Absorption und Emission von Ir
III( ppy)
2(acac)
Bachelorarbeit von Adrian Heil
Absorption und Emission von Ir
III( ppy)
2(acac)
Bachelorarbeit von Adrian Heil Düsseldorf, August 2012
Durchgeführt am
Institut für Theoretische Chemie und Computerchemie Mathematisch-Naturwissenschaftliche Fakultät
Heinrich-Heine-Universität Düsseldorf
1. Gutachterin: Frau Prof. Dr. Christel M. Marian 2. Gutachter: Herr Jun.-Prof. Dr. Jörg Tatchen
der angegebenen Quellen und Hilfsmittel angefertigt und die den benutzten Quellen wörtlich oder inhaltlich entnommenen Stellen als solche kenntlich gemacht habe. Diese Arbeit hat in gleicher oder ähnlicher Form noch keiner Prüfungsbehörde vorgelegen.
Düsseldorf, 22.8.2012
Unterschrift (Adrian Heil)
Im nachfolgenden wurde der Einfluss von Fluorsubstituenten auf die Absorption und Emission und Geometrie eines OLED-Farbstoffs untersucht. Es konnte gezeigt werden, dass durch die Substituti- on sowohl in Absorption als auch in Emission eine Blauverschiebung stattfindet, diese wurde auch von diversen Arbeitsgruppen im Experiment beobachtet. Anregungen mit hoher Helligkeit bleiben erhalten. Ebenfalls wurde deutlich, dass die Auswirkungen auf die Geometrie relativ gering sind, hier ist nur die unmittelbare Nähe der Substituenten betroffen, d.h. hauptsächlich die Phenylringe, wobei sich Bindungslängen- und winkel um 2 pm oder weniger bzw. 2.5° oder weniger ändern, die meisten bleiben jedoch gleich.
Zudem wurde gezeigt, dass sich die verwendeten def-SV(P)- und def-TZVP-Basissätze in den Ergeb- nissen, also in den Bildungslängen und -winkeln sowie in Anregungsspektren nur wenig unterschei- den und somit für dieses Molekül der von der Rechenzeit her bessere def-SV(P)-Basissatz gewählt werden kann
Abbildungsverzeichnis IV
Tabellenverzeichnis VII
Abkürzungsverzeichnis VIII
1 Einleitung 1
1.1 Ir(ppy)2(acac)-Ir(F2ppy)2(acac) . . . 1
1.2 Organische LEDs . . . 1
1.3 Iridium Komplexe . . . 2
1.4 Singulett- und Triplett-Anregungen . . . 2
1.5 Das Franck-Condon Prinzip . . . 3
2 Methoden 4 2.1 Dichtefunktionaltheorie (DFT) . . . 4
2.1.1 Das BH-LYP Funktional . . . 6
2.1.2 Das PBE0 Funktional . . . 6
2.2 Multireference Configuration Interaction (MRCI) . . . 6
2.3 DFT/MRCI . . . 7
2.4 Verwendete Software . . . 7
2.5 Technische Details der Rechnungen . . . 8
2.5.1 Technische Angaben . . . 8
2.5.2 Vibes . . . 8
2.5.3 Der def-SV(P) Basissatz . . . 8
2.5.4 Der def-TZVP Basissatz . . . 9
3 Auswertung 9 3.1 Die Geometrie . . . 9
3.1.1 Vergleich zwischen def-SV(P) und def-TZVP beiIr(ppy)2(acac) . . . 9
3.1.2 Vergleich zwischenIr(ppy)2(acac)undIr(F2ppy)2(acac) . . . 9
3.1.3 Vergleich zwischen Singulett- und Triplett-Geometrie . . . 10
3.2 Schwingungsfrequenzen . . . 10
3.3 Anregungen und Übergänge . . . 11
3.3.1 Ir(ppy)2(acac) . . . 11
3.3.2 Ir(F2ppy)2(acac) . . . 19
4 Diskussion 25 4.1 Ergebnis des Basissatzvergleichs . . . 25
4.1.1 Geometrie . . . 25
4.1.2 Absorption . . . 25
4.2 Vergleich der Geometrie mit experimentellen Daten . . . 26
4.2.1 Ir(ppy)2(acac) . . . 26
4.2.2 Ir(F2ppy)2(acac) . . . 26
4.3 Auswirkungen der Fluorsubstituenten auf die Anregungen . . . 26
4.4 Absorption/ Emission im Experiment und berechnet . . . 27
4.4.1 Ir(ppy)2(acac) . . . 27
4.4.2 Ir(F2ppy)2(acac) . . . 29
5 Zusammenfassung und Ausblick 31
Literatur i
B Anhang Tabellen xvi
C 10 Wurzeln xlv
C.1 def-SV(P) . . . xlv C.2 def-TZVP . . . l C.3 Auswirkungen des Basissatzes auf die Anregungen an der Singulett-Geometrie
liv
D Testspektren liv
1.1 Bezeichnungen inIr(F2ppy)2(acac) . . . 1
1.2 Jablonski-Schema[4] . . . 3
3.1 MO-Diagramm vonIr(ppy)2(acac)mit def-SV(P) in der Singulett-Geometrie. Als Funktional wurde BH-LYP verwendet. . . . 12
3.2 Singulett-Absorptionsspektrum vonIr(ppy)2(acac)mit 50 Wurzeln und def-SV(P) . . . 14
3.3 MO-Diagramm vonIr(ppy)2(acac)mit def-TZVP in der Singulett-Geometrie. Als Funktional wurde BH-LYP verwendet. . . . 15
3.4 Singulett-Absorptionsspektrum vonIr(ppy)2(acac)mit 50 Wurzeln . . . 17
3.5 MO-Diagramm für die Triplett-Geometrie vonIr(ppy)2(acac).Als Funktional wurde BH-LYP verwendet. 17 3.6 Lage der Zustände beiIr(ppy)2(acac). . . 19
3.7 Anregung, Emission und adiabatische Energie vonIr(ppy)2(acac)mit Basissatz SV(P) . . . 19
3.8 MO-Diagramm vonIr(F2ppy)2(acac)in der Singulett-Geometrie . . . 20
3.9 Singulett-Absorption vonIr(F2ppy)2(acac) . . . 22
3.10 MO-Diagramm vonIr(F2ppy)2(acac)in der Triplett-Geometrie . . . 23
3.11 Lage der Zustände beiIr(ppy)2(acac). . . 24
3.12 Anregung, Emission und adiabatische Energie vonIr(F2ppy)2(acac)mit Basissatz SV(P) . . . 25
4.1 Vergleich der Singulett-Absorption beiIr(ppy)2(acac)mit Bassissatz def-SV(P) und def-TZVP. Als Peakverbreiterung wurde 4250cm−1gewählt. . . . 26
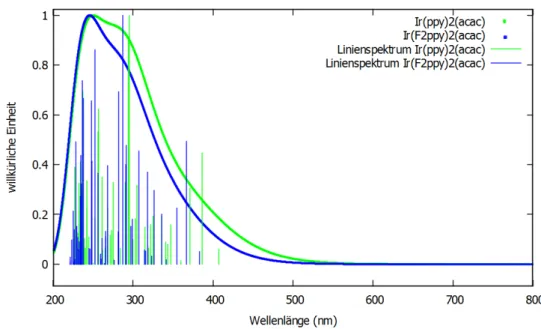
4.2 Vergleich der Absorption zwischenIr(ppy)2(acac)undIr(F2ppy)2(acac) . . . 27
4.3 Berechnetes Absorptionsspektrum vonIr(ppy)2(acac)mit einer Halbwertsbreite von 4250cm−1 im Vergleich mit dem Experiment. Für das experimentelle Spektrum [29] wurde ein Molekül mit der glei- chen Isomerie verwendet, statt acac-Ligand hat es jedoch einen 2,2,6,6-Tetramethyl-3,5-Heptandion- Liganden. . . . 28
4.4 Berechnete Emissionspektren vonIr(ppy)2(acac)im Vergleich mit dem Experiment. Für das experi- mentelle Spektrum [29] wurde ein Molekül mit der gleichen Isomerie verwendet, statt acac-Ligand hat es jedoch einen 2,2,6,6-Tetramethyl-3,5-Heptandion-Liganden. . . . 29
4.5 Berechnetes Absorptionsspektrum vonIr(F2ppy)2(acac)mit einer Halbwertsbreite von 4250cm−1im Vergleich mit dem Experiment. Das experimentelle Spektrum wurde mit dem gleichen Molekül anderer Isomerie aufgenommen [24]. . . . 30
4.6 Berechnete Emissionspektren vonIr(F2ppy)2(acac)im Vergleich mit dem Experiment. Das experimen- telle Spektrum wurde mit dem gleichen Molekül anderer Isomerie aufgenommen [24].. . . 30
A.1 HOMO-12 (links), HOMO-11 (mitte), HOMO-10 (rechts) vonIr(ppy)2(acac)mit def-SV(P) in der Singulett-Geometrie und BH-LYP als Funktional. . . . iii
A.2 HOMO-9 (links), HOMO-8 (rechts) vonIr(ppy)2(acac)mit def-SV(P) in der Singulett-Geometrie und BH-LYP als Funktional . . . iii
A.3 HOMO-7 (links), HOMO-6 (mitte links), HOMO-5 (mitte rechts), HOMO-4 (rechts) vonIr(ppy)2(acac) mit def-SV(P) in der Singulett-Geometrie und BH-LYP als Funktional. . . iii
A.4 HOMO-3 (links), HOMO-2 (mitte links), HOMO-1 (mitte rechts), HOMO (rechts) vonIr(ppy)2(acac) mit def-SV(P) in der Singulett-Geometrie und BH-LYP als Funktional. . . iii
A.5 LUMO (links), LUMO+1 (mitte links), LUMO+2 (mitte rechts), LUMO+3 (rechts) vonIr(ppy)2(acac) mit def-SV(P) in der Singulett-Geometrie und BH-LYP als Funktional. . . iv
A.6 LUMO+4 (links), LUMO+5 (mitte links), LUMO+6 (mitte rechts), LUMO+12 (rechts) vonIr(ppy)2(acac) mit def-SV(P) in der Singulett-Geometrie und BH-LYP als Funktional. . . iv
A.7 HOMO-12 (links), HOMO-9 (mitte links), HOMO-8 (mitte rechts), HOMO-7 (rechts) vonIr(ppy)2(acac) mit def-TZVP in der Singulett-Geometrie und BH-LYP als Funktional. . . v
A.8 HOMO-6 (links), HOMO-5 (mitte links), HOMO-4 (mitte rechts), HOMO-3 (rechts)vonIr(ppy)2(acac) mit def-TZVP in der Singulett-Geometrie und BH-LYP als Funktional. . . v
A.9 HOMO-2 (links), HOMO-1 (mitte links), HOMO (mitte rechts), LUMO (rechts)vonIr(ppy)2(acac) mit def-TZVP in der Singulett-Geometrie und BH-LYP als Funktional. . . v
mit def-TZVP in der Singulett-Geometrie und BH-LYP als Funktional. . . v
A.11 LUMO+5 (links), LUMO+6 (rechts)vonIr(ppy)2(acac)mit def-TZVP in der Singulett-Geometrie
und BH-LYP als Funktional . . . vi
A.12 HOMO-5 (links), HOMO-4 (mitte), HOMO-3 (rechts) vonIr(ppy)2(acac) mit def-SV(P) in der
Triplett-Geometrie und BH-LYP als Funktional . . . vii
A.13 HOMO-2 (links), HOMO-1 (mitte), HOMO (rechts) vonIr(ppy)2(acac)mit def-SV(P) in der Triplett-
Geometrie und BH-LYP als Funktional . . . vii
A.14 LUMO (links), LUMO+1 (mitte), LUMO+2 (rechts) vonIr(ppy)2(acac)mit def-SV(P) in der Triplett-
Geometrie und BH-LYP als Funktional . . . vii
A.15 LUMO+3 (links), LUMO+4 (rechts) vonIr(ppy)2(acac)mit def-SV(P) in der Triplett-Geometrie
und BH-LYP als Funktional . . . vii
A.16 HOMO-10 (links), HOMO-9 (mitte links), HOMO-8 (mitte rechts), HOMO-7 (rechts) vonIr(F2ppy)2(acac)
mit def-SV(P) in der Singulett-Geometrie und BH-LYP als Funktional. . . viii
A.17 HOMO-6 (links), HOMO-5 (mitte links), HOMO-4 (mitte rechts), HOMO-3 (rechts) vonIr(F2ppy)2(acac)
mit def-SV(P) in der Singulett-Geometrie und BH-LYP als Funktional. . . viii
A.18 HOMO-2 (links), HOMO-1 (mitte links), HOMO (mitte rechts), LUMO (rechts) vonIr(F2ppy)2(acac)
mit def-SV(P) in der Singulett-Geometrie und BH-LYP als Funktional. . . viii
A.19 LUMO+1 (links), LUMO+2 (mitte links), LUMO+3 (mitte rechts), LUMO+4 (rechts) vonIr(F2ppy)2(acac)
mit def-SV(P) in der Singulett-Geometrie und BH-LYP als Funktional. . . viii
A.20 LUMO+5 (links), LUMO+6 (rechts) vonIr(F2ppy)2(acac)mit def-SV(P) in der Singulett-Geometrie
und BH-LYP als Funktional . . . ix
A.21 HOMO-5 (links), HOMO-4 (mitte), HOMO-3 (rechts) vonIr(F2ppy)2(acac)mit def-SV(P) in der
Triplett-Geometrie und BH-LYP als Funktional . . . x
A.22 HOMO-2 (links), HOMO-1 (mitte), HOMO (rechts) vonIr(F2ppy)2(acac)mit def-SV(P) in der
Triplett-Geometrie und BH-LYP als Funktional . . . x
A.23 LUMO (links), LUMO+1 (mitte), LUMO+2 (rechts) vonIr(F2ppy)2(acac)mit def-SV(P) in der
Triplett-Geometrie und BH-LYP als Funktional . . . x
A.24 LUMO+3 (links), LUMO+4 (rechts) vonIr(F2ppy)2(acac)mit def-SV(P) in der Triplett-Geometrie
und BH-LYP als Funktional . . . x
A.25 Franck-Condon Spektrum für Absoption (S0 -> T1) und Emission (T1 -> S0) vonIr(ppy)2(acac)bei 0K xi A.26 Franck-Condon Spektrum für Absoption (S0 -> T1) und Emission (T1 -> S0) vonIr(ppy)2(acac)bei
298K . . . xi A.27 Franck-Condon Spektrum für Absoption (S0 -> T1) und Emission (T1 -> S0)vonIr(F2ppy)2(acac)bei
0K . . . xii A.28 Franck-Condon Spektrum für Absoption (S0 -> T1) und Emission (T1 -> S0)vonIr(F2ppy)2(acac)bei
298K . . . xii
A.29 Experimentelles Spektrum mit Temperaturänderung vonIr(ppy)2(acac)[23].Bei dem Isomer han-
delt es sich nicht um das des berechneten Moleküls, sondern um das aus Abbildung A.33. . . . xiii A.30 experimentelles Spektrum vonIr(ppy)2(acac)(olivgrün) inCHCl3bei RT[29] Bei dem Isomer han-
delt es sich um das des berechneten Molekül, allerdings mit 2,2,6,6-Tetramethyl-3,5-Heptandion-Ligand
statt acac-Ligand . . . xiv
A.31 Experimentelles Absorptionsspektrum vonIr(F2ppy)2(acac)inCH2Cl2bei RT[24]. Bei dem Isomer handelt es sich nicht um das des berechneten Moleküls. Das Isomer ist in seiner unfluorierten Struktur
in Abbildung A.33 gezeigt. . . . xiv
A.32 experimentelles Emissionsspektrum vonIr(F2ppy)2(acac)inCH2Cl2 bei RT [24]. Bei dem Isomer handelt es sich nicht um das des berechneten Moleküls. Das Isomer ist in seiner unfluorierten Struktur
in Abbildung A.33 gezeigt. . . . xv
A.33 Struktur vonIr(ppy)2(acac)mit anderer Isomerie für Strukturvergleich[23] . . . xv
A.34 Struktur von Ir(tpy)3(acac) für Strukturvergleich (andere Nomenklatur)[16] . . . xvi
C.1 HOMO-5 (links), HOMO-4 (mitte), HOMO-3 (rechts). . . xlvi
C.3 LUMO (links), LUMO+1 (mitte), LUMO+2 (rechts) . . . xlvi
C.4 LUMO+3 (links), LUMO+4 (rechts) . . . xlvi
C.5 MO-Diagramm zuIr(ppy)2(acac) . . . xlvii
C.6 Absorptionsspektrum Singulett vonIr(ppy)2(acac) . . . xlix
C.7 HOMO-5 (links), HOMO-4 (mitte links), HOMO-3 (mitte rechts), HOMO-2 (rechts) . . . li
C.8 HOMO-1 (links), HOMO (mitte links), LUMO (mitte rechts), LUMO+1 (rechts) . . . li
C.9 LUMO+2 (links), LUMO+3 (mitte), LUMO+4 (rechts) . . . li
C.10 Absorptionsspektrum Singulett vonIr(ppy)2(acac) . . . liii
D.1 Vergleich zwischen def-SV(P) und def-TZVP mit Peakverbreiterung 2250cm−1 . . . liv
D.2 Vergleich zwischenIr(ppy)2(acac)undIr(F2ppy)2(acac)mit Peakverbreiterung 2250cm−1 . . . lv
D.3 Berechnetes Absorptionsspektrum vonIr(ppy)2(acac)mit einer Halbwertsbreite von 2250cm−1 im
Vergleich mit dem Experiment. Für das experimentelle Spektrum [29] wurde ein Molekül mit der glei- chen Isomerie verwendet, statt acac-Ligand hat es jedoch einen 2,2,6,6-Tetramethyl-3,5-Heptandion-
Liganden. . . . lv
D.4 Berechnetes Absorptionsspektrum vonIr(F2ppy)2(acac)mit einer Halbwertsbreite von 2250cm−1im Vergleich mit dem Experiment. Das experimentelle Spektrum wurde mit dem gleichen Molekül anderer
Isomerie aufgenommen [24]. . . . lvi
1.1 Anregungen und Übergänge in OLEDs [31] . . . 2
2.1 Anzahl der CSFs . . . 8
2.2 Input-Parameter von Vibes . . . 8
3.1 Nullpunktschwingungsenergien vonIr(ppy)2(acac). . . 11
3.2 Nullpunktschwingungsenergie vonIr(F2ppy)2(acac)mit def-SV(P) . . . 11
3.3 Lokalisation der Ladungsdichte in den Orbitalen vonIr(ppy)2(acac)mit def-SV(P) . . . 13
3.4 Auswahl besonders heller Übergänge vonIr(ppy)2(acac)mit def-SV(P) . . . 14
3.5 Lokalisation der Ladungsdichte in den Orbitalen vonIr(ppy)2(acac)mit def-TZVP . . . 16
3.6 Auswahl besonders heller Übergänge vonIr(ppy)2(acac)mit def-TZVP . . . 16
3.7 Lokalisation der Elektronendichte in den Orbitalen vonIr(ppy)2(acac) . . . 18
3.8 Lokalisation der Elektronendichte in den Orbitalen vonIr(F2ppy)2(acac) . . . 21
3.9 Auswahl besonders heller Singulett-Übergänge vonIr(F2ppy)2(acac) . . . 22
3.10 Lokalisation der Elektronendichte in den Orbitalen vonIr(F2ppy)2(acac) . . . 23
B.1 Singulett-Anregungen vonIr(ppy)2(acac)mit def-SV(P) und 50 Wurzeln, Konfiguration, Ladungsver- teilung und Anteile sowie Energie und Helligkeit . . . xvii
B.2 Triplett-Anregungen an Singulett-Geometrie vonIr(ppy)2(acac)mit def-SV(P) und 49 Wurzeln, Kon- figuration, Ladungsverteilung und Anteil sowie Energie und Helligkeit . . . xxi
B.3 Singulett-Anregungen vonIr(ppy)2(acac)mit def-TZVP und 50 Wurzeln, Konfiguration, Ladungsver- teilung und Anteile sowie Energie und Helligkeit . . . xxiv
B.4 Singulett-Anregungen vonIr(F2ppy)2(acac)mit def-SV(P) und 50 Wurzeln, Konfiguration, Ladungs- verteilungen und Anteile. . . xxviii
B.5 Triplett-Anregungen vonIr(F2ppy)2(acac)mit def-SV(P) und 49 Wurzeln, Konfiguration, Ladungsver- teilung und Anteile . . . xxxi
B.6 Singulett-Anregungen vonIr(ppy)2(acac) . . . xxxiv
B.7 Triplett-Anregungen vonIr(ppy)2(acac) . . . xxxiv
B.8 Singulett-Anregungen vonIr(F2ppy)2(acac). . . xxxv
B.9 Triplett-Anregungen vonIr(F2ppy)2(acac) . . . xxxv
B.10 Bindungslängen vonIr(ppy)2(acac)im elektronischen Grundzustand, Vergleich experimentell und be- rechnet . . . xxxvi
B.11 Bindungswinkel vonIr(ppy)2(acac), Vergleich experimentell und berechnet . . . xxxvii
B.12 Vergleich Bindungslängen vonIr(ppy)2(acac)undIr(F2ppy)2(acac)mit Basissatz def-SV(P) . . . . xxxviii
B.13 Vergleich der Bindungswinkel vonIr(ppy)2(acac)undIr(F2ppy)2(acac)mit Basissatz def-SV(P) . . xxxix
B.14 Vergleich der Bindungslängen von Singulett- und Triplett-Geometrie beiIr(ppy)2(acac) . . . xl
B.15 Vergleich der Bindungswinkel der Singulett- und Triplett-Geometrie vonIr(ppy)2(acac) . . . xli
B.16 Vergleich der Bindungslängen zwischen Singulett- und Triplett-Geometrie beiIr(F2ppy)2(acac) . xlii B.17 Vergleich der Bindungswinkel zwischen Singulett- und Triplett-Geometrie beiIr(F2ppy)2(acac) . xliii B.18 Vergleich der Bindungslängenunterschiede fürIr(ppy)2(acac)undIr(F2ppy)2(acac) . . . xliv B.19 Vergleich der Bindungswinkelunterschiede fürIr(ppy)2(acac)undIr(F2ppy)2(acac) . . . xlv
C.1 Lokalisation der Ladungsdichte in den Orbitalen vonIr(ppy)2(acac) . . . xlvii
C.2 Anteil der Ladungsdichte im d-Orbital von Ir. . . xlviii
C.3 Singulett-Anregungen vonIr(ppy)2(acac) . . . xlviii
C.4 Energie und Helligkeit der Singulett-Übergänge vonIr(ppy)2(acac) . . . xlix
C.5 Triplett-Anregungen vonIr(ppy)2(acac) . . . l
C.6 Energie und Helligkeit der Triplett-Übergänge vonIr(ppy)2(acac) . . . l
C.7 Lokalisation der Ladungsdichte in den Orbitalen vonIr(ppy)2(acac) . . . lii
C.8 Singulett-Anregungen vonIr(ppy)2(acac) . . . lii
C.9 Energie und Helligkeit der Singulett-Übergänge vonIr(ppy)2(acac) . . . liii
C.10 Vergleich der Wellenlängen und Oszillatorstärken . . . liv
B88 . . . Becke 1988 Austauschfunktional BH–LYP . . . . Becke Half and Half Lee–Yang–Parr
CASSCF . . . . Complete Active Space Self-Consistent Field CI . . . Configuration Interaction
CSF . . . Configuration State Function DFT . . . Density Functional Theory E . . . Energie
ECP . . . Effective Core Potential
eV . . . Elektronenvolt, Einheit der Energie
FWHM . . . Full width at half maximum (Halbwertsbreite) HF. . . Hartree-Fock
HOMO . . . Highest Occupied Molecular Orbital
Ir(F2ppy)2(acac) (Bis(2-(2,4-difluorophenyl)pyridin))(acetylacetonato)iridium(III) Ir(ppy)2(acac) Bis(2-phenylpyridin)(Acetylacetonato)iridium(III)
Ir(ptpy)3. . . . Tris (p-Toluol)pyridin Iridium(III) K . . . Kelvin - Einheit der Temperatur LCAO . . . Linear combinations of atomic orbitals
lm/W . . . lumen/Watt, Einheit für die Lichtausbeute pro Leistung LUMO. . . Lowest Unoccupied Molecular Orbital
LYP . . . Lee–Yang–Parr Korrelationsfunktional MCSCF . . . Multi-configurational self-consistent field MRCI . . . Multi Reference Configuration Interaction nm. . . Nanometer
OLED . . . Organic Light Emitting Diode
PBE0 . . . Perdew, Burke and Ernzerhof, hybridisiert durch Adamo pm. . . Picometer
RAS . . . Restricted Active Space RT . . . Raumtemperatur (298 K) UV . . . Ultraviolett
Å . . . Ångström - Längeneinheit. Entspricht 100 pm.
1 Einleitung
1.1 Ir(ppy)2(acac) - Ir(F2ppy)2(acac)
Im nachfolgenden wird das MolekülIr(ppy)2(acac)(Bis(2-phenylpyridin)(acetylacetonato)iridium(III)) behandelt. Des Weiteren wird die Auswirkung von Fluorsubstituenten in 2,4-Position an den Phenyl- ringen, alsoIr(F2ppy)2(acac) (Bis(2-(2,4-difluorophenyl)pyridin)(acetylacetonato)iridium(III)) be- trachtet und mit den Ergebnissen des unfluorierten Moleküls verglichen.
Das Molekül besitzt einen Singulett-Grundzustand. DerT1-Zustand resultiert aus einer HOMO→ LUMO-Anregung.
Die im weiteren Text verwendeten Bezeichnungen werden in Abbildung 1.1 erläutert. Aus Über- sichtsgründen wurden die Wasserstoffatome ausgeblendet sowie nicht weiter betrachtete Atome nicht nummeriert. Da beide Moleküle mit Ausnahme der vier Fluoratome identisch sind, wurde hier zur Erklärung der BezeichnungenIr(F2ppy)2(acac)gewählt.
Abbildung 1.1:Bezeichnungen inIr(F2ppy)2(acac)
So beschreibt ph(1) den Phenylring des (im Bild) linken Phenylpyridins, py(1) beschreibt den Py- ridinring und ppy(1) meint den gesamten Phenylpyridin-Liganden. Gleiches gilt für den rechten Phenylpyridin-Liganden.
1.2 Organische LEDs
Organic Light Emitting Diodes (OLEDs) sind eine Weiterentwicklung der bereits seit langem ver- wendeten LEDs. Zuvor verwendete man beispielsweise AlGaAs für rote, GaP für grüne sowie InGaN für blaue LEDs [22].
Bei den organischen LEDs werden organische Liganden an ein Zentralatom koordiniert. Durch eine Delokalisierung derπ Elektronen wird das organische Molekül elektrisch leitfähig. Durch die ange- legte Spannung bewegen sich Elektronen von der Kathode zur Anode, hierbei werden die Elektronen ins LUMO an der Kathode angeregt, während im HOMO an der Anode Löcher entstehen. Durch elektrostatische Kräfte werden die Löcher und Elektronen zueinander geführt und bilden hierbei ein Exziton. Der Zerfall dieses angeregten Zustands resultiert in der Abgabe von Energie [19].
Man unterscheidet hier zwischen Ligand Centered (LC), Metal Centered (MC) und Metal-Ligand Charge Transfer (MLCT) Anregungen.
Tabelle 1.1:Anregungen und Übergänge in OLEDs [31]
Anregung Übergang
LC π→π?
MC d→d∗
MLCT d→π?
Durch die Koordination des/ der Liganden an ein Zentralatom wird das Elektronensystem beeinflusst und es kommt zu einer Rotverschiebung. [31]
Die Vorteile der organischen LEDs gegenüber ihren anorganischen Vorgängern sind eine höheres lm/W Verhältnis. Dies liegt daran, dass sowohl Singulett- als auch Triplett-Zustände zur Lichtemissi- on beitragen, wobei das Verhältnis von Singulett zu Triplett bei 1:3 liegt. Theoretisch kann die interne Quantenausbeute bei 100% liegen [19].
1.3 Iridium Komplexe
Iridium Komplexe bieten eine Vielzahl an Möglichkeiten zur Lichtemission. Die Wahl des Liganden beeinflusst hierbei das Emissionsspektrum stark, so dass sowohl rote als auch blaue Phosphoreszenz möglich ist.
Durch das Übergangsmetall (hier also Iridium) ist ein eigentlich formal verbotenener Übergang des niedrigsten angeregten Triplett-Zustands in den Singulett-Grundzustand durch Spin-Bahn-Kopplung möglich. [31]
Der acac-Ligand sorgt für eine leichte Rotverschiebung, von 517 zu 525 nm. Des Weiteren hat das Ir(ppy)2(acac)-Spektrum eine geringere Halbwertsbreite (FWHM) als das vonIr(ppy)3[25].
1.4 Singulett- und Triplett-Anregungen
Bei der Absorption von Photonen, die zu einer elektronischen Anregung führt, wird ein Molekül aus dem Grundzustand vertikal angeregt, wobei es in einen höheren Singulett-Zustand gerät. Da die hö- heren Singulett-Zustände bezüglich ihrer Nullpunktschwingungskoordinate gegenüber dem Grund- zustand verschoben sind, muss das Molekül erst relaxieren, um in dem angeregten Zustand das Mini- mum zu erreichen. Die dabei freigesetzte Energie wird als thermische Energie an die Umgebung ab- gegeben. Von hier kann das Molekül in einen tieferen Singulett-Zustand, einen Triplett-Zustand oder den Grundzustand (in der Regel auch Singulett) übergehen. Wird beim Übergang in den Singulett- Zustand Strahlung abgegeben (Emission), wird dies als Fluoreszenz bezeichnet. Den strahlungslosen Übergang in einen Zustand gleicher Multiplizität (hier Singulett) bezeichnet man als Interne Kon- version (IC), den strahlungslosen Übergang in einen Zustand anderer Multiplizität (hier Tripett) als Intersystem Crossing (ICS).
Die Emission beim Übergang von T1 in S0 wird als Phosphoreszenz bezeichnet. Dieser Effekt ist eigentlich aufgrund verschiedener Spin-Multiplizität verboten, wird jedoch durch die Spin-Bahn- Kopplung ermöglicht. Hierbei koppeln Singulett- und Triplett-Zustand, so dass der Triplett-Zustand Eigenschaften des Singulett-Zustands und umgekehrt aufweist und damit eine Übergangswahrschein- lichkeit bekommt [5].
Abbildung 1.2:Jablonski-Schema[4]
Die Helligkeit eines Übergangs bei der Absorption resultiert aus dem Übergangsdipolmoment einer Anregung. Hierbei wechselwirkt das Dipolmoment mit dem E-Feld der elektromagnetischen Strah- lung. Folglich ist eine Absorption (oder Emission) von elektromagnetischer Strahlung nur von Ato- men oder Molekülen möglich, die im Übergang ein Dipolmoment aufweisen.
Dabei gibt Gleichung (1) mit|−→
Mmn|2als Übergangsdipolmoment,ν als Frequenz der elektromagne- tischen Strahlung,meals Masse eines Elektrons und e als Elementarladung
f= 8π2meν 3he2 |−→
Mmn|2 (1)
den Zusammenhang zwischen Oszillatorstärke f und Übergangsdipolmoment an [6].
1.5 Das Franck-Condon Prinzip
Das Franck-Condon-Prinzip besagt, dass ein Molekül bei elektronischer Anregung in den höheren Schwingungszustand übergeht, dessen Schwingungswellenfunktion der Schwingungswellenfunktion im Schwingungsgrundzustand des niedrigeren elektronischen Zustand am ähnlichsten ist. Diese Ähn- lichkeit lässt sich durch das Überlappungsintegral bestimmen.
Der Gesamtzustand eines Moleküls besteht aus einem elektronischen TeilΨε und einem Vibrations- Teil Ψν. Innerhalb der Born-Oppenheimer-Approximation folgt daher für das Übergangsdipolmo- ment
µf i= ˆ
Ψ?εfΨ?νf (
−e
∑
i
ri+e
∑
I
ZIRI )
ΨεiΨνidτ (2)
µf i=−e
∑
i
ˆ
Ψ?εfriΨεidτε ˆ
Ψ?νfΨνidτν+e
∑
I
ZI
ˆ
Ψ?εfΨεidτε ˆ
Ψ?νfRIΨνidτν (3) Da zwei unterschiedliche elektronische Zustände orthogonal sind, ist der zweite Term auf der rechten Seite von Gleichung (3) null. Daraus folgt
µf i=−e
∑
i
ˆ
Ψ?εfriΨεidτε ˆ
Ψ?νfΨνidτν=µεfεiS(νf,νi) (4)
mit
µε=−e
∑
i
ˆ
Ψ?εfriΨεidτε (5) und
S(νf,νi) = ˆ
Ψ?ν
fΨνidτν (6)
Dabei gibt Gleichung (5) das elektrische Übergangsdipolmoment (aus Gleichung (1)) und Gleichung (6) das Überlappungsintegral, auch Franck-Condon-Faktor genannt, an [9].
2 Methoden
2.1 Dichtefunktionaltheorie (DFT)
Die Dichtefunktionaltheorie basiert auf dem Hohenberg-Kohn-Theorem [18]. Dieses zeigte, dass die Eigenschaften im Grundzustand eines Vielteilchensystems von seiner Elektronendichte abhängt. Die Elektronendichte hängt nur von 3 Koordinaten ab.
Das Integral der Elektronendichte über den gesamten Raum entspricht der Anzahl der Elektronen.
N= ˆ
ρ(r)dr (7)
Darüber hinaus lässt sich, da die Kerne als Punktladungen aufgefasst werden können und somit ihre Positionen ein lokales Maximum an Elektronendichte darstellen, die für die Aufstellung des Hamil- tonoperators benötigten Kernladungszahlen über die Dichte nach Gleichung (8) ermitteln mit Z als Kernladungszahl von A,rAals radiale Entfernung von A undρ die sphärische mittlere Dichte.
∂ ρ(rA)
∂rA |rA=0=−2ZAρ(rA) (8) Die Energie des Systems lässt sich in eine kinetische und eine potentielle Komponente aufteilen.
Hierbei wird das System als klassisch betrachtet. Die Anziehung zwischen Kern und Elektronen lässt sich als
Vne[ρ(r)] =
nuclei
∑
k
ˆ Zk
|r−rk|ρ(r)dr (9)
darstellen und die Abstoßung einer klassischen Ladungsverteilung als Vee[ρ(r)] = 1
2
ˆ ˆ ρ(r1)ρ(r2)
|r1−r2| dr1r2 (10)
mitr1undr2als Integrationsvariablen über den gesamten Raum. Hierbei werden jedoch die energe- tischen Beiträge von Korrelation und Austausch ignoriert.
Für die kinetische Energie nimmt man ein homogenes Elektronengas an, welches eine konstante Dichte ungleich 0 hat. Nach Thomas und Fermi lässt sich für dieses System die Gleichung (11) aufstellen.Tuegsteht hier für uniform electron gas.
Tueg[ρ(r)] = 3
10 3π223 ˆ
ρ
5
3(r)dr (11)
Da Gleichung (10) u.a. die Austauschwechselwirkung fehlt, schlugen Bloch und Dirac für ein homo- genes Elektronengas die Austauschenergie
EX[ρ(r)] =−3 4
3 π
13ˆ ρ
4
3(r)dr (12)
vor. Zusammen mit den Gleichungen (9) bis (11) wurde hieraus das Thomas-Fermi-Dirac-Modell, das aufgrund seiner Ungenauigkeiten jedoch nie Anwendung fand.[13]
Auf dieser Grundlage haben Kohn und Sham [21] eine Theorie entwickelt, die die Gesamtenergie des Systems weitgehend beschreibt.
Sie postulierten, dass der Hamiltonoperator für ein System nicht wechselwirkender (non-interacting) Elektronen einfacher aufstellbar ist. Der Hamiltonoperator lässt sich als Summe von Einelektro- nenoperatoren darstellen, deren Eigenfunktionen Slater-Determinanten der einzelnen Einelektronen- Funktionen und dessen Eigenwerte der Summe der Einelektronen-Eigenwerte entspricht.
Dies lässt sich erreichen, indem ein fiktives System mit nicht wechselwirkenden Elektronen gewählt wird, dessen Dichte der Dichte des realen Systems entspricht. Anschließend wird die Gesamtenergie des Systems in Einzelenergien
E[ρ(r)] =Tni[ρ(r)] +Vne[ρ(r)] +Vee[ρ(r)] +4T[ρ(r)] +4Vee[ρ(r)] (13) aufgeteilt. Diese beziehen sich auf die kinetische Energie der nicht wechselwirkenden Elektronen, die Kern-Elektron-Wechselwirkung (Gl. 9), die klassischen Elektron-Elektron-Abstoßung (Gl. 10), einen Korrekturterm für die kinetische Energie der wechselwirkenden Elektronen sowie einen Korrektur- term für alle nicht-klassischen Elektron-Elektron-Abstoßungen.
Drückt man die Dichte durch Orbitale aus und fasst alle Korrekturterme unterEXC, lässt sich Glei- chung (13) schreiben als
E[ρ(r)] =
N
∑
i
hχi| −1
2∇2i|χii − hχi|
nuclei
∑
k
Zk
|ri−rk|χii
+
N
∑
i
hχi|1 2
ˆ ρ(r0)
|ri−r0|dr0|χii+Exc[ρ(r)]
(14) mit der Dichte für ein N-Elektronen System
ρ(r) =
N
∑
i=1
|χi(r)|2 (15)
Die Orbitale die E minimieren, genügen der Bedingung
hKSi χi=εiχi (16)
wobei der Kohn-Sham-Einelektronen-Operator definiert ist als
hKSi =−1 2∇2i −
nuclei
∑
k
Zk
|ri−rk|+
ˆ ρ(r0)
|ri−r0|dr0+Vxc (17)
mit
Vxc=δExc
δ ρ (18)
Excist hierbei eine Zusammenführung der schwierigen Terme4T und4Vee.[13]
2.1.1 Das BH-LYP Funktional
Becke-Half and Half Austausch mit LYP-Korrelation. Hierbei wird die Austauschwechselwirkung zu 50% durch B88 und zu 50% durch HF beschrieben [10].
Dabei kommt es zu der Austausch-Korrelations-Energie in Gleichung (19) [3].
Exc=0.5ExHF+0.5EXB88+EcLY P (19) 2.1.2 Das PBE0 Funktional
Kombination von PBE mit einem vordefinierten exakten Austauschterm.
Hierbei wird das Perdew-Wang (PW) Korrelationsfunktional mit einer neuen Austauschgröße kom- biniert zu Gleichung (20) [7].
ExPBE= bx2
1+ax2 (20)
mitb=0.00336,a=0.00449,x= |∇ρ|
ρ3/4
Dies führt zu einer Austausch-Korrelations-Energie in Gleichung 21 [3]
EXC=0.25EXHF+0.75EXPBE+ECmPW91 (21) 2.2 Multireference Conguration Interaction (MRCI)
Führt man eine CI-Berechnung für alle Elektronen sowie alle Orbitale im kompletten aktiven Raum durch, so nennt sich dies “full configuration CI”. Für einen bestimmten Basissatz ist dies die best- mögliche Berechnung, da es jede mögliche CSF berücksichtigt. Eine full-CI-Rechnung mit einem unendlichen großen Basissatz führt daher zu einer exakten Lösung der nichtrelativistischen zeitunab- hängigen Schrödingergleichung.
Problematisch hier ist jedoch die Anzahl der CSFs, die eine full CI Rechnung mit großem Basissatz nur auf sehr kleinen Molekülen erlauben.
Für größere Moleküle muss ein kleiner Basissatz gewählt werden, was jedoch diese Methode weniger interessant macht. Eine Lösung hier ist die Nutzung des RAS, wobei nur eine begrenzte Anzahl an Anregungen betrachtet wird [13]
Die full-CI-Wellenfunktion zeigt Gleichung (22).Ψ0 ist hierbei eine closed-shell restricted HF De- terminante, hierzu lässt sich eine große Anzahl von anderen N-Elektronen Determinanten aus 2K Spin Orbitalen konstruieren. Hierzu beschreibt man lediglich, worin sich diese von|Ψ0iunterschei- den bis zur N-Tupel angeregten Determinante. Diese Wellenfunktion lässt sich erweitern zur exakten Viel-Elektronen-Wellenfunktion|Φ0i.
|Φ0i=c0|Ψ0i+
∑
ar
cra|Ψrai+
∑
a<b r<s
crsab|Ψrsabi+
∑
a<b<c r<s<t
crstabc|Ψrstabci+
∑
a<b<c<d r<s<t<u
crstuabcd|Ψrstuabcdi+· · · (22)
Die Einschränkungen der Summationsindizes stellen sicher, dass jede Anregung nur einmal gezählt wird. Gleichung (24) zeigt einen Auszug der Struktur der CI-Matrix. Aus Übersichtgründen wurde die Gleichung (22) in eine symbolische Form überführt
|Φ0i=c0|Ψ0i+cS|Si+cD|Di+cT|Ti+cQ|Qi+. . . (23)
wobei|Sifür die einfachen,|Difür die doppelten Anregungen und so weiter steht [28].
|Ψrai |Ψrsabi |Ψrstabci |Ψrstuabcdi . . .
|Ψ0i |Si |Di |Ti |Qi . . .
hΨ0| hS|
hD|
hT| hQ|
...
hΨ0|H|Ψˆ 0i 0 hΨ0|H|Diˆ 0 0 . . .
hS|H|Siˆ hS|H|Diˆ hS|H|Tˆ i 0 . . .
hD|H|Diˆ hD|H|Tˆ i hD|H|Qiˆ . . . hT|H|Qiˆ . . . hQ|H|Qiˆ . . . ...
(24)
Bei der MRCI wird ähnlich vorgegangen, sie ist also vergleichbar mit der single-reference CI, mit dem Unterschied, dass MCSCF-Wellenfunktionen oder andere Wellenfunktionen mit Einfach- und Zweifachanregungen anstelle der HF-Wellenfunktionen verwendet werden [13].
2.3 DFT/MRCI
Aufgrund des großen Rechenaufwands, insbesondere für große Systeme sind ab initio Methoden wie MRCI, die statische sowie dynamische Korrelationseffekte einschließen, wenig praktikabel. Die auf der Theorie von Kohn-Sham basierende DFT hingegen zeigt zuverlässige Ergebnisse bei geringem Rechenaufwand zur Beschreibung der dynamischen Elektronenkorrelation.
Die Idee hinter einer Kombination von DFT und CI ist, dass die dynamische Elektronenkorrelation aus der DFT und die statische Elektronenkorrelation aus der CI-Rechnung genommen wird.
Die Nutzung von einigen Tausend der wichtigsten CSF und der Parametrisierung des verwendeten Hamiltonoperators führt zu einem Fehler von unter 0.2 - 0.3 eV bei relativen Energien.[17].
2.4 Verwendete Software
Alle quantenchemischen Berechnungen wurden mit Turbomole 5.10 durchgeführt [2].
Dabei wurde aoforce zur Berechnung der Schwingungsfrequenzen genutzt [30].
(U)DFT-Berechnungen wurden mit dscf durchgeführt.
DFT/MRCI-Berechnungen wurden mit DFT/MRCI durchgeführt [17, 20].
Oszillatorstärken wurden mit proper bestimmt [11].
Franck-Condon Spektren wurden mit vibes berechnet [15].
Die Gaußverbreiterung wurde mit plotter durchgeführt.
Die experimentellen Spektren wurden mit Origin 8.5 digitalisiert [1].
2.5 Technische Details der Rechnungen 2.5.1 Technische Angaben
2.5.1.1 DFT/MRCI Für die DFT/MRCI-Rechnungen wurde festgelegt, dass zwei Elektronen aus den untersten fünf besetzten Orbitale in die untersten fünf unbesetzten Orbitale angeregt wurden.
In einem ersten Durchlauf wurde der Selektionsschwellwert auf 0.8 gesetzt. Von den hier erhalte- nen Referenzkonfigurationen wurden alle mit einem Gewicht von mehr als 0.003 in einem weiteren Durchlauf mit Selektionsschwellwert von 1.0 verwendet.
FürIr(ppy)2(acac)mit def-SV(P) Basissatz wurden die untersten 64 Rumpfelektronen (in 32 Orbi- talen) sowie die obersten 106 Orbitale eingefroren.
FürIr(ppy)2(acac)mit def-TZVP Basissatz wurden die untersten 64 Rumpfelektronen (in 32 Orbi- talen) sowie die obersten 238 Orbitale eingefroren. Diese Anzahl ergibt sich aus der energetischen Übereinstimmung mit dem ersten eingefrorenen Orbital der def-SV(P) Rechnung.
FürIr(F2ppy)2(acac)mit def-SV(P) Basissatz wurden die untersten 72 Rumpfelektronen (in 36 Or- bitalen) sowie die obersten 106 Orbitale eingefroren.
Tabelle 2.1 zeigt dabei die Anzahl der resultierenden CSFs für die beiden Moleküle. Zu beachten ist, dass die Triplett-Geometrie-Rechnungen mit 10 Wurzeln durchgeführt wurden und die Singulett- Geometrie-Rechnungen mit 50 Wurzeln.
Tabelle 2.1:Anzahl der CSFs Molekül
Singulett-Geometrie Triplett-Geometrie
def-SV(P) def-TZVP def-SV(P)
Singulett-Zustand Triplett-Zustand Singulett-Zustand Singulett-Zustand Triplett-Zustand
Ir(ppy)2(acac) 25583273 37704379 49485666 10729919 14706183
Ir(F2ppy)2(acac) 20916088 28743064 - 9338401 12308125
2.5.2 Vibes
Tabelle 2.2 gibt die Input-Parameter wieder, die für die Erstellung der Franck-Condon-Spektren mit Vibes verwendet wurden. Für beide Moleküle wurden die gleichen Parameter gewählt.
Tabelle 2.2: Input-Parameter von Vibes 0 K 298 K Anzahl d. Punkte 8192
Interval 300 fs Dämpfungη 600cm−1 Temperature 0 K 298 K 2.5.3 Der def-SV(P) Basissatz
Split Valence mit Polarisation.
Rumpf-Atomorbitale werden durch eine kontrahierte Gaußfunktion dargestellt, Valenz-Atomorbitale durch zwei, erweitert durch einen Satz von Polarisationsfunktionen für alle Nicht-Wasserstoffatome[26, 14].
Für Ir wird ein ECP hinzugefügt. Hierbei werden die Rumpfelektronen durch ein Pseudopotential ersetzt [8].
2.5.4 Der def-TZVP Basissatz Triple Zeta Valence mit Polarisation.
Hierbei werden drei Gaußfunktionen pro Valenz-Atomorbital verwendet. Ebenfalls ist die Darstel- lung der Rumpf-Atomorbitale besser als bei SV(P). Für H-Atome wird eine p-Polarisationsfunktion verwendet [27, 14].
Wie auch bei def-SV(P) wird für Ir ein ECP hinzugefügt [8].
3 Auswertung 3.1 Die Geometrie
Die Geometrie des Moleküle wurde mit DFT berechnet, hierbei wurde PBE0 als Funktional und def-SV(P) in der einen, def-TZVP in der anderen Rechnung als Basissatz gewählt.
Sowohl für Ir(ppy)2(acac) als auch fürIr(F2ppy)2(acac) lässt sich der Trend erkennen, dass die Bindungslängen zwischen den Liganden und Iridium von ihrer Position abhängen. Dies liegt am trans-Effekt, der dafür sorgt, dass eine trans-Metall-Bindung abhängig von der Art des trans-ständigen Liganden geschwächt werden kann [12]. So scheint ein Kohlenstoffatom von Phenyl in trans-Stellung zu einer stärkeren Bindung zu führen als acac oder Pyridin.
3.1.1 Vergleich zwischen def-SV(P) und def-TZVP beiIr(ppy)2(acac)
Die Bindungswinkel (s. Tab.B.11) der N-Ir-C-, O-Ir-O-, N-Ir-O und C-Ir-O-Bindungen weichen um 0.2 – 0.6° ab. Im acac-Liganden gibt es ebenfalls Abweichungen von bis zu 0.6°. Innerhalb der Phe- nylpyridinringe sind die Abweichungen minimal, sie betragen 0.0 – 0.1°, so dass hier die Größe des Basissatzes keine große Rolle zu spielen scheint.
Die Bindungslängen (s. Tab.B.10) weichen entweder um 0 oder um 1 pm ab. Es gibt Abweichungen und gleiche Bindungslängen sowohl in den Phenylpyridinringen als auch bei den Ligand-Iridium- Bindungen, im acac Liganden sind die C-C und O-C Bindungslängen sogar alle identisch.
Dies deutet darauf hin, dass bereits der kleinere def-SV(P) Basissatz ausreichend ist für die Geome- trieoptimierung.
3.1.2 Vergleich zwischenIr(ppy)2(acac) und Ir(F2ppy)2(acac)
Beim Vergleich zwischen dem fluorierten und nichtfluorierten Molekül zeigen sich über weite Be- reiche hohe Übereinstimmung. Abweichungen in den Bindungslängen (s. Tab. B.12) finden sich bei den Bindungen des fluorierten Kohlenstoffs. Die maximale Abweichung beträgt 2 pm, die meisten Bindungslängen sind identisch.
Bei den Bindungswinkeln (s. Tab. B.13) finden sich größere Abweichungen. Die geringsten Abwei- chungen bilden hier die Winkel der Liganden am Iridium zueinander. Die deutlichsten Abweichungen finden sich im fluorierten Phenylring der ppy-Liganden. Hier beträgt die Abweichung bis zu 2.5◦, ist also auch verhältnismäßig gering.
3.1.3 Vergleich zwischen Singulett- und Triplett-Geometrie 3.1.3.1 Ir(ppy)2(acac)
Die Bindungslängen (s. Tab. B.14) unterscheiden sich zwischen Singulett- und Triplett-Geometrie nur in der Ligand-Iridium-Bindungslänge. Hier gibt es Abweichungen, die von +2 pm (Ir-N1-Bindung) bis -4 pm (Ir-C11*-Bindung) reichen. Bei der Ir-N1-Bindung ist dies vor allem darauf zurückzufüh- ren, dass im HOMO eine stark verdrehte bindende Wechselwirkung auftritt. Da bei derT1-Geometrie ins LUMO angeregt wurde und dort nahezu keine Ladung am Iridium vorliegt, fällt diese Wechsel- wirkung weg, was in einer Verlängerung der Bindung um 2 pm resultiert.
Gegenteiliges ist bei der Ir-C11*- und Ir-N1*-Bindung des ppy(1)-Liganden der Fall. Hier gibt es im HOMO eine antibindende Wechselwirkung mit Knoten zwischen Iridium und dem entsprechenden Atom am Liganden. Durch die Anregung ins LUMO fällt diese Wechselwirkung wie auch bei der Ir-N1-Bindung weg, was in einer Verkürzung der Bindungslänge um 2 pm (für Ir-N1*) bzw. 4 pm (für Ir-C11*) resultiert.
Die Bindungswinkel (s. Tab. B.15) zeigen nur geringe Abweichungen von 2.2° oder weniger.
3.1.3.2 Ir(F2ppy)2(acac)
Die Bindungslängen (s. Tab. B.16) unterscheiden sich zwischen Singulett- und Triplett-Geometrie nur in der Ligand-Iridium Bindungslänge. Hier gibt es Abweichungen, die von +2 pm (Ir-N1-Bindung) bis -4 pm (Ir-N1*-Bindung) reichen. Bei der Ir-N1-Bindung des ppy(2)-Liganden ist dies vor allem darauf zurückzuführen, dass im HOMO eine (verdrehte) bindende Wechselwirkung auftritt. Da bei derT1-Geometrie ins LUMO angeregt wird und dort nahezu keine Ladung am Iridium vorliegt, fällt diese Wechselwirkung weg, was in einer Verlängerung der Bindung um 2 pm resultiert.
Gegenteiliges ist bei der Ir-C11*- und Ir-N1*-Bindung des ppy(1)-Liganden der Fall. Hier gibt es im HOMO eine antibindende Wechselwirkung mit Knoten zwischen Iridium und dem entsprechenden Atom am Liganden. Durch die Anregung ins LUMO fällt diese Wechselwirkung wie auch bei der Ir-N1-Bindung weg, was in einer Verkürzung der Bindungslänge um 4 pm (für Ir-N1*) bzw. 1 pm (für Ir-C11*) resultiert.
Die Bindungswinkel (s. Tab. B.17) zeigen nur geringe Unterschiede von 1.6° oder weniger.
3.1.3.3 Vergleich der Unterschiede zwischen Ir(ppy)2(acac) undIr(F2ppy)2(acac)
Hier wurde untersucht, wie groß die Unterschiede zwischen den Veränderungen bei Singulett- und Triplett-Geometrie beiIr(ppy)2(acac)undIr(F2ppy)2(acac)sind.
Die Bindungslängenunterschiede (s. Tab. B.18) treten überwiegend bei der Iridium-Ligand-Bindung auf, innerhalb der Liganden liegen sie bei 0-1 pm. Bei der Iridium-Ligand-Bindung treten die Unter- schiede lediglich bei C11* und N1* auf, also nur bei einem der beiden Liganden.
Die Bindungswinkel (s. Tab. B.19) unterscheiden sich über weite Bereiche des Moleküls nicht oder nur um unter 0.3°. Ausnahme hier bilden die Winkel der Liganden am Iridium, hier gibt es Abwei- chungen, die jedoch nicht als groß bezeichnet werden können. Hier erkennt man, dass die Fluorierung nur wenig Auswirkungen auf die Unterschiede der Singulett- und Triplett-Geometrien hat, nur unmit- telbar am Zentralatom sind die Abweichungen in einem Bereich von bis zu 1.8◦feststellbar.
3.2 Schwingungsfrequenzen
Die optimierten Singulett- und Triplett-Geometrien stellen Minima auf der Potentialfläche dar.
Wie in Tabelle 3.1 zu sehen ist, nimmt die Nullpunktschwingungsenergie beiIr(ppy)2(acac)in der Triplett-Geometrie gegenüber der Singulett-Geometrie ab.
Ebenfalls ist sie in der def-TZVP-Rechnung geringer als in der def-SV(P)-Rechnung.
Tabelle 3.1: Nullpunktschwingungsenergien vonIr(ppy)2(acac)
Geometrie def-SV(P) Energie/ hartree def-TZVP Energie/ hartree
Singulett 0.437 0.435
Triplett 0.433 -
Tabelle 3.2 zeigt den gleichen Trend beiIr(F2ppy)2(acac), wie auch schon beiIr(ppy)2(acac)zu be- obachten war, nämlich eine Abnahme der Nullpunktschwingungsenergie in der Triplett-Geometrie.
Tabelle 3.2: Nullpunktschwingungsenergie vonIr(F2ppy)2(acac)mit def-SV(P)
Geometrie Energie/ hartree
Singulett 0.406
Triplett 0.401
3.3 Anregungen und Übergänge 3.3.1 Ir(ppy)2(acac)
3.3.1.1 Singulett-Geometrie
3.3.1.1.1 def-SV(P)
Die vorgenommenen Rechnungen mit 50 Wurzeln für den Singulett-Zustand und 49 Wurzeln für den Triplett-Zustand zeigen Anregungen, die von HOMO-12 bis LUMO+12 für den Singulett- Zustand und HOMO-10 bis LUMO+12 für den Triplett-Zustand reichen.
Die Abbildungen A.1, A.2, A.3, A.4, A.5, A.6 zeigen die beteiligten Orbitale.
Das entsprechende MO-Diagramm zeigt Abbildung 3.1. Aus Übersichtlichkeitsgründen sind HOMO- 10 sowie LUMO+7 bis LUMO+11 zwar eingetragen, jedoch ohne bildliches Molekülorbital, da sie in der Singulett-Anregung im betrachteten Anregungsbereich keine Rolle spielen.
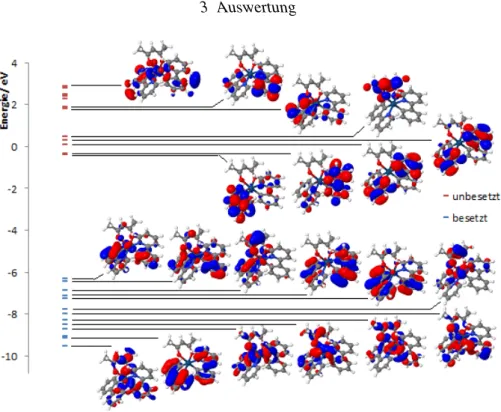
Abbildung 3.1:MO-Diagramm vonIr(ppy)2(acac)mit def-SV(P) in der Singulett-Geometrie. Als Funktional wurde BH-LYP verwendet.
Die Energiedifferenz zwischen HOMO und LUMO beträgt 5.91 eV.
Eine genauere Auflistung zeigt sich in Tabelle 3.3
Auffällig bei der Betrachtung der MOs ist, dass die Ladung bei den energetisch tieferen Orbitalen eher auf dem acac-Liganden vorzufinden ist. Es zeigt sich ebenfalls im HOMO-11 (Abbildung A.1), dass hier keine Ladung am Iridium lokalisiert ist, bei den Anregungen aus diesem Orbital handelt es sich also um reine LC-Übergänge. Darüber hinaus sind Anregungen ausσ-Orbitalen zu sehen.
Betrachtet man den Anteil der Ladung im d-Orbital des Iridium-Zentralatom an der Gesamtladung im jeweiligen Orbital (Tabelle 3.3), so zeigt sich, dass insbesondere Übergänge aus HOMO, HOMO-1, HOMO-5, HOMO-6 und HOMO-8 einen hohen MLCT Anteil haben. Die Ladungsanteile wurden mit der Mullikan Populationsanalyse durchgeführt. Diese basiert auf der LCAO-Methode. Aufgrund des geringen Ladungsanteils am Iridium in den unbesetzten Orbitalen wurde für diese keine Popula- tionsanalyse durchgeführt.
Tabelle 3.3:Lokalisation der Ladungsdichte in den Orbitalen vonIr(ppy)2(acac)mit def-SV(P)
Molekülorbital Art Ladungsverteilung Energie/ eV Ladungsanteil im d-Orbital (%)
HOMO-12 d/π/σ Ir/py(1)/ph(2)/acac -9.54 9.7
HOMO-11 π ppy(1+2) -9.16 0.4
HOMO-10 π ppy(1+2) -9.07 0.5
HOMO-9 d/σ Ir/ph(1)/py(2)/acac -8.71 12.55
HOMO-8 d/π/σ Ir/ph(1)/ppy(2)/acac -8.49 38.55
HOMO-7 d/π/σ Ir/ph(1+2)/acac -8.28 12.3
HOMO-6 d/π/σ Ir/py(1)/ph(2)/acac -8.01 28.45
HOMO-5 d/π Ir/ acac -7.77 57.35
HOMO-4 d/π Ir/ph(2)/ ppy(1) -7.25 4.65
HOMO-3 d/π Ir/ ph(1)/ ppy(2) -7.12 7.15
HOMO-2 d/π Ir/ ph(1)/ acac -6.87 14.70
HOMO-1 d/π Ir/ ph(1+2)/acac -6.45 29.60
HOMO d/π Ir/ ph(1+2)/acac -6.31 32.10
LUMO π* ppy(1) -0.4 -
LUMO+1 π* ppy(2) -0.34 -
LUMO+2 π* py(1+2) 0.09 -
LUMO+3 π* ppy(1+2) 0.3 -
LUMO+4 π* acac 0.46 -
LUMO+5 π* ph(1+2) 1.81 -
LUMO+6 π* ph(1+2) 1.87 -
LUMO+12 d*/σ* Ir/ph(1+2) 2.87 -
Tabelle B.1 zeigt die Übergänge sowie die genaue Ladungsverteilung vor und nach der Anregung, sowie die dazugehörigen Energien in Elektronenvolt und Wellenlänge sowie Oszillatorstärke.
Die Mehrheit der Übergänge hat aufgrund von Beteiligungen von Orbitalen mit hohem d-Anteil eine Mischung aus MLCT- und LC-Übergängen. Eine Besonderheit stellt derS0 →S32 Anregung dar, hier liegt durch den d*-Anteil im LUMO+12 zum Teil ein MC-Übergang vor. Dieser Übergang war sonst bei keiner anderen Anregung in relevantem Anteil (über 10%) zu beobachten, auch beim Ir(F2ppy)2(acac)mit gleichem Basissatz sowie beiIr(ppy)2(acac)mit def-TZVP-Basissatz war die- ser Übergang nicht anzutreffen.
Auch lässt sich kein Trend ablesen bezüglich der Helligkeit, ob sie durch einen hohen Anteil von MLCT-oder LC-Übergängen beeinflusst wird. So zeigt sich, dass die hellste Anregung, nämlich S0→S16zumindest in relevantem Anteil, überwiegend aus Orbitalen mit sehr wenig d-Anteil kommt und somit eher ein LC-Übergang ist, andere Anregungen mit hohem MLCT-Anteil sind eher dunkel.
Jedoch ist auch der gegenteilige Trend zu beobachten.
Einfluss auf die Helligkeit hat jedoch das Übergangsdipolmoment (siehe 1.4). Eine genauere Betrach- tung erfolgt daher für die wichtigen AnregungenS0→S1 undS0→S2 und für die beiden hellsten AnregungenS0→S16undS0→S17.
Der Übergangsdipolvektor derS0→S1 Anregung zeigt durch py(1). Dies liegt an der Ladungsver- schiebung von ph(2) zu py(1).
Der Übergangsdipolvektor derS0→S2Anregung zeigt durch ppy(2). Dies liegt an der Ladungsver- schiebung von ph(1) zu py(2).
Der Übergangsdipolvektor derS0→S16Anregung zeigt in den Raum zwischen ph(1) und acac. Dies liegt hauptsächlich an Ladung, die von ph(2) zu ppy(1) übertragen wird.
Der Übergangsdipolvektor derS0→S17 Anregung verläuft zwischen ph(1) und ph(2) und zeigt in Richtung des Pyridinteils. Dies liegt an der Ladung, die aus den Phenylteilen auf den gesamten Phe- nylpyridinliganden verteilt wird.
Eine Auswahl besonders heller Übergänge (f(L) > 0.05) mit der dazugehörigen Energie sowie dem S0→S1Übergang zeigt Tabelle 3.9.
Tabelle 3.4:Auswahl besonders heller Übergänge vonIr(ppy)2(acac)mit def-SV(P)
Anregung Energiedifferenz/ eV Wellenlänge/ nm Oszillatorstärke f(L)
S0→S1 3.05 406.9 0.01331
S0→S2 3.21 386.1 0.09754
S0→S3 3.34 371.0 0.06636
S0→S13 4.07 304.5 0.06895
S0→S16 4.21 294.7 0.2179
S0→S17 4.22 293.5 0.19682
S0→S18 4.28 289.8 0.07113
S0→S20 4.51 275.1 0.07153
S0→S23 4.63 268.0 0.04993
S0→S27 4.74 261.5 0.07591
S0→S28 4.82 257.0 0.1359
S0→S29 4.84 256.0 0.11595
S0→S33 5.12 242.1 0.07311
S0→S37 5.22 237.3 0.15181
S0→S38 5.25 236.3 0.06297
S0→S41 5.3 233.7 0.08953
S0→S43 5.35 231.9 0.07084
S0→S48 5.46 226.9 0.08498
Hier sieht man, dass besonders viele helle Übergänge im Bereich von 295 nm und 250 nm sind.
Ebenfalls liegen alle Übergänge im UV-Bereich.
Abbildung 3.2 zeigt das dazugehörige Spektrum aller Übergänge aus Tabelle B.1.
Abbildung 3.2:Singulett-Absorptionsspektrum vonIr(ppy)2(acac)mit 50 Wurzeln und def-SV(P)
Analog dazu zeigt Tabelle B.2 die entsprechenden Triplett-Übergänge an der Singulett-Geometrie, und die dazugehörigen Energien und Helligkeiten. Die Helligkeiten sind ausgehend vom T1, also T1→Tn.
3.3.1.1.2 def-TZVP
Die Rechnungen wurden mit dem def-TZVP-Basissatz durchgeführt. Da hier nur der Vergleich zum def-SV(P) Basissatz entscheidend ist, wurden nur die Singulett-Anregungen mit 50 Wurzeln berechnet. Hierbei reichen die Anregungen von HOMO-12 bis LUMO+6.
Die Abildungen A.7, A.8, A.9, A.10, A.20 zeigen die entsprechenden Orbitale.
Das dazugehörige MO-Diagramm zeigt Abbildung 3.1.
Abbildung 3.3:MO-Diagramm vonIr(ppy)2(acac)mit def-TZVP in der Singulett-Geometrie. Als Funktional wurde BH-LYP verwendet.
Der energetische Abstand zwischen HOMO und LUMO beträgt 5.90 eV.
Es zeigt sich hier eine große Übereinstimmung zu den Ergebnissen der def-SV(P)-Rechnung. Auch hier nimmt der Ladungsanteil auf dem acac-Liganden innerhalb der betrachteten besetzten Orbitale ab. Bei allen betrachteten Orbitalen ist Ladung im d-Orbital des Iridiums zu beobachten. Ausnahme bildet wie bei def-SV(P) das HOMO-10 und HOMO-11, die keine Ladung im d-Orbital haben, jedoch hier in den 50 untersten Zuständen an keiner Anregung beteiligt sind. Bei def-SV(P) gibt es hingegen Anregungen aus HOMO-11.
Eine genaue Auflistung zeigt Tabelle 3.5.