dar (vgl. VOLLHARDT et al. 2007, S. 1130). Methanol dient dabei lediglich als Lösungsmittel und nimmt keinen Einfluss auf den Ablauf der Reaktion. Dieser Abschnitt der Reaktion erfolgt analog zur Synthese des Polyamids Nylon 6.6 und dient neben der Einstellung eines äquimolaren Verhältnisses von Adipinsäure- und 1,6-Diaminohexan- Ionen der Aufreinigung der Edukte (vgl. SPECHT 1998, S. 11).
Im 2. Abschnitt des Versuchs erfolgt der parallele Ablauf zweier unterschiedlicher Reaktionen.
Die erste Reaktion stellt eine säurekatalysierte Additions-Eliminierungs-Reaktion von Adipinsäure und 1,4-Butandiol zu einer Ester-Repetiereinheit dar (vgl. HUNTEMANN 2000, S. 20). Der Mechanismus dieser Reaktion gleicht der in Kapitel 4.2.2.2. analysierten Veresterung und wird daher an dieser Stelle nicht näher erläutert.
Die zweite Reaktion findet zwischen den Bestandteilen des im 1. Abschnitt des Versuchs hergestellten Hexamethylendiaminadipats statt (vgl. HUNTEMANN 2000, S. 20). In der Schmelze des Hexamethylendiaminadipats liegt ein Gleichgewicht zwischen den Salzionen und ihren Ausgangsstoffen (vgl. SENS 1988, S. 6). Letztere reagieren, ebenfalls in einer Additions-Eliminierungs-Reaktion, zu einer Amid-Repetiereinheit.
Der Mechanismus dieser Reaktion weist eine gewisse Analogie zur säurekatalysierten Esterbildung auf. Im 1. Schritt der Reaktion erfolgt die Protonierung einer Carboxygruppe der Adipinsäure. Für das entstehende Zwischenprodukt können 3 mesomere Grenzstrukturen ermittelt werden.
Anschließend findet im 2. Schritt der Reaktion ein nucleophiler Angriff einer Aminogruppe des 1,6-Diaminohexans auf die protonierte Carboxygruppe der Adipinsäure statt, aus dem ein tetraedisches Zwischenprodukt hervorgeht.
Im 3. Schritt des Reaktionsmechanismus entsteht schließlich unter Wasserabspaltung ein Amid.
Analog zur Esterbildung ist auch die Amidbildung reversibel (vgl. VOLLHARDT et al. 2007, S. 994).