Synthesis and Coordination Chemistry of Anionic Pnictogenylborane Derivatives
242
0
0
Volltext
(2)(3)
(4)(5)
(6)(7)
(8)
(9)
(10)(11)
(12)(13)
(14)
(15)
(16)(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
(39)
(40)
(41)
(42)
(43)
Abbildung
![Figure S 3.2. Molecular structure of 3 in the solid state. Selected bond lengths [Å] and angles [°]: As-B:](https://thumb-eu.123doks.com/thumbv2/1library_info/3737445.1509093/47.892.159.735.363.741/figure-molecular-structure-solid-state-selected-lengths-angles.webp)
![Figure S 3.5. Molecular structure of the anion of 6 in the solid state. Selected bond lengths [Å] and angles[°]:](https://thumb-eu.123doks.com/thumbv2/1library_info/3737445.1509093/50.892.283.617.301.695/figure-molecular-structure-anion-solid-selected-lengths-angles.webp)
+7
![Figure S 3.6. Molecular structure of the cation of 6 in the solid state. Selected bond lengths [Å] and angles [°]:](https://thumb-eu.123doks.com/thumbv2/1library_info/3737445.1509093/51.892.253.641.111.498/figure-molecular-structure-cation-solid-selected-lengths-angles.webp)
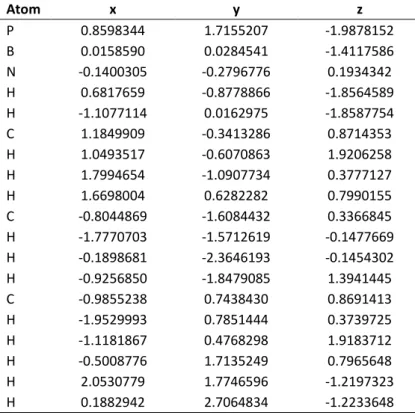
![Table S 3.8. Cartesian coordinates of the optimized geometry of [H 2 P-BH 2 -AsH 2 ] - at the B3LYP/def2-TZVP level of theory](https://thumb-eu.123doks.com/thumbv2/1library_info/3737445.1509093/64.892.111.789.377.766/table-cartesian-coordinates-optimized-geometry-tzvp-level-theory.webp)
![Table S 3.12. Cartesian coordinates of the optimized geometry of [H 2 As-BH 2 -AsH 2 -BH 2 -PH 2 ] - at the B3LYP/def2- B3LYP/def2-TZVP level of theory](https://thumb-eu.123doks.com/thumbv2/1library_info/3737445.1509093/66.892.117.529.235.566/table-cartesian-coordinates-optimized-geometry-tzvp-level-theory.webp)
![Table S 3.15. Cartesian coordinates of the optimized geometry of [PH 2 ] - at the B3LYP/def2-TZVP level of theory](https://thumb-eu.123doks.com/thumbv2/1library_info/3737445.1509093/68.892.109.787.803.1065/table-cartesian-coordinates-optimized-geometry-tzvp-level-theory.webp)
ÄHNLICHE DOKUMENTE
In order to get the 3-chloropropyl-substituted o- carborane derivatives in a one-step reaction starting from o-carborane, we tried the reaction of dilithio- o-carborane (formed in
Kantlehner and coworkers (Stuttgart/Aalen) give an account of their studies on the reaction of N, N, N , N - tetramethyl-chloroformamidinium chloride with sodium,
After N-galactosylation and subsequent O-sil- ylation, nucleophilic addition of organometallic reagents proceeded with high regio- and stereo- selectivity at 4-position. Substituents
The 1 : 1 iminium intermediate, generated by the addition of a secondary amine to acetalde- hyde is trapped by the (N-isocyanimino)triphenylphosphorane in the presence of
b Chemistry Department, Faculty of Science, Menoufi a University, Shebin El-Koam, Egypt.
The antimicrobial activity of the synthesized compounds was evaluated against three micro- organisms; Bacillus subtilis (ATCC 6633) (Gram- positive), Pseudomonas aeruginosa
Compared to an identical library generated by conventional parallel synthesis, a microwave- assisted procedure dramatically decreased reaction times from hours to minutes, and yields
The use of base required in such reactions was obviated by performing the re- action in water which not only avoided the use of base but also gave good yields (75 – 90 %) within 4.0
