Identification and functional characterization of mPDCA-1 as a novel antigen-uptake receptor
on murine plasmacytoid dendritic cells
enabling (cross-) priming of naïve CD4 + and CD8 + T cells.
I NAUGURAL -D ISSERTATION
zur
Erlangung des Doktorgrades
der Mathematisch-Naturwissenschaftlichen Fakultät der Universität zu Köln
vorgelegt von Jens A. A. Fischer
aus Leverkusen
Bergisch Gladbach, 2008
Berichterstatter/in:
Erstgutachter:
Prof. Dr. Manolis Pasparakis
Zweitgutachterin:
Prof. Dr. Dagmar Knebel-Mörsdorf
Tag der mündlichen Prüfung: 24. Oktober 2008
Für Virginia
ZUSAMMENFASSUNG
Plasmazytoide Dendritische Zellen (PDCs) repräsentieren eine Subpopulation Dendritischer Zellen und sind die Hauptproduzenten von Typ I Interferonen nach viraler oder mikrobieller Stimulation. Dadurch beeinflussen und verbinden sie die angeborene und adaptive Immunabwehr. Obwohl es immer mehr Anzeichen einer Beteiligung von PDCs an der Entstehung und Aufrechterhaltung von Autoimmunerkrankungen oder Krebs gibt, und ihnen auch eine Rolle bei der Induktion von Toleranz zugeschrieben wird, ist wenig über ihre exakte immunologische Funktion bekannt. Besonders ihre Rolle als Antigen-präsentierende Zellen bei der Induktion einer T-Zell-Antwort wird kontrovers diskutiert. Die funktionelle Charakterisierung von murinen PDCs wurde durch das Fehlen eines spezifischen Oberflächenrezeptors erschwert. PDCs wurden identifiziert anhand der Co-Expression von B220, Ly-6C, und CD11c.
In dieser Arbeit wurden verschiedene monoklonale Antikörper generiert, die alle ein Oberflächenantigen erkannten, das spezifisch auf PDCs in naïven Mäusen exprimiert war und das als „murines PDC Antigen 1“ (mPDCA-1) bezeichnet wurde. Mithilfe differentieller Genexpressionsanalyse konnte gezeigt werden, daß die anti-mPDCA-1 Antikörper das „Bone marrow stromal antigen 2“ (BST2) erkennen.
Weitere Experimente zeigten, daß Ligation des mPDCA-1 Rezeptors eine Toll-like Rezeptor- induzierte Produktion von Typ I Interferonen in PDCs inhibierte. Die Kreuzvernetzung des Rezeptors mit anti-mPDCA-1 Antikörpern resultierte in einer intrazellulären Kalzium- Mobilisierung sowie in einer allgemeinen Phosphorylierung von Proteintyrosinresten. Des Weiteren führte die Kreuzvernetzung sowohl in vitro als auch in vivo zu einer schnellen und effizienten Internalisierung des Rezeptor-Antikörperkomplexes.
Als nächstes wurde die potentielle Funktion des mPDCA-1 Moleküls als PDC-spezifischer Antigen-Aufnahmerezeptor untersucht. Da die Applikation des vollständigen anti-mPDCA-1 Antikörpers in vivo in Fc-vermittelter oder ADCC-abhängiger Depletion der PDCs resultierte, wurde Ovalbuminprotein kovalent an ein nichtdepletierendes F(ab’)
2-Fragment des anti-PDCA-1 Antikörpers konjugiert. Somit konnten PDCs spezifisch in vitro und in vivo angefärbt werden.
Über mPDCA-1 aufgenommenes Antigen wurde prozessierte und auf Klasse I und II MHC- Molekülen präsentiert. Dabei waren PDCs in der Lage, naïve CD4
+and CD8
+T-Lymphozyten in vitro effizient zu primen. Sowohl das Priming als auch das cross-priming antigenspezifischer T-Zellen war abhängig von einer Aktivierung der PDCs, die mit einer verstärkten Expression costimulatorischer und MHC-Moleküle einherging. Dieser zusätzliche Stimulus schien auch die Antigenprozessierungs- und präsentationsmaschinerie zu aktivieren.
Letztlich wurde eine heterogene Expression des „Stem cell antigen 1“ (Sca-1) auf PDCs
beobachtet, die organabhängig variierte. Sca-1
-PDCs erschienen früher in der Entwicklung der
Zellen und produzierten mehr IFN α nach Stimulation. Aktivierte PDCs regulierten die
Expression von Sca-1 hoch.
Zusammengefasst ist mPDCA-1 ein spezifischer Marker für die Identifizierung von PDCs. Die
direkte Interaktion mit naïven T-Zellen unterstreicht die Rolle der PDCs in der Koordination von
angeborener und adaptiver Immunantwort. Die Ergebnisse dieser Arbeit zeigen, daß PDCs ein
viel versprechendes Ziel für die Entwicklung neuartiger Therapiemöglichkeiten und deren
Untersuchung im Mausmodel sind, vor allem für die Behandlung von Tumor- oder
Autoimmunerkrankungen, z.B. von SLE.
ABSTRACT
Plasmacytoid dendritic cells (PDCs) represent a distinct subset of dendritic cells in humans and mice. In the murine system PDCs were characterized by the co-expression of B220, Ly-6C, and CD11c. Due to their ability to produce large amounts of interferon (IFN)-alpha upon microbial challenge and due to their stimulatory capacity they are believed to link innate and adaptive immune responses. Although there is growing evidence of their contribution in the induction of anti-viral immune responses, autoimmune disorders and tolerance, less is known about their exact function. In particular their role as antigen-presenting cells in the induction of T cell responses is still controversially discussed.
In the present study, a panel of monoclonal antibodies (mAb) was generated, all recognizing a cell surface antigen specifically expressed on PDCs in naïve mice. The antigen was termed mPDCA-1. Differential gene expression analysis revealed that mPDCA-1 is identical to the bone marrow stromal antigen 2 (BST2). Triggering of mPDCA-1/BST2 with the mAb resulted in calcium mobilization and overall protein-tyrosine phosphorylation, which inhibited TLR-induced IFN-alpha production in PDCs. Cross-linking of mPDCA-1 also resulted in rapid internalization of the antibody-receptor complex in vitro and in vivo. Since the administration of the complete anti-mPDCA-1 mAb resulted in Fc-mediated or ADCC-dependent depletion of PDCs in vivo, Ovalbumin protein was covalently conjugated to a non-depleting anti-mPDCA-1-F(ab’)
2fragment. When targeted via mPDCA-1, antigens entered the MHC class I and II processing and presentation pathway and PDCs were shown to efficiently prime naïve CD4
+and CD8
+T cells in vitro. Interestingly, this process was dependent on stimulation of PDCs leading to the activation of their antigen processing and presentation machinery. In contrast, without activation PDCs failed to stimulate naïve T cells.
In summary, mPDCA-1/BST2 is a novel specific marker for PDCs in mice, influencing their innate and adaptive functions. Further experiments including the identification of the natural ligand of mPDCA-1/BST2 in mice will be needed for better understanding its in vivo function.
The effect of IFN-alpha abrogation after mPDCA-1 triggering could be investigated in murine models of autoimmune disease (e.g. SLE) or in viral infections. Furthermore, targeting antigen via mPDCA-1 would be a promising system to study the role of PDCs in adaptive immunity including the initiation of cytotoxic T cell responses in vivo.
In the second part of the work, two subpopulations of PDC characterized by the differential
expression of the stem cell antigen 1 (Sca-1) could be identified. Sca-1- PDCs produced large
amounts of IFN-alpha after stimulation with TLR9 ligands, appeared earlier in the development
and were predominantly present in the bone marrow. In contrast, Sca-1
+PDCs were poor IFN-
alpha producers, represented the majority of PDCs in the lymph nodes and seemed to develop
from Sca-1
-PDCs upon in vitro activation or adoptive transfer. Further work will be necessary to
elucidate whether Sca-1 expression characterizes developmental/activation stages or two
different subpopulations of PDCs.
ABBREVIATIONS
aa Amino acid
Ab antibody
ADCC Antibody-dependent cell-mediated cytotoxicity
Ag Antigen
APC Antigen-presenting cell APC Allophycocyanin
BDCA Blood dendritic cell antigen, e.g. BDCA-2, -3, -4
BM Bone marrow
bp Base pair
BrdU 5-bromo-2-desoxyuridin (thymidine analoge) BSA Bovine serum albumin
BST2 Bone marrow-stromal antigen 2 [Ca
2+]
iIntracellular calcium concentration CD Cluster of Differentiation
cDC conventional (myeloid) dendritic cell CCL Chemokine (CC) motif ligand CDS Protein coding sequence CLR Ca
+2-dependent lectin receptor CLSM Confocal laser scanning microscopy
CpG ODNs Cytosine-phosphate-guanine oligodeoxynucleotides CRM cysteine-rich motifs
CTL Cytotoxic T lymphocyte
DAMP Danger-associated molecular pattern DC Dendritic cell
DCIR DC Immunoreceptor
DC-SIGN Dendritic Cell-specific Intercellular Adhesion Molecule 3 (ICAM-3)-grabbing Nonintegrin (CD209)
Dectin-1/-2 DC-associated C type lectin 1 and 2 dH
20 Deionized water
DNA Deoxyribonucleic acid
ds RNA Double-stranded ribonucleic acid EEA-1 Early endosomes antigen 1
ELISA Enzyme-linked immunosorbent assay ER Endoplasmic reticulum
F(ab’)
2Fragment antigen binding region, based on two combined Fab domains, each composed of the variable and constant region of light and heavy chain (only CH
1)
FACS Flow cytometric cell sorting
Fc Fragment crystallizable region of an antibody, constant part, based on the CH
2and CH
3domain of the heavy chain FCS Fetal calv serum
FITC Fluorescein isothiocyanate
FLT-3L (FL) FMS-related tyrosine kinase 3 ligand Foxp3 Forkhead box p3
FSC Forward scatter
GITR Glucocorticoid-induced tumor necrosis factor receptor GOC Gene ontology clustering
HA Hemagglutinin
HEV High endothelial venule HRP Horseradish peroxidase HSV Herpex simplex virus i.p. Intraperitoneally i.v. Intravenously
ICOS Inducible T cell costimulator IDO Indoleamine 2,3-dioxygenase IFN Interferon
IFN-I Type I interferon
Ig Immunoglobulin
imDC Immature DC
IKK Inhibitor of NF-kB kinase
IL Interleukin
IP10 IFN-inducible 10kDa protein
ITAM/ITIM Immunoreceptor tyrosine-based activation/inhibitory motif IPC Interferon-producing cells (syn. PDC)
IRAK1/4 IL1-receptor-associated kinase 1/4 IRF Interferon regulatory factor
LCMV Lymphocytic choriomeningitis virus
LN Lymph node
LPS Lipopolysaccharide LRR Leucine rich repeat mAb Monoclonal antibody mar mAb Mouse anti-rat mAb MACS Magnetic cell separation
MB MIcrobead
MCMV Murine cytomegalovirus
MHC Major histocompatibility complex
MFI Mean fluorescence intensity
MIP Macrophage inflammatory protein
MMR Macrophage mannose receptor
Mock control Cells transfected with empty vector (without gene of interest) mPDCA-1 Mouse plasmacytoid dendritic cell antigen 1
MPG1 Macrophage specific gene 1 (syn. MSP1, MPEG1) MyD88 Myeloid differentiation primary-response protein 88 NEMO NF-kB essential modulator
NF-kB Nuclear factor-kappa B NK cell Natural killer cell
ODN Oligodeoxynucleotide ORF Open readding frame
OVA Ovalbumin (Hen egg white protein) pAb Polyclonal antibody
PAGE Polyacryl-amide gel electrophoresis PAMP Pathogen-associated molecular pattern PBMC Peripheral blood mononuclear cells PBS Phosphate buffered saline
PCR Polymerase chain reaction PDC Plasmacytoid dendritic cell
PD-1L Programmed cell death receptor 1 ligand PE R-phycoerythin
PerCP Peridinin chlorophyll (A) protein
PIQOR Parallel Identification and quantification of RNA PMA Phorbol-12-myristate-13-acetate
PMF Peptide mass fingerprinting (phorbol ester) PRR Pattern recognition receptor
PVDF Polyvinylidene Fluoride ram mAb rat anti-mouse mAb
RPMI Rosewell Park Memorial Institute Medium RSV Respiratory syncytial virus
RT Room temperature
RT-PCR Reverse transcriptase PCR s.c. Subcutaneous
SDS Sodium dodecyl sulfate
SEM Standard error of measurement or mean Siglec-H Sialic acid binding Ig-like lectin H
SLE Systemic lupus erythematosus SSC Side scatter
ss RNA single-stranded ribonucleic acid
STAT-1 Signal transducer and activator of transcription-1
TAP Transporters associated with antigen processing
Tcm Central memory T cell TCR T cell receptor
Tem Effector memory T cell
TGF Transforming growth factor beta
T
HT helper cell
TIL Tumor infiltrating lymphocyte TIR Toll/IL-1R
TLR Toll-like receptor
TMD Trans-membrane domain TNF Tumor necrosis factor Th T helper cell type Tr1/reg Regulatory T cell
TRAF Tumor necrosis factor receptor-associated factor TRIS Tris(hydroxymethyl)aminomethane
UTR Untranslated region
LIST OF FIGURES
Fig.1.1 Professional antigen-presenting cells process intracellular and extracellular
pathogens differently. 5
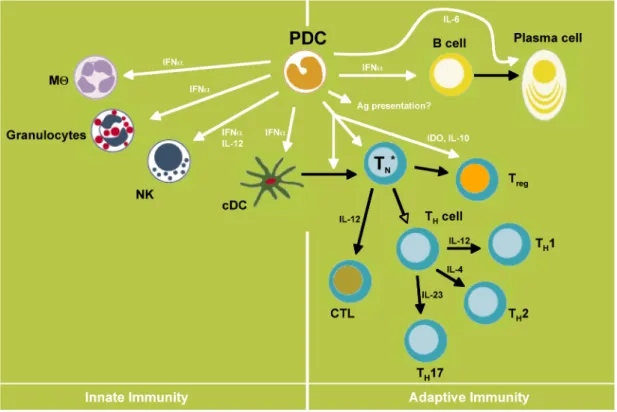
Fig. 1.2 The role of PDCs bridging innate and adaptive immune responses. 11