AUS DEM LEHRSTUHL FÜR INNERE MEDIZIN III PROF. DR. MED. WOLFGANG HERR
DER FAKULTÄT FÜR MEDIZIN DER UNIVERSITÄT REGENSBURG
Die Entwicklung des Homingrezeptorprofils isolierter regulatorischer T-Zellen unter in vitro Stimulation
Inaugural – Dissertation zur Erlangung des Doktorgrades
der Medizin
der
Fakultät für Medizin der Universität Regensburg
vorgelegt von
Johanna Franziska Kerschbaum
2020
AUS DEM LEHRSTUHL FÜR INNERE MEDIZIN III PROF. DR. MED. WOLFGANG HERR
DER FAKULTÄT FÜR MEDIZIN DER UNIVERSITÄT REGENSBURG
Die Entwicklung des Homingrezeptorprofils isolierter regulatorischer T-Zellen unter in vitro Stimulation
Inaugural – Dissertation zur Erlangung des Doktorgrades
der Medizin
der
Fakultät für Medizin der Universität Regensburg
vorgelegt von
Johanna Franziska Kerschbaum
2020
Dekan: Prof. Dr. Dirk Hellwig
1. Berichterstatter: Prof. Dr. Matthias Edinger
2. Berichterstatter: Prof. Dr. Hinrich Abken
Tag der mündlichen Prüfung 29.05.2020
Abkürzungsverzeichnis
Abb Abbildung
Ag Antigen
ANOVA One-Way Analysis of Variance
APC Allophycocyanin
APCs Antigenpräsentierende Zellen
ATRA All-trans-Retinsäure (all-trans-retinoic acid)
bzw beziehungsweise
CCR Chemokinrezeptor
CD Cluster of differentiation
CLA kutanes Lymphozyten Antigen “cutaneous lymphocyte Ag”
d Tag
DAPI 4′,6-Diamidin-2-phenylindol DC dendritische Zellen
DGZ Dichtegradientenzentrifugation DLI Donor Lymphozyten Infusion exp polyklonal expandierte Zellen exv ex vivo isolierte Zellen
FACS Durchflusszytometrie (fluorescence-activated cell sorting) FITC Fluorescein Isothiocyanat
FOXP3 Forkhead Box Protein P3
FSC Vorwärts-Streulicht (forward scatter)
GCS-F Granulozyten Kolonie stimulierender Faktor (Granulocyte-Colony Stimulating Factor)
GMP Gute Herstellungsbedingungen (good manufacturing practice) GVL Graft-versus-Leukämie
GVHD Graft-versus-Host-Erkrankung (graft-versus-host-disease)
HSZT Hämatopoetische Stammzelltransplantation
ICOS Induzierbarer Kostimulator IDO Indoleamine-2,3-dioxygenase
Ig Immunglobulin
IL2 Rekombinantes humanes Interleukin 2
ITAMs Immunoreceptor tyrosine-based acivation motifs
IPEX immune dysregulation, polyendocrinopathy, enteropathy, X-linked
KG Körpergewicht
KLF2 Kruppel-ähnlicher Faktor 2 (Kruppel-like factor 2)
LK Lymphknoten
MACS Magnetische Zellseparation
MFI Mittlere Intensität der Fluoreszenz (mean-fluorescence-intensity) MHC Haupthistokompatibilitätskomplex
mLK Mesenteriale Lymphknoten (mesenterial lymph nodes) MNC Mononukleäre Zellen
mRNA Boten-Ribonucleinsäure (messenger ribonucleic acid)
MW Mittelwert
NK-Zellen natürliche Killer-Zellen n.s. Nicht signifikant
p Irrtumswahrscheinlichkeit
PBMC Mononukleäre Zellen des peripheren Blutes PerCP Peridininchlorophyll
PHA Phytohämagglutinin
PI Propidiumiodid
RNA Ribonukleinsäure (ribonuclein acid)
SEM Standardfehler des Mittelwertes (standard error of the mean) SLO Sekundäre lymphatische Organe
SSC „Seitwärts-Streulicht“ (sidewards scatter)
TCR T-Zellrezeptor
Tkonv Konventionelle CD4
+T-Zelle
Treg Regulatorische T-Zelle
tTreg im Thymus gereifte regulatorische T-Zelle iTreg in vitro gereifte regulatorische T-Zelle
pTreg in der Peripherie gereifte regulatorische T-Zelle TSDR Treg spezifische, demethylierte Region
z.B. Zum Beispiel
Inhaltsverzeichnis
Inhaltsverzeichnis ... 3
1 Einleitung ... 6
1.1 Die T-Zell vermittelte Immunantwort ... 6
1.2 Regulatorische T-Zellen ... 10
1.2.1 Der Transkriptionsfaktor FOXP3 ... 13
1.2.2 Phänotypische Differenzierung ... 14
1.2.3 Funktionelle Eigenschaften ... 16
1.2.4 Subgruppen regulatorischer T-Zellen ... 17
1.3 Hämatopoetische Stammzelltransplantation ... 18
1.3.1 Die Graft-versus-Host-Erkrankung ... 20
1.3.2 GVHD und regulatorische T-Zellen ... 24
1.4 Die Migration von T-Zellen ... 28
1.4.1 Molekulare Bestandteile der Migration ... 29
1.4.2 Die zelluläre Extravasation ... 33
1.4.3 Das Migrationsverhalten regulatorischer T-Zellen ... 34
1.4.4 Induktion von gewebespezifischem Homing bei Aktivierung ... 35
2 Zielsetzung ... 37
3 Material und Methoden ... 38
3.1 Material ... 38
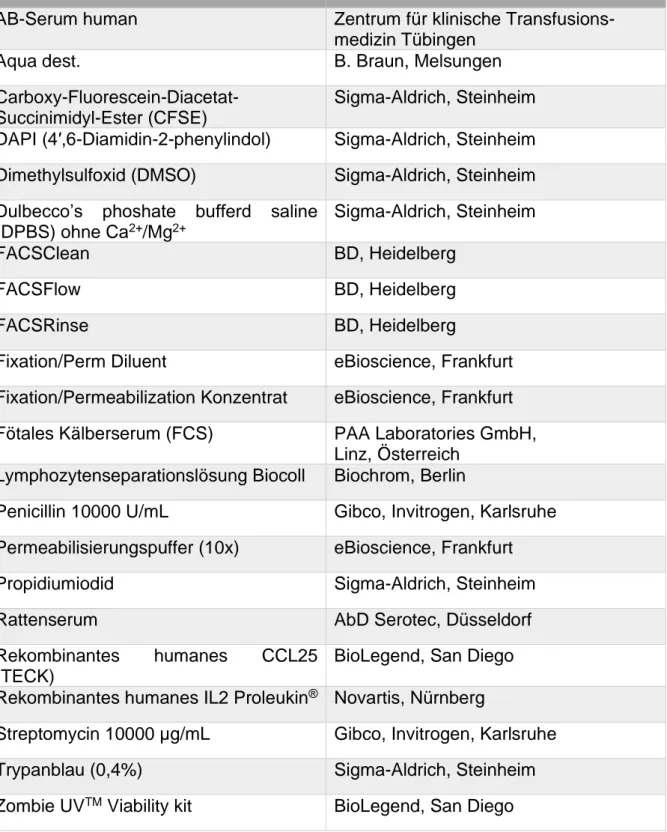
3.1.1 Chemikalien und Reagenzien ... 38
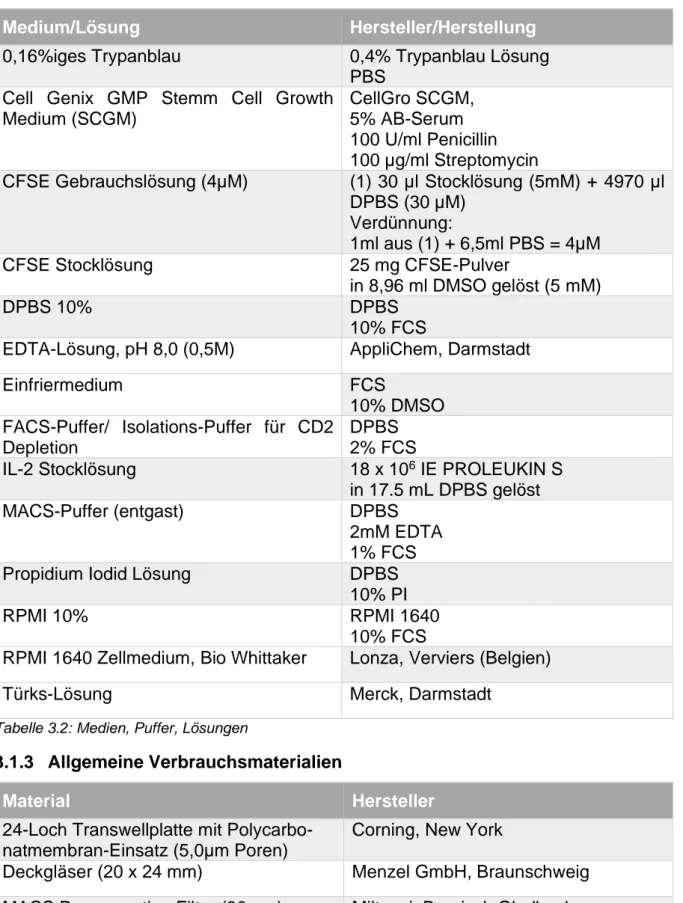
3.1.2 Medien, Puffer, Lösungen ... 39
3.1.3 Allgemeine Verbrauchsmaterialien ... 39
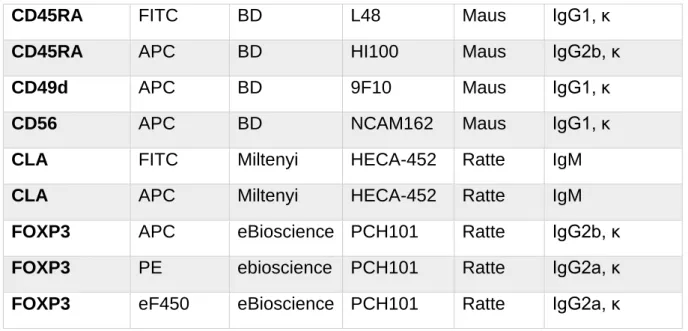
3.1.4 Antikörper für die Durchflusszytometrie (FACS) ... 40
3.1.5 Magnetische Beads ... 41
3.1.6 Geräte ... 41
3.1.7 Software und Programme ... 42
3.2 Methoden ... 42
3.2.1 Bestimmung der Zellzahl ... 42
3.2.2 Isolation der verwendeten Zellpopulationen ... 43
3.2.2.1 Dichtegradientenzentrifugation ...43
3.2.2.2 Auftauen von Zellen ...43
3.2.2.3 Magnetische Separation ...44
3.2.3 Antikörperbasierte Zellfärbung zur durchflusszytometrischen Analyse ... 44
3.2.3.1 Oberflächenfärbung ...44
3.2.3.2 Intranukleäre Foxp3 Färbung ...45
3.2.3.3 CFSE Färbung ...46
3.2.4 Isolation und Sortierung humaner T-Zellen ... 47
3.2.4.1 Magnetbasierte Anreicherung ...47
3.2.4.2 Fluoreszenzbasiertes Sorting...47
3.2.5
In vitro Methoden ... 483.2.5.1 Polyklonale Expansion von Treg- und Tkonv-Populationen ...48
3.2.5.2 Gemischte Lymphozyten Reaktion ...49
3.2.5.3 Migrationsassay ...50
3.2.6 Statistik ... 50
4 Ergebnisse ... 51
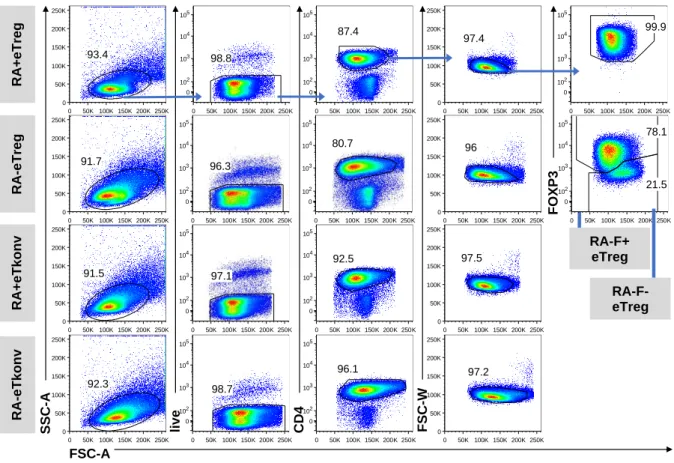
4.1 Homingrezeptorprofile ex vivo isolierter CD4
+T-Zellen ... 51
4.2 Analyse des Homingrezeptorprofils CD4
+T-Zellen im Rahmen der polyklonalen Expansion ... 55
4.2.1 Isolation und Expansion der Zielpopulationen ... 55
4.2.2 Homingrezeptorexpression expandierter CD4
+T-Zellen ... 59
4.2.3 Vergleich des Homingrezeptorprofils CD4
+T-Zellen vor und nach polyklonaler Expansion ... 61
4.2.4 Analyse der Homingrezeptoren und der FOXP3-Expression von CD4
+T-Zellen nach polyklonaler Expansion mit ATRA ... 66
4.2.4.1 Homingrezeptorprofil CD4
+T-Zellen bei Kultivierung mit ATRA ...66
4.2.4.2 Stabilisierung und Steigerung der FOXP3 Expression bei Kultivierung mit ATRA ...70
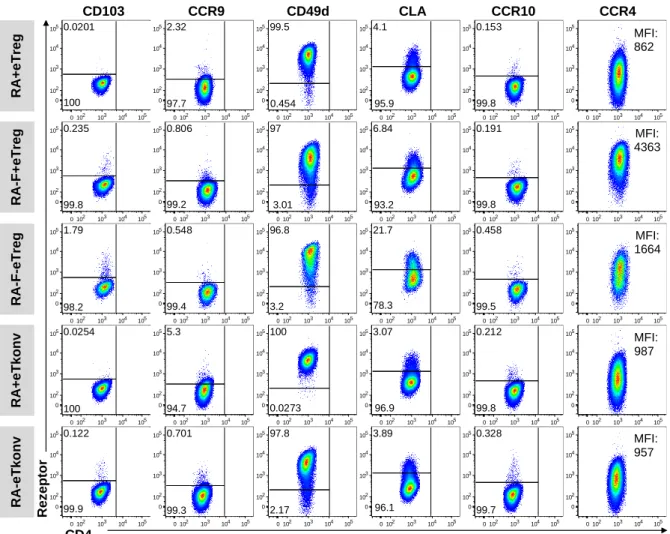
4.3 Homingrezeptorprofil von CD4
+T-Zellen nach allogener in vitro Stimulation ... 71
5 Diskussion ... 75
5.1 Homingrezeptorprofile ex vivo isolierter CD4
+T-Zellen ... 76
5.2 Homingrezeptorprofile von CD4
+T-Zellen nach polyklonaler Expansion und Vergleich zu ex vivo isolierten Zellen ... 83
5.2.1 Homingrezeptorprofile CD4
+T-Zellen nach polyklonaler Expansion mit ATRA
... 95
5.3 Homingrezeptorprofile CD4
+T-Zellen nach allogener in vitro Stimulation ... 100
6 Zusammenfassung ... 102
7 Anhang ... 104
7.1 Tabellenverzeichnis ... 104
7.2 Abbildungsverzeichnis ... 104
8 Literaturverzeichnis ... 105
9 Danksagung ... 129