Mathematisch-Naturwissenschaftliche Fakultät
Themenstellerin: Prof. Dr. Christel M. Marian Zweitgutachter: Jun.-Prof. Dr. Jörg Tatchen
Bachelorarbeit
Einfluss der Liganden auf die
elektronische Struktur von Iridiumkomplexen:
fac-Ir(thpy)
3im Vergleich zu fac-Ir(ppy)
3vorgelegt von:
Student: Gerd Schrörs
Studiengang: Wirtschaftschemie
Fachsemester: 7
Matrikelnummer: 1895243
Geburtsdatum: 04. Oktober 1988
Adresse: Ilvericher Str. 9, 47809 Krefeld
Telefon-Nr.: 02151 571243
E-Mail: gerd.schroers@uni-duesseldorf.de
Düsseldorf, den 03. Januar 2013
1 EINLEITUNG...1
1.1 ORGANISCHE LEDS...1
2 THEORETISCHE GRUNDLAGEN UND METHODEN...1
2.1 IRIDIUM-KOMPLEXE...1
2.1.1 fac-Ir(thpy)3...1
2.1.2 fac-Ir(ppy)3 ...2
2.2 FRANCK-CONDON-PRINZIPUND OSZILLATORSTÄRKE...3
2.3 ELEKTRONISCHE ANREGUNGEN...3
2.4 DICHTEFUNKTIONAL-THEORIE ...6
2.5 MULTIREFERENCE CONFIGURATION INTERACTION...7
2.6 DFT / MRCI ...8
2.7 SOFTWARE ...8
2.8 ANGABENZU RECHNUNGSDURCHFÜHRUNG ...8
2.8.1 Basissatz...8
2.8.2 Effektives Rumpf-Potenzial...8
2.8.3 Verwendete Funktionale...8
2.8.4 DFT/MRCI...8
3 ERGEBNISSE UND DISKUSSION...9
3.1 SINGULETT-GEOMETRIE: STRUKTUR...9
3.1.1 fac-Ir(thpy)3...9
3.1.2 fac-Ir(ppy)3 ...10
3.2 SINGULETT-GEOMETRIE: MOLEKÜLORBITALE ...11
3.2.1 fac-Ir(thpy)3...11
3.2.2 fac-Ir(ppy)3 ...14
3.3 SINGULETT-GEOMETRIE: DFT-MRCI...15
3.3.1 fac-Ir(thpy)3...15
3.3.2 fac-Ir(ppy)3 ...18
3.4 TRIPLETT-GEOMETRIE: STRUKTUR ...19
3.4.1 fac-Ir(thpy)3...19
3.4.2 fac-Ir(ppy)3 ...21
3.5 TRIPLETT-GEOMETRIE: MOLEKÜLORBITALE ...22
3.5.1 fac-Ir(thpy)3...22
3.5.2 fac-Ir(ppy)3 ...25
3.6 TRIPLETT-GEOMETRIE: DFT-MRCI...26
3.6.1 fac-Ir(thpy)3...26
3.6.2 fac-Ir(ppy)3 ...28
4 FAZIT UND AUSBLICK ...30
5 ANHANG...I
6 LITERATURVERZEICHNIS...XXVI
Abb. 1: Molekülstruktur fac-Ir(thpy)3...2
Abb. 2: Molekülstruktur fac-Ir(ppy)3 ...2
Abb. 3: Franck-Condon-Prinzip...3
Abb. 4: Jablonski-Diagramm...4
Abb. 5: MO-Schema eines oktaedrisch koordinierten Komplexes...5
Abb. 6: Übergänge in Phosphoreszenz-Emittern...6
Abb. 7: MO-Diagramm von fac-Ir(thpy)3 in Singulett-Geometrie...11
Abb. 8: Molekülorbital LUMO+10 von fac-Ir(thpy)3 in Singulett-Geometrie...12
Abb. 9: MO-Diagramm von fac-Ir(ppy)3 in Singulett-Geometrie...14
Abb. 10: Normierte Singulett-Absorption von fac-Ir(thpy)3, Vergleich berechnet zum Experiment...18
Abb. 11: Berechnete normierte Singulett-Absorptionen, Vergleich fac-Ir(thpy)3 zu fac-Ir(ppy)3...18
Abb. 12: MO-Diagramm von fac-Ir(thpy)3 in Triplett-Geometrie...23
Abb. 13: MO-Diagramm von fac-Ir(ppy)3 in Triplett-Geometrie...25
Abb. 14: Lage der Zustände in Singulett- und Triplett-Geometrie bei fac-Ir(thpy)3...28
Abb. 15: fac-Ir(thpy)3 Anregung, Emission und adiabatische Energie ...28
Abb. 16: Lage der Zustände in Singulett- und Triplett-Geometrie bei fac-Ir(thpy)3 und fac-Ir(ppy)3....29
Abb. 17: fac-Ir(thpy)3 LUMO+10 (Singulett-Geom.)...VI Abb. 18: fac-Ir(thpy)3 LUMO+9 (Singulett-Geom.)...VI Abb. 19 bis 30: fac-Ir(thpy)3 LUMO+5 bis HOMO-5 (Singulett-Geometrie)...VI-VIII Abb. 31 bis 42: fac-Ir(thpy)3 LUMO+5 bis HOMO-5 (Triplett-Geometrie)...VIII-X Abb. 43 bis 54: fac-Ir(ppy)3 LUMO+5 bis HOMO-5 (Singulett-Geometrie)...X-XII Abb. 55 bis 66: fac-Ir(ppy)3 LUMO+5 bis HOMO-5 (Triplett-Geometrie)...XII-XIV
Verzeichnis der Tabellen
Tab. 1: Übergänge im Jablonski-Diagramm ...4Tab. 2: Anregungen und Übergänge in Übergangsmetall-Komplexen...5
Tab. 3: fac-Ir(thpy)3 ausgewählte Abweichungen der Bindungslängen in Singulett-Geometrie...9
Tab. 4: fac-Ir(thpy)3 ausgewählte Bindungswinkel in Singulett-Geometrie...9
Tab. 5: fac-Ir(thpy)3 ausgewählte Abweichungen der Diederwinkel in Singulett-Geometrie ...10
Tab. 6: fac-Ir(ppy)3 ausgewählte Abweichungen der berechneten Bindungslängen in Singulett- Geometrie im Vergleich zu fac-Ir(thpy)3 ...10
Tab. 7: Charakteristik, MO-Energien und Elektronendichte-Verteilung von fac-Ir(thpy)3 in Singulett- Geometrie...13
Tab. 8: Charakteristik, MO-Energien und Elektronendichte-Verteilung von fac-Ir(ppy)3 in Singulett- Geometrie...15
Tab. 9: Energiedifferenzen wichtiger Übergänge von fac-Ir(thpy)3 in Singulett-Geometrie ...15
Tab. 10: Auswahl der Singulett-Anregungen aus der Singulett-Geometrie von fac-Ir(thpy)3...16
Tab. 12: Auswahl der Singulett-Anregungen aus der Singulett-Geometrie von fac-Ir(ppy)3...19
Tab. 13: fac-Ir(thpy)3 ausgewählte Bindungslängen der Triplett-Geometrie...20
Tab. 14: fac-Ir(thpy)3 ausgewählte Bindungswinkel der Triplett-Geometrie ...21
Tab. 15: fac-Ir(ppy)3 ausgewählte Abweichungen der berechneten Bindungslängen in Triplett- Geometrie im Vergleich zu fac-Ir(thpy)3 ...22
Tab. 16: Charakteristik, MO-Energien und Elektronendichte-Verteilung von fac-Ir(thpy)3 in Triplett- Geometrie...24
Tab. 17: Charakteristik, MO-Energien und Elektronendichte-Verteilung von fac-Ir(ppy)3 in Triplett- Geometrie...26
Tab. 18: Energiedifferenzen wichtiger Übergänge von fac-Ir(thpy)3 in Triplett-Geometrie ...26
Tab. 19: Ausgewählte Singulett-Anregungen aus der Triplett-Geometrie von fac-Ir(thpy)3...27
Tab. 20: Ausgewählte Triplett-Anregungen aus der Triplett-Geometrie von fac-Ir(thpy)3...27
Tab. 21: Singulett-Anregungen aus der Triplett-Geometrie von fac-Ir(ppy)3...29 Tab. 22: fac-Ir(thpy)3 Bindungslängen in Singulett-Geometrie, Vergleich berechnet und experimentellI Tab. 23: fac-Ir(thpy)3 Bindungswinkel in Singulett-Geometrie, Vergleich berechnet und experimentell ...I Tab. 24: fac-Ir(ppy)3 Bindungslängen in Singulett-Geometrie...II Tab. 25: fac-Ir(thpy)3 Bindungslängen in Triplett-Geometrie, Vergleich zur Singulett-Geometrie ...III Tab. 26: fac-Ir(thpy)3 Bindungswinkel in Triplett-Geometrie...IV Tab. 27: fac-Ir(thpy)3 Diederwinkel in Triplett-Geometrie...IV Tab. 28: fac-Ir(ppy)3 Bindungslängen in Triplett-Geometrie und Abweichung von der Singulett- Geometrie...V Tab. 29: Singulett-Anregungen aus der Singulett-Geometrie von fac-Ir(thpy)3 mit 21 Wurzeln...XVI Tab. 30: Triplett-Anregungen aus der Singulett-Geometrie von fac-Ir(thpy)3 mit 20 Wurzeln...XVII Tab. 31: Singulett-Anregungen aus der Singulett-Geometrie von fac-Ir(ppy)3 mit 40 Wurzeln...XIX Tab. 32: Triplett-Anregungen aus der Singulett-Geometrie von fac-Ir(ppy)3 mit 40 Wurzeln...XX Tab. 33: Singulett-Anregungen aus der Triplett-Geometrie von fac-Ir(thpy)3 mit 21 Wurzeln...XXI Tab. 34: Triplett-Anregungen aus der Triplett-Geometrie von fac-Ir(thpy)3 mit 20 Wurzeln...XXII Tab. 35: Singulett-Anregungen aus der Triplett-Geometrie von fac-Ir(ppy)3 mit 40 Wurzeln...XXIV Tab. 36: Triplett-Anregungen aus derTriplett-Geometrie von fac-Ir(ppy)3 mit 40 Wurzeln...XXV
Verzeichnis des Anhangs
Anhang I: Geometrie-Daten...I Anhang II: Molekülorbitale...VI Anhang III: DFT-MRCI-Daten...XV
A Anregung
CI Configuration Interaction
CSF Configuration State Function
DFT Dichtefunktional-Theorie
ECP Effective Core Potential
F Fluoreszenz
fac-Ir(ppy)3 fac-Tris-(2-phenylpyridin)iridium(III) fac-Ir(thpy)3 fac-Tris-[2-(2-thienyl)-pyridin]iridium(III)
HF Hartree-Fock
HOMO Highest Occupied Molecular Orbital
IC Interne Konversion
ISC Intersystem-Crossing
LC Ligand Centered
LMCT Ligand-to-Metal-Charge Transfer LUMO Lowest Unoccupied Molecular Orbital
MC Metal Centered
MCSCF Multi Configuration Self-Consistent Field MLCT Metal-to-Ligand Charge Transfer
MO Molekülorbital
MRCI Multireference Configuration Interaction (O)LED (Organic) Light-Emitting Diode
P Phosphoreszenz
S Singulett
SVP Spit Valence Polarization
T Triplett
UDFT Unrestricted Density Functional Theory
VR Schwingungsrelaxation
1 Einleitung
1.1 Organische LEDs
Während bereits seit den 60er Jahren kristalline anorganische LEDs zum Einsatz kommen, konnten sich organi- sche und metallorganische Emitter erst seit einiger Zeit in breiter technischer Anwendung durchsetzen, da nun die Stromversorgung über transparente leitfähige Polymere ermöglicht wurde (Im Jahr 2000 erhielten Alan Heeger, Alan MacDiarmid und Hideki Shirakawa hierfür den Nobelpreis in Chemie) [1][2].
In den letzten Jahren wird die Entwicklung potentieller Emitterfarbstoffe auch von staatlicher Seite zusehends forciert [3]. Dieses große Interesse ist evident, betrachtet man die zahlreichen Vorteile dieser Technologie ge - genüber bisherigen. Flächige Anwendungen, hohe Effizienz und die gezielte Beeinflussbarkeit des Emissions- spektrums. Es gibt jedoch auch Nachteile gegenüber bisherigen Leuchtmitteln wie geringe Lebensdauer und ge- ringe Leuchtdichte [4].
Ein Anspruch der theoretischen Chemie ist es daher, über Berechnungen die Vorgänge in der Elektronenstruktur entsprechender Verbindungen zu verstehen.
2 Theoretische Grundlagen und Methoden
2.1 Iridium-Komplexe
Iridiumkomplexe eignen sich, wie andere Organo-Übergangsmetall-Komplexe, gut für Phosphoreszenz-Emitter aufgrund ihrer starken Spin-Bahn-Kopplung. Hierdurch werden auch Übergänge zugängig, welche durch die Auswahlregeln als grundsätzlich spinverboten eingestuft werden. Die Emissionsspektren der Iridium-Komplexe lassen sich in der Praxis über die Ligandenwahl und über Umgebungseffekte einstellen, sodass ein Großteil des sichtbaren Spektrums abgedeckt werden kann [5]. Die hier vorgestellten Komplexe emittieren im grünen Ener- giebereich [6].
2.1.1 fac-Ir(thpy)3
Der Phosphoreszenz-Emitter fac-Ir(thpy)3 [fac-Tris-[2-(2-thienyl)-pyridin]iridium(III)] und seine elektronische Struktur steht im Fokus dieser Arbeit, wobei der Komplex fac-Ir(ppy)3 als Vergleich herangezogen wird und auf bereits bestehende Daten zurückgegriffen wird [7]. Die Eigenschaften der Liganden werden durch das elekro- nenreiche Thiophen und das elektronenarme Pyridin beeinflusst. Insgesamt ist eine Elektronen-Donor-Akzep- tor-Wechselwirkung innerhalb der einzelnen (thpy)-Liganden zu erwarten. Da für fac-Ir(thpy)3 in Phosphores- zenzspektren vibronische Kopplung und Lösungsmitteleffekte beobachtet wurden, postulieren Tsuboyama et al., dass die Phosphoreszenz vornehmlich durch den niedrigsten Triplettzustand charakterisiert werde, welcher
3π-π* Charakteristik aufweise [5]. Hierbei handele es sich folglich um einen ligandenzentrierten LC Übergang, worauf weiter unten in 2.3 genauer eingegangen werden wird. Für den sehr ähnlichen Komplex fac-Ir(ppy)3, in welchem die Thionyl-Bereiche durch Phenyl ersetzt sind, wird dies jedoch nicht gefunden [8]. Daher soll im Folgenden besonders die Elektronendichte-Verteilung in den Molekülorbitalen untersucht werden.
Der Komplex fac-Ir(thpy)3 ist von besonderem Interesse, da seine Eigenschaften als Emitter-Farbstoff leicht zu beeinflussen sind. Hierzu können große konjugierte funktionelle Gruppen oder Donor- bzw. Akzeptor-Gruppen eingeführt werden [5].
In Abbildung 1 werden die Atome des Komplexes unter Ausblendung des Wasserstoffs nummeriert dargestellt.
Im Folgenden wird zur leichteren Vergleichbarkeit immer die selbe Projektion des Komplexes gewählt.
Element Farbe
Iridium dunkelblau
Kohlenstoff grau
Schwefel gelb
Stickstoff hellblau
Abb. 1: Molekülstruktur Ir(thpy)3
2.1.2 fac-Ir(ppy)3
Der zum Vergleich dienende Komplex fac-Ir(ppy)3 [fac-Tris-(2-phenylpyridin)iridium(III)] emittiert im Grünen und ist seit langem als starker Phosphoreszenz-Emitter bekannt [9]. Hier sind in den Liganden Phenyl-Gruppen- statt des Thionphenyl im fac-Ir(thpy)3 zu finden. Dank seiner hohen Zersetzungstemperatur von 413 °C ist der Komplex für die Verarbeitung durch Vakuum-Bedampfung geeignet [5]. Die Emission aus dem niedrigsten Tri- plett-Zustand T1 findet unter Elektronendichte-Verschiebung vom Metall hin zum Liganden, also durch einen
3MLCT-Übergang, statt [8].
Element Farbe
Iridium dunkelblau
Kohlenstoff grau
Stickstoff hellblau
Abb. 2: Molekülstruktur Ir(ppy)3
2.2 Franck-Condon-Prinzip und Oszillatorstärke
Bei der Wechselwirkung von Materie mit elektromagnetischen Wellen ist die Wahrscheinlichkeit des Übergangs zu höher liegenden elektronischen Zuständen proportional zum Überlappungsintegral der Schwingungszustände des Grundzustandes und des Ziel-Zustandes. Anschaulich ist hierbei ein Übergang im elektronischen Anre- gungsspektrum besonders wahrscheinlich, wenn sich die Schwingungswellenfunktionen bei konstanten Kernpo- sitionen ähnlich sind, das heißt ihre Phasen nah beieinander liegen [10].
Das Überlappungsintegral S zweier Wellenfunktionen ist dabei wie folgt definiert:
S=∫ Ψ1*Ψ2 (1) Bei völlig identischen Funktionen ist S = 1, bei Orthogonalität gilt S = 0. Sein Betragsquadrat wird als Franck- Condon-Faktor bezeichnet. Deutlich wird das Franck-Condon-Prinzip im eindimensionalen Fall, da sich hier die Energie als Potentialkurve abbilden lässt (Abb. 3). Da hierbei in der Regel die elektronische Anregung mit einer Anregung in höher liegende Schwingungszustände einhergeht, wird die überschüssige Energie an die Um- gebung abgegeben, bis ein thermisch relaxierter angeregter Zustand erreicht wird. Folglich ist in diesem Fall die Emission gegenüber der Absorption rotverschoben [11].
Abb. 3: Franck-Condon-Prinzip [11]
Während das Franck-Condon-Prinzip die Wahrscheinlichkeit der Übergänge beschreibt, ist ihre Intensität pro- portional zur Oszillatorstärke f. Dabei ist μ das Dipolmoment des Überganges bei der Frequenz ν [11].
f = 8 π m
eν
∣μ
∣23 h e
2 (2)2.3 Elektronische Anregungen
Ist der elektronische Grundzustand ein Singulett-Zustand, wie es bei fac-Ir(thpy)3 und fac-Ir(ppy)3 der Fall ist, so können bei elektronischer Anregung verschiedene Prozesse ablaufen. Unter Energieaufnahme wird ein Elek- tron aus dem energetischen Grundzustand S0 in einen angeregten Singulett-Zustand angehoben, von wo es in den niedrigsten angeregten Singulett-Zustand S1 relaxiert. Es folgt unter Beibehaltung der Spinausrichtung ent- weder Fluoreszenz oder nach Interner Konversion strahlungslose Schwingungsrelaxation zurück in den Grund- zustand. Liegt Spin-Bahn-Kopplung vor, ist der Übergang in den niedrigsten angeregten Triplett-Zustand T1
Abb. 4: Jablonski-Diagramm [12]
mittels Intersystem-Crossing unter Spininversion zugängig. Nach Schwingungsrelaxation in die energetisch günstigste T1-Kerngeometrie ist eine direkte Relaxation in den S0-Zustand entweder über Phosphoreszenz oder nach erneutem Intersystem-Crossing über Schwingungsrelaxation möglich [11].
Strahlungslose Übergänge zwischen angeregten Zuständen erfolgen meist schnell, sodass längere Lebensdauern nur bei S1 und T1 zu finden sind und höhere angeregte Zustände nur marginal besetzt sind [14].
spin-erlaubt spin-verboten
strahlend Fluoreszenz (F) Phosphoreszenz (P)
strahlungslos Absorption (A)
Interne Konversion (IC) Schwingungsrelaxation (VR)
Intersystem - Crossing (ISC)
Tab. 1: Übergänge im Jablonski-Diagramm
Bei der Ligandenfeldtheorie geht man davon aus, dass die Liganden mit dem Zentralatom über kovalente Bin- dungen verknüpft sind. Betrachtet man Liganden mit freien σ-Elektronenpaaren, so lassen sich in oktaedrischer Anordnung über sechs symmetrieadaptierte Linearkombinationen sechs Liganden-MOs mit den Symmetrieklas- sen a1g, eg und t1u bilden. Bei der Wechselwirkung zweier der insgesamt fünf d-Orbitale eines zentralen Ions mit den beiden eg-Orbitalen der Liganden ergeben sich zwei bindende und zwei antibindende Molekülorbitale. Die drei verbleibenden d-Orbitale der Klasse t2g können mit keinem der Ligandenorbitale wechselwirken und ver- bleiben als nichtbindende Orbitale am Zentralatom. Abbildung 5 zeigt das dazugehörige schematische MO-Dia- gramm. Besitzen die Liganden Orbitale mit π-Symmetrie, so können diese mit den t2g -Orbitalen des zentralen Atoms wechselwirken. Sind diese Ligandenorbitale unbesetzt und liegen energetisch höher als die t2g-Orbitale (a), kommt es zu einer Vergrößerung von Δ. Diesen Effekt, der z.B. bei CO, NO+ und CN- auftritt, bezeichnet man als π-Rückbindung. Im umgekehrten Fall (b) kommt es zu einer Verringerung von Δ.
Abb. 5: MO-Schema eines oktaedrisch koordinierten Komplexes, nur Ligandenorbitale mit lokaler σ- Symmetrie werden berücksichtigt (links) [11]; Mögliche Auswirkungen von π-Bindungen auf Δ: (a) bei
unbesetzten π-Ligandenorbitalen, (b) bei besetzten π-Ligandenorbitalen [13]
Wenn die Energiedifferenz zwischen t2g und eg gering ausfällt, gilt das Aufbauprinzip nicht streng. Es kann zu High-Spin- und Low-Spin-Besetzungen kommen. Welche Elektronenkonfiguration bevorzugt wird, hängt von der Größe dieses Abstandes Δ ab. Für starke Ligandenfelder ist Δ größer als die Spinpaarungsenergie P und Low-Spin wird bevorzugt. Für schwache Ligandenfelder gilt Δ < P, die High-Spin-Besetzung wird bevorzugt [15][16].
Anregungen können anhand der Lokalisation im Komplex charakterisiert werden. Man unterscheidet MC- (Me- tal Centered), LC- (Ligand Centered), MLCT- (Metal-to-Ligand-Charge Transfer) und LMCT-Anregungen (Li- gand-to-Metal-Charge Transfer).
Anregung MC LC MLCT LMCT
Übergang d → d* π → π* d → π* π → d*
Tab. 2: Anregungen und Übergänge in Übergangsmetall-Komplexen [17]
Die Entstehung von Anregungszuständen speziell in OLEDs lässt sich über die Rekombination zweier entge- gengesetzter Ladungsträger beschreiben, welche in die Emitter-Schicht injiziert werden: Einerseits Elektronen durch kathodische Reduktion und andererseits Defekt-Elektronen („Elektronen-Löcher“) durch anodische Oxi- dation. Beide wandern im äußeren elektrischen Feld durch die Trägermatrix in Richtung der jeweils gegensätz- lich geladenen Elektrode. Die Coulombkräfte zwischen der negativen Ladung des Elektrons und dem formell positiven Loch bewirken eine gegenseitige Attraktion. Treffen Elektron und Loch aufeinander, kombinieren bei- de zu einem nach außen neutralen, wasserstoffähnlichen Quasiteilchen, dem Exziton. Elektron und Defekt- Elektron besitzen einen halbzahligen Spin. Sie kombinieren schon auf weite Entfernung zu 25 % Singulett und 75 % Triplett-Exzitonen [18][19]. Die vier möglichen Spin-Kombinationen sind alle gleich wahrscheinlich, da für große Elektron-Elektronenloch-Abstände energetische Entartung vorliegt. Erst bei weiterer Annäherung führt die kurzreichweitige Austauschwechselwirkung zur Energieaufspaltung von T1 und S1. Bei weiterer Annä-
herung geht das Elektron auf das lochtragende Emitter-Molekül über, welches sich nun wie ein regulär pho- to-angeregtes Molekül verhält. Dies kann auch als Metall-zu-Ligand Ladungsübertragung (MLCT) aufgefasst werden [20]. Bei der Rekombination des Elektron-Loch-Paars kann es zur Emission eines Photons kommen:
M* → M + hν
Aber auch zahlreiche strahlungslose Prozesse können ablaufen, welche zur Deaktivierung führen und der Effi- zienz der Lumineszenzdiode abträglich sind. Unimolekulare, wie Dissoziation, Assoziation, Isomerisierung, Homolyse und reduktive Eleminierung sind denkbar. Bimolekulare Prozesse können in der Praxis durch die Gestalt der Trägermatrix gesteuert werden. Hierzu zählen Spin-Gitter-Relaxation, thermische Stoßdesaktivie- rung, elektronischer Energietransfer, Elektronentransfer unter Ionenbildung und Assoziation von Zweitmolekü- len [21].
Die hier beschriebenen Moleküle fac-Ir(thpy)3 und fac-Ir(ppy)3 zählen zu den Triplett-Emittern. In Ir-Komple- xen gilt das Spinverbot elektrischer Dipolübergänge nicht streng, da Spin-Bahn-Kopplung auftritt. Dies macht Phosphoreszenzübergänge zugänglich und führt zu kurzen Emissions-Abklingzeiten, sodass Quenchingprozesse aus T1 und Sättigungseffekte eingeschränkt werden können. Aufgrund der schnellen ISC-Prozesse von S1 nach T1 wird Fluoreszenz unterdrückt. Dieser Effekt, das Triplett-Harvesting, bedeutet eine Steigerung der Effizienz, da so bis zu 100% der Exzitonen zur Emission zur Verfügung stehen, statt der nur zu 25% populierten S1-Zu- stände bei Fluoreszenz-Emittern. Des Weiteren ist eine Nullfeldaufspaltung der T1-Subzustände zu erwarten, deren Ursprung in der Spin-Bahn-Kopplung und der Jahn-Teller-Verzerrung liegt. Ein Nachteil der Triplett- Emitter sind die im Vergleich zu S1 langen Lebensdauern des T1-Zustandes, was in der OLED-Anwendung zu Sättigungseffekten und beschleunigter Degradation führt [14].
Abb. 6: Übergänge in Phosphoreszenz-Emittern unter potentieller Ausnutzung aller gebildeten Exzitonen [14]
2.4 Dichtefunktional-Theorie
Die Skalierung reduziert sich in der DFT von 3N Koordinaten auf 3 Koordinaten gegenüber wellenfunktionsba- sierten Methoden. Wird die Elektronendichte über den gesamten Raum integriert, erhält man die Gesamtladung aller Elektronen des Systems. Diese entspricht in atomaren Einheiten der Anzahl N der Elektronen im System:
N = ∫ ρ( r) δ r
(3) Im Hohenberg-Kohn-Formalismus formuliert man die Energie des Grundzustandes eines Mehrteilchensystems als Funktional der Elektronendichte, um sie dann iterativ zu optimieren. Die Energie E [ρ] wird nun in die kine- tischen Energie T [ ]ρ, die Coulomb-Wechselwirkung von Kern und Elektron Ene [ ]ρ, die Elektron-Elektron-Ab-stoßung J [ ]ρ und in die Austauschwechselwirkung K [ ] ρ aufgeteilt:
E [ρ(r)] = T [ρ( r)]+ E
ne[ρ(r )] + J [ρ( r)]+ K [ ρ( r)]
(4) Hierbei lässt sich die Coulomb-Wechselwirkung zwischen Kern und Elektron und zwischen Elektron und Elek- tron gut beschreiben, nicht jedoch die kinetische Energie und die Austauschwechselwirkung.Mit Hilfe des Kohn-Sham-Formalismus lassen sich kinetische Energie und Austauschwechselwirkung besser beschreiben. Es wird der Ansatz gemacht, die kinetische Energie nicht über die Elektronendichte zu konstruie- ren. Statt dessen wird ein Modellproblem nicht wechselwirkender Teilchen behandelt: Analog zur Störungs- theorie wird hierin der Hamiltonoperator in einen exakten Tni und einen Störterm ΔT aufgeteilt. Für den exakten Anteil werden antisymmetrierte Produkte von Orbitalen benötigt, ähnlich einer HF-Berechnung. Hierbei ist für ein System nicht wechselwirkender Elektronen die kinetische Energie die Summe der einzelnen kinetischen Energien der Elektronen. Alle übrigen Anteile werden mit dem Austausch-Korrelationsfunktional Exc zusam- mengefasst behandelt:
E[ρ(r)] =Tni[ρ(r)] +Ene[ρ(r)] +J[ρ(r)]+ ΔT[ρ(r)] +Exc[ρ(r)] (5) Die Kohn-Sham-DFT als Einfachreferenzmethode ist nicht zur Bestimmung von Multikonfigurationszuständen geeignet. Rechnungen unter Verwendung lokaler Dichtefunktionale überschätzen die Bindungsenergie tendenzi- ell. Gut beschrieben wird die dynamische Elektronenkorrelation. Die statische Korrelations-Energie wird nicht beschrieben. Hierfür wäre ein Mehrdeterminanten-Ansatz notwendig [10][22]. Die Korrelationsenergie beträgt in ihrer Größenordnung zwar nur ca. 1% der Gesamtenergie, bewegt sich damit jedoch in der Größenordnung von chemischen Bindungen. Ihre Berücksichtigung ist folglich von großer Wichtigkeit.
2.5 Multireference Configuration Interaction
Die Configuration Interaction (CI)-Methode macht den Ansatz, die Wellenfunktion als Linearkombination ver- schiedener Konfigurationen darzustellen:
Ψ =a0ΨHF+ ∑iocc.∑rvir.airΨir+ ∑i<occ.j∑r<svir. aijrsΨijrs+... (6) Der erste Summand entspricht hierbei der HF-Grundzustands-Wellenfunktion. Die nachfolgenden Summanden (Einzel-, Doppelanregungen etc.) werden über die Koeffizienten a gewichtet einbezogen. Werden alle denkba- ren Konfigurationen einbezogen, so spricht man von Full CI. Je nach chemischer Fragestellung können so ein- zelne Summanden mit null gewichtet werden, um die Kosten der Rechnung gering zu halten.
Die Multireference Configuration Interaction (MRCI) macht einen ganz ähnlichen Ansatz. Hier kann eine MCSCF-Wellenfunktion als Ausgangspunkt dieser ab initio-Methode dienen, um ausgewählte relevante ange- regte Zustände zu beschreiben. Die Startorbitale werden entsprechend einer der jeweiligen Fragestellung ange- passten Kombination von Konfigurationen optimiert. In diesen Configuration State Functions (CSFs) sind In- formationen bezüglich der Besetzungszahl und des Spins enthalten. Dynamische wie auch statische Elektronen- korrelation werden hiermit beschrieben [22]. Diese Methode wurde bereits in den 1970er Jahren von Peyerimhoff und Buenker beschrieben [23].
2.6 DFT / MRCI
Ziel dieser Methode ist es, die dynamische Elektronenkorrelation aus einer DFT-Rechnung um die statische ei- ner MRCI-Rechnung zu ergänzen. Eine Doppelzählung der dynamischen Elektronenkorrelation wird durch die Parametrisierung der Hamilton-Matrix der MRCI-Rechnung verhindert. Insgesamt können so auch große Syste- me mit geringem Kostenaufwand in angeregten Zuständen genau beschrieben werden, ohne große Abweichun- gen von einer reinen ab initio MRCI Berechnung in Kauf nehmen zu müssen. Die Eignung der Methode zur Beschreibung von Singulett- und Triplettzuständen von Übergangsmetallverbindungen konnte hierbei nachge- wiesen werden [24].
2.7 Software
Erste Geometriedaten werden mit Spartan08 erstellt, welche als Ausgangspunkt nachfolgender Rechnungen die- nen [25]. Für die Geometrieoptimierung, Schwingungsfrequenzen und die DFT-Berechnungen wird Turbomole verwendet [26]. Die Berechnung der angeregten Zustände erfolgt mit dem DFT/MRCI-Programm [27].
Die Visualisierung der Orbitale und Geometrien erfolgt über jmol [28], die Bestimmung der Geometriedaten über molden [29].
2.8 Angaben zu Rechnungsdurchführung
2.8.1 Basissatz
Für Iridium wurde ein def-SVP-Basissatz verwendet, für alle übrigen Atome ein SVP-Basissatz. Hierin werden die Orbitale des Rumpfes über eine, Valenz-Orbitale über zwei kontrahierte Gaußfunktion beschrieben. Nicht- Wasserstoffatome werden hierbei um Polarisationsfunktionen ergänzt [30].
2.8.2 Effektives Rumpf-Potenzial
Zur Verringerung des Rechenaufwands in den Berechnungen wird beim Iridium das Effektive Rumpf-Potenzial (def-ECP) ecp-60mwb (Multifit Wood-Boring) verwendet. Hierbei wird die Wechselwirkung von 60 Rumpf- Elektronen untereinander und mit den Valenzelektronen über das ECP beschrieben [31]. Auch relativistische Effekte werden so berücksichtigt. Die Semicore-Elektronen werden nicht gut über den ECP-Basissatz beschrie- ben und werden daher nicht mitkorreliert, sondern eingefroren. Sie dienen dazu, dass die 6s und die 6p-Orbitale eine Knotenebene erhalten und die räumlich benachbarten 5d-Elektronen angemessene, nicht lokale Austausch- Wechselwirkung mit den 5s und 5p-Elektronen erfahren.
2.8.3 Verwendete Funktionale
Zur Geometrieoptimierung der Singulett-Geometrie mittels DFT und der Triplett-Geometrie mittels UDFT wur- de das PBE0-Funktional genutzt [32]. Für die MRCI-Entwicklungen wird das „half-and-half“ Hybrid-Funktio- nal BH-LYP von Becke verwendet. Darin wird die Austauschwechselwirkung zu 50% über das Funktional B88 und zu 50% durch HF-Austauschterme beschrieben [33]. Hinzu kommt ein LYP-Korrelationsfunktional. Die Anwendbarkeit dieses Funktionals zur Beschreibung von Übergangsmetall-Komplexen in DFT/MRCI-Rech- nungen konnte von Grimme und Waletzke gezeigt werden [24].
2.8.4 DFT/MRCI
Die Geometrieoptimierung für die Singulett-Geometrie erfolgt in C3-Symmetrie, für die Triplett-Geometrie in C1-Symmetrie. Die DFT/MRCI-Rechnungen werden in C1-Symmetrie durchgeführt, da die Punktgruppe C3 kei- ne Abelsche Gruppe darstellt.
Die ersten DFT/MRCI-Berechnungen werden mit einem Selektionsschwellwert (esel-Wert) von 0,8 durchge- führt, die endgültigen mit 1,0.
In der ersten DFT/MRCI-Berechnung in C1-Symmetrie werden keine Orbitale eingefroren, in der in C3-Symme- trie die untersten Orbitale (1-49). Um die Kosten der endgültigen DFT/MRCI-Rechnungen gering zu halten, werden in beiden Symmetrien die untersten Orbitale (1-49) sowie die obersten (464-589) Orbitale eingefroren.
In den Singulett-Anregungen werden 21, in den Triplett-Anregungen 20 Wurzeln berechnet.
3 Ergebnisse und Diskussion
3.1 Singulett-Geometrie: Struktur
3.1.1 fac-Ir(thpy)3
Sowohl die experimentell bestimmten als auch die in Singulett-Geometrie berechneten Geometriedaten der Li- ganden untereinander sind jeweils identisch. Der Komplex weist hier C3-Symmetrie auf.
Die berechneten Geometriedaten stimmen nahezu mit den im Röntgenbeugungs-Experiment (Einkristall 0,45x0,35x0,45 mm, Enraf-Nonius CAD-4 Diffraktometer) gefundenen überein (ausführliche Tabellen finden sich im Anhang) [34]. Daher sollen hier nur die jeweils größten Abweichungen sowie die Bindungen zum Iridi- um-Atom aufgeführt werden. Die Bindungslänge zwischen Zentralatom und Stickstoff wird leicht überschätzt, wie auch zwischen den Kohlenstoffatomen C3,C6 und C7 innerhalb des Pyridyl-Teils der Liganden. Diese ge- ringen Abweichungen können auf die inhärente Ungenauigkeit der Berechnungen zurückgeführt werden.
fac-Ir(thpy)3 Bindungslängeberechnet [Å] Bindungslängeexperimentell [Å] Abweichungberechnet-experimentell [Å]
Ir1-N2 2.19 2.1355 0.06
Ir1-C39 2.01 2.0066 0.00
C7-C3 1.40 1.3649 0.03
C3-C6 1.39 1.3530 0.03
Tab. 3: fac-Ir(thpy)3 ausgewählte Abweichungen der Bindungslängen in Singulett-Geometrie, Vergleich berechnet und experimentell
Auch die Bindungswinkel der Berechnungen in Singulett-Geometrie stimmen nahezu mit den im Experiment gefundenen überein. Die größte Abweichung findet sich beim Winkel zwischen dem Pyridyl- und Thiophenyl- Bereich der Liganden mit 2,1°. Interessant sind die Winkel, welche das zentrale Iridium-Atom als Scheitelpunkt besitzen. Der Winkel N2-Ir1-C32 würde in einer perfekt oktaedrischen Umgebung 180°, N2-Ir1-N12 und N2- Ir1-C46 je 90° betragen, was aber nicht der Fall ist. Die verzerrt-oktaedrische Komplexstruktur des Experi- ments wird mit Abweichungen unter einem Grad in der Berechnung wiedergefunden.
fac-Ir(thpy)3 Winkelberechnet [°] Winkelexperimentell [°] Abweichungberechnet-experimentell [°]
C3-C6-C5 119.6 121.111 -1.6
C6-C5-C43 126.4 128.170 -1.7
N2-C5-C43 113.3 111.198 2.1
N2-Ir1-C32 172.3 172.858 -0.6
N2-Ir1-N12 96.3 96.035 0.3
N2-Ir1-C46 90.1 89.999 0.1
Tab. 4: fac-Ir(thpy)3 ausgewählte Bindungswinkel in Singulett-Geometrie, Vergleich berechnet und experimentell
Größere Abweichungen der Diederwinkel sind mit 5,0° zwischen den Liganden zu finden. Der Winkel N2-C5- C43-C39 wäre in einem planaren Liganden 0,0°. Es findet sich aber, wie auch im Experiment, eine leichte Ver- drillung des Pyridyl- zum Thiophenyl-Bereich um ca. 1°.
fac-Ir(thpy)3 Diederwinkelberechnet [°] Diederwinkelexperimentell [°] Abweichungberechnet-experimentell [°]
N2-Ir1-C46-C47 84.0 88.995 5.0
N2-Ir1-N22-C25 88.7 87.667 -1.0
N2-C5-C43-C39 1.0 0.915 -0.1
Tab. 5: fac-Ir(thpy)3 ausgewählte Abweichungen der Diederwinkel in Singulett-Geometrie, Vergleich berechnet und experimentell
Insgesamt stimmen die Geometriedaten der Singulett-Geometrie weitestgehend mit den im Röntgenbeugungs- experiment gefundenen überein. Dabei können Differenzen aus Ungenauigkeiten der Berechnungen resultieren.
Ferner kann die Einbettung der Komplexe in einen Kristallverband zu Geometrieverzerrungen führen, welche in der Einzelmolekül-Berechnung im Vakuum nicht gefunden werden.
3.1.2 fac-Ir(ppy)3
Auch in fac-Ir(ppy)3 sind in der Singulett-Geometrie alle drei Liganden identisch, daher erfolgt der Vergleich exemplarisch für einen von ihnen. An den in beiden Komplexen vorkommenden Bindungen treten teilweise Differenzen der Bindungslängen auf.
fac-Ir(thpy)3 Bindungslänge [Å] fac-Ir(ppy)3 Bindungslänge [Å] Abweichung gegnüber fac-Ir(ppy)3 [Å]
Ir1-N2 2.19 Ir1-N13 2.17 0.02
Ir1-C39 2.01 Ir1-C2 2.02 -0.01
C5-C43 1.44 C7-C8 1.46 -0.02
N2-C5 1.36 N13-C8 1.36 0.00
N2-C4 1.34 N13-C12 1.34 0.00
C4-C7 1.39 C11-C12 1.39 0.00
C7-C3 1.40 C10-C11 1.40 0.00
C3-C6 1.39 C9-C10 1.39 0.00
C5-C6 1.41 C8-C9 1.41 0.00
Tab. 6: fac-Ir(ppy)3 ausgewählte Abweichungen der berechneten Bindungslängen in Singulett-Geometrie im Vergleich zu fac-Ir(thpy)3
Die Iridium-Stickstoff-Bindung ist in fac-Ir(thpy)3 um 0,02 Å länger, während die Ir-Kohlenstoff-Bindung um 0,01 Å kürzer ist als bei fac-Ir(ppy)3. Auch die verbrückende C-C Bindung ist in fac-Ir(thpy)3 kürzer. Die Bin- dungen in den Pyridyl-Bereichen der Liganden sind in beiden Komplexen identisch.
3.2 Singulett-Geometrie: Molekülorbitale
3.2.1 fac-Ir(thpy)3
Zur besseren Übersicht sind vergrößerte Abbildungen der Molekülorbitale aus den nachfolgenden MO-Dia- grammen dem Anhang beigefügt (Vgl. Abb. 17-66). Die Visualisierung aller Molekülorbitale erfolgt bis zu ei- ner minimalen Elektronendichte von 0,03.
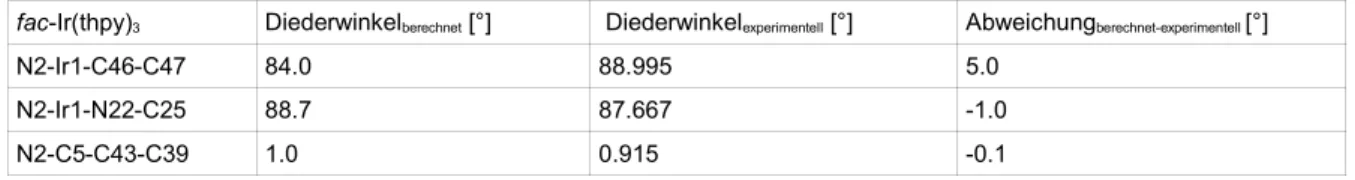
Abb. 7: MO-Diagramm von Ir(thpy)3 in Singulett-Geometrie
Um in den Molekülorbitalen die Lage der Koordinatenachsen festlegen zu können, ist es vorteilhaft, ein MO mit dz2-Charakter als Referenz auszuwählen. Zwar erscheint das HOMO zunächst geeignet. Hierbei käme die z- Achse entlang der C3-Drehachse zu liegen. Versucht man jedoch die d-Elektronendichte der Molekülorbitale HOMO-1 bis -5 dem so gewonnenen Koordinatensystem zuzuordnen, so kommen die Orbitallappen nicht in- nerhalb der Ebenen der Achsen zu liegen. Daher soll hier ein höheres MO mit dz2-Charakter, LUMO+10, als Referenz dienen.
Abb. 8: Molekülorbital LUMO+10 von Ir(thpy)3 in Singulett-Geometrie
Die z-Achse liegt folglich entlang der Bindungsachse N2-Ir1-C32. Die x-Achse wird über die Bindungsachse N22-Ir1-C39 und die y-Achse entlang C46-Ir1-N12 festgelegt. Der Charakter der MOs HOMO und HOMO-5 wird als verzerrte xy-Orbitale interpretiert, statt des augenscheinlichen dz2-Charakters. Diese Verzerrung kommt durch die symmetrische π-Elektronendichte-Verteilung auf den Liganden zu Stande. Den Molekülorbi- talen LUMO bis LUMO+5 fehlt eine d-Charakteristik, da kaum Elektronendichte am Zentralatom lokalisiert ist.
In allen Molekülorbitalen ist Elektronendichte auf Liganden zu finden, so dass sie π- bzw. π*-Charakter haben.
Die hieraus resultierende Charakterisierung der Molekülorbitale ist dem MO-Diagramm zu entnehmen. Folg- lich führen alle denkbaren Anregungen bei den hier betrachteten Orbitalen von im Grundzustand besetzten zu unbesetzten Orbitalen zu einem d/π → π*-Übergang, also einer Mischung aus MLCT- und LC-Anregung. Bei Übergängen in höhere MOs mit d-Charakter (zum Beispiel LUMO+9 und +10) können auch MC-Anteile auf- treten.
Betrachtet man die Details der π-Elektronendichte-Allokation auf den Liganden, so fällt auf, dass diese in den energetisch nicht entarteten MOs HOMO-5, HOMO, LUMO und LUMO+3 symmetrisch ist. HOMO-5 weist besonders hohe Dichten in den Thiophenyl-Bereichen und am Schwefel selbst auf. Das HOMO zeigt auch er- höhte Dichte im Thiophenyl-Bereich, der Schwefel selbst bleibt aber frei und auch im Pyridyl-Bereich ist Elek- tronendichte lokalisiert. Im LUMO ist sie gleichmäßig über alle Atome der drei Liganden verteilt, in LUMO+3 nur auf die Pyridyl-Segmente.
Die entarteten MOs weisen asymmetrische Elektronendichte-Verteilungen an den Liganden auf. HOMO-4 und HOMO-3 ähneln sich sehr stark: Ein Ligand bleibt nahezu frei, die beiden anderen haben mit unterschiedlicher Intensität hohe Dichten in der Umgebung des Schwefels. In HOMO-2 und HOMO-1 sind größere Unterschiede zu erkennen, dennoch sind sie beinah entartet. HOMO-2 besitzt einen Liganden mit gleichmäßig verteilter ho- her Elektronendichte und zwei fast identische Liganden mit geringerer Dichte, welche vor allem am Stickstoff und den Atomen C43 bzw. C50 zu finden ist. In HOMO-1 bleibt ein Ligand frei, die beiden übrigen unterschei- den sich nur in ihren Phasen. Hier ist die Elektronendichte fast gleichmäßig verteilt, leichte Erhöhungen sind im Bereich der Schwefelatome zu finden. Auch die unbesetzten entarteten Orbitale ähneln sich stark. LUMO+1 und LUMO+2 besitzen je einen Liganden frei von Elektronendichte. Von den beiden verbleibenden Liganden
hat der eine höhere, der andere geringere Elektronendichte, innerhalb der Liganden ist die Verteilung homogen über alle Atome. Mit LUMO+4 und LUMO+5 verhält es sich genau so, wie in LUMO+1 und LUMO+2, mit dem Unterschied, dass an den beiden Liganden mit Elektronendichte der Thiophenyl-Bereich frei bleibt. Diese Daten werden in der nachfolgenden Tabelle zusammengefasst.
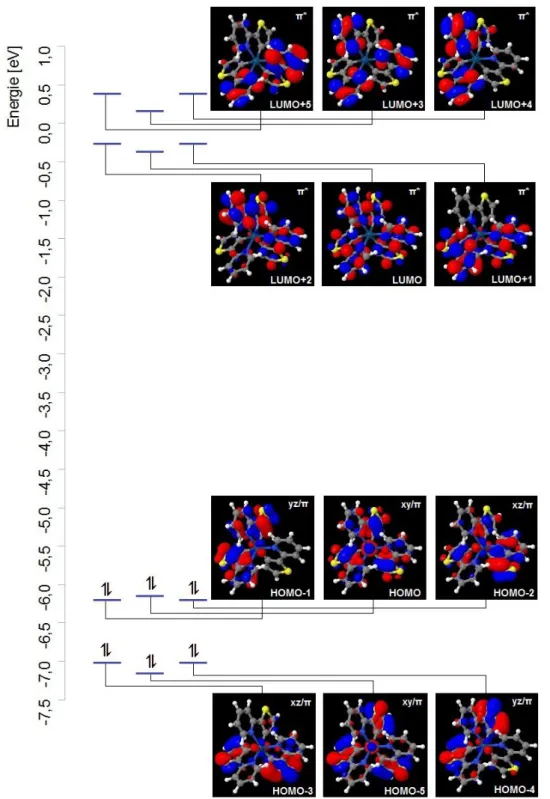
Molekülorbital Charakteristik Energie [eV] Elektronendichte-Verteilung
LUMO +5 π* 0.38 py, (th)
LUMO +4 π* 0.38 py, (th)
LUMO +3 π* 0.15 py, (th)
LUMO +2 π* -0.27 thpy
LUMO +1 π* -0.27 thpy
LUMO π* -0.37 thpy
HOMO d/π -6.16 Ir, thpy
HOMO -1 d/π -6.21 Ir, th, (py)
HOMO -2 d/π -6.21 Ir, th, (py)
HOMO -3 d/π -7.02 Ir, th
HOMO -4 d/π -7.02 Ir, th
HOMO -5 d/π -7.16 Ir, th
Tab. 7: Charakteristik, MO-Energien und Elektronendichte-Verteilung von Ir(thpy)3 in Singulett-Geometrie (py= Pyridyl, th= Thionyl, thpy= Thionylpyridyl)
Im Vergleich der Molekülorbitale HOMO und LUMO in Singulett-Geometrie beobachtet man eine starke Ver- ringerung der d-Elektronendichte am Iridium-Atom und bei allen Liganden symmetrische Dichteveränderungen innerhalb der Liganden selbst. Zwischen C41-S42, C35-S34 und C48-S49 wird aus der bindenden Wechselwir- kung im HOMO eine antibindende im LUMO. Daher erwartet man eine Bindungsverlängerung dieser Bindun- gen, was für C41-S42 auch bestätigt werden kann. Ferner fällt die Dichteverringerung an den Bindungen C43- 39, C50-46 und C32-33 auf. Dementsprechend erwartet man eine Verlängerung dieser Bindungen bei Anregung vom HOMO zum LUMO. Auch dies lässt sich bestätigen, da die Bindung zwischen Atom 43 und 39 verlängert wird. Allerdings gilt dies nicht für die übrigen Liganden, da beim Übergang zur Triplett-Geometrie Jahn-Teller- Verzerrung auftritt. Die Bindungen zum Zentralatom verändern sich nur marginal, aber auch sie unterscheiden sich an den einzelnen Liganden. Eine genaue Diskussion der Triplett-Geometrie wird in Abschnitt 3.4 erfolgen.
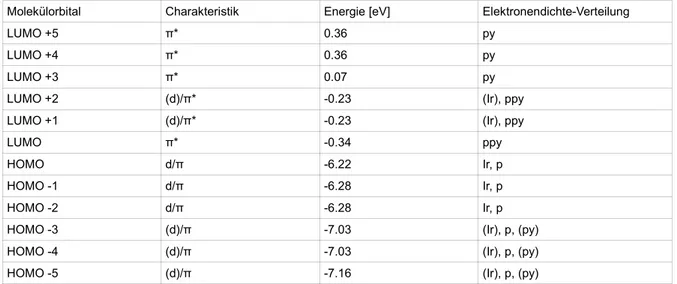
3.2.2 fac-Ir(ppy)3
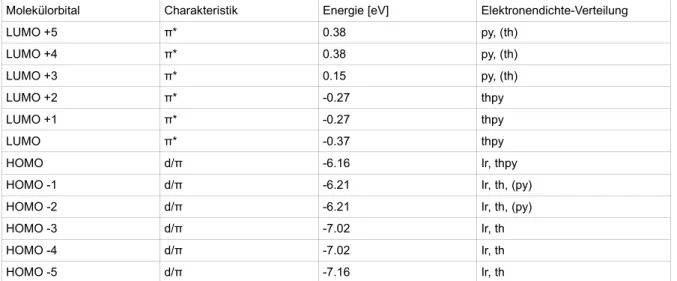
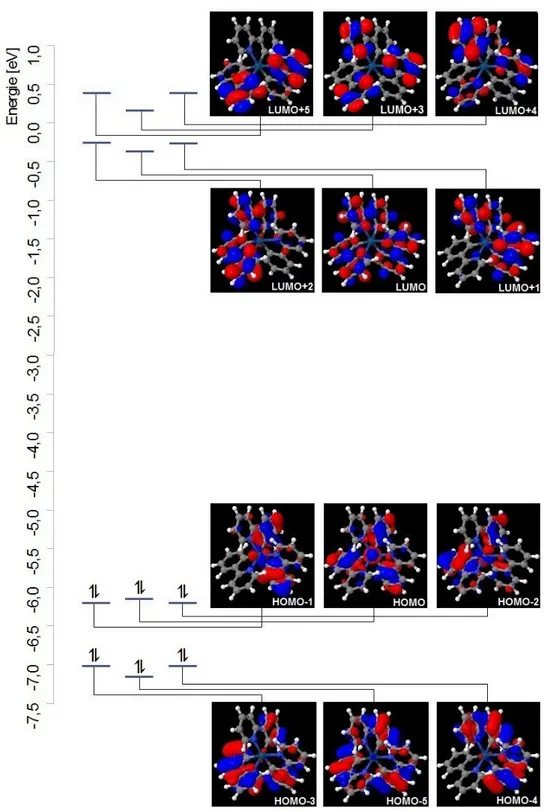
Die qualitative Anordnung der MO-Energien ist in beiden Komplexen identisch. Eine größere Abweichung fin- det sich zwischen den Orbitalen LUMO+3 beider Komplexe mit 0,08 eV. Hinsichtlich der d-Charakteristik fin- den sich Unterschiede. Es findet sich keine klare Trennung bezüglich der d-Elektronendichte zwischen HOMO(-n) und LUMO(+n). LUMO+1 und +2 und HOMO-3 bis -5 weisen jeweils geringe d-Elektronendichte auf. Bei den höchsten besetzten MOs besitzen folglich nur HOMO bis HOMO-2 klaren d-Charakter. Die Über- gänge zwischen diesen Grenzorbitalen können also MLCT-, LC- und MC-Anteile besitzen.
Abb. 9: MO-Diagramm von Ir(ppy)3 in Singulett-Geometrie
Auch quantitativ liegen die MO-Energien nah bei denen des Ir(thpy)3, wie die nachfolgende Tabelle zeigt:
Molekülorbital Charakteristik Energie [eV] Elektronendichte-Verteilung
LUMO +5 π* 0.36 py
LUMO +4 π* 0.36 py
LUMO +3 π* 0.07 py
LUMO +2 (d)/π* -0.23 (Ir), ppy
LUMO +1 (d)/π* -0.23 (Ir), ppy
LUMO π* -0.34 ppy
HOMO d/π -6.22 Ir, p
HOMO -1 d/π -6.28 Ir, p
HOMO -2 d/π -6.28 Ir, p
HOMO -3 (d)/π -7.03 (Ir), p, (py)
HOMO -4 (d)/π -7.03 (Ir), p, (py)
HOMO -5 (d)/π -7.16 (Ir), p, (py)
Tab. 8: Charakteristik, MO-Energien und Elektronendichte-Verteilung von Ir(ppy)3 in Singulett-Geometrie (py= Pyridyl, p= Phenyl, ppy= Phenylpyridyl)
3.3 Singulett-Geometrie: DFT-MRCI
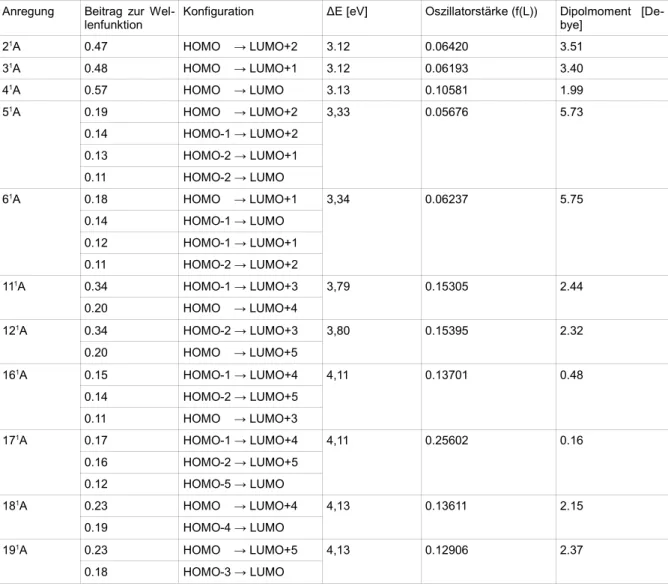
3.3.1 fac-Ir(thpy)3
Anhand des MO-Diagramms ergibt sich folgende energetische Reihenfolge der Energiedifferenzen bei den Konfigurationen zwischen den Grenzorbitalen HOMO-2 bis LUMO+2. Theoretisch erwartet man für diese rei- ne Konfigurationen von S0 → S1 bis S0 → S9:
Konfiguration Δ MO-Energie [eV] Energetische Reihenfolge in DFT/MRCI
Singulett-Anregungen Triplett-Anregungen
HOMO → LUMO 5.78 41A 13A
HOMO -1 → LUMO 5.83 61A 23A
HOMO -2 → LUMO 5.83 51A 33A
HOMO → LUMO +1 5.89 31A 23A
HOMO → LUMO +2 5.89 21A 33A
HOMO -1 → LUMO +1 5.94 61A 23A
HOMO -2 → LUMO +1 5.94 51A 33A
HOMO -1 → LUMO +2 5.94 51A 13A
HOMO -2 → LUMO +2 5.94 61A 13A
Tab. 9: Energiedifferenzen wichtiger Übergänge von Ir(thpy)3 in Singulett-Geometrie
Diese Reihenfolge findet sich nicht in den Ergebnissen der DFT/MRCI-Rechnung wieder, da diese Konfigura- tionen jeweils nur anteilig zu jeder Anregung beitragen. Ferner wird nicht berücksichtigt, dass die Anregungs- energien neben den Orbitalenergiedifferenzen noch Beiträge von Zweielektronentermen, also den Coulomb- und Austauschtermen, enthalten. Aus Gründen der Übersichtlichkeit wird an dieser Stelle darauf verzichtet, alle Anregungen aufzulisten, zu denen die Konfigurationen beitragen. Betrachtet man jeweils die energetisch nied- rigste Anregung, in der die Konfigurationen mit großem Beitrag zur Wellenfunktion auftreten, so ergibt sich statt dessen die energetische Reihenfolge, wie in der dritten Spalte von Tabelle 9 angegeben.
Aus dem MO-Diagramm erwartet man die beinahe entartete energetische Lage der Konfigurationen HOMO-1
→ LUMO+1, HOMO-2 → LUMO+2, HOMO-1 → LUMO+2 und HOMO-2 → LUMO+2. Dies lässt sich be- stätigen, da jeweils die Konfigurationen HOMO-1 → LUMO+1 und HOMO-2 → LUMO+2 sowie HOMO-2
→ LUMO+2 und HOMO-1 → LUMO+2 zu den selben Anregungen in Singulett-Geometrie beitragen.
Bei der Auswahl der verschiedenen Konfigurationen der Anregungen werden diejenigen mit einem Beitrag zur Wellenfunktion über 0,10 ausgewählt. Weisen alle Konfigurationen einer Anregung geringere Beiträge auf, so wird die größte ausgewählt. Die Oszillatorstärken der Triplett-Anregungen beziehen sich jeweils auf den Über- gang T0 → Tn.
Helle Übergänge treten besonders bei (111A) 3,79 und (121A) 3,80 eV (30568 bis 30616 cm-1) und von (161A) 4,11 bis (191A ) 4,13 eV (33121 bis 33311 cm-1) auf. Sie werden das Anregungsspektrum prägen.
Anregung Beitrag zur Wel-
lenfunktion Konfiguration ΔE [eV] Oszillatorstärke (f(L)) Dipolmoment [De- bye]
21A 0.47 HOMO → LUMO+2 3.12 0.06420 3.51
31A 0.48 HOMO → LUMO+1 3.12 0.06193 3.40
41A 0.57 HOMO → LUMO 3.13 0.10581 1.99
51A 0.19 HOMO → LUMO+2 3,33 0.05676 5.73
0.14 HOMO-1 → LUMO+2
0.13 HOMO-2 → LUMO+1
0.11 HOMO-2 → LUMO
61A 0.18 HOMO → LUMO+1 3,34 0.06237 5.75
0.14 HOMO-1 → LUMO
0.12 HOMO-1 → LUMO+1
0.11 HOMO-2 → LUMO+2
111A 0.34 HOMO-1 → LUMO+3 3,79 0.15305 2.44
0.20 HOMO → LUMO+4
121A 0.34 HOMO-2 → LUMO+3 3,80 0.15395 2.32
0.20 HOMO → LUMO+5
161A 0.15 HOMO-1 → LUMO+4 4,11 0.13701 0.48
0.14 HOMO-2 → LUMO+5
0.11 HOMO → LUMO+3
171A 0.17 HOMO-1 → LUMO+4 4,11 0.25602 0.16
0.16 HOMO-2 → LUMO+5
0.12 HOMO-5 → LUMO
181A 0.23 HOMO → LUMO+4 4,13 0.13611 2.15
0.19 HOMO-4 → LUMO
191A 0.23 HOMO → LUMO+5 4,13 0.12906 2.37
0.18 HOMO-3 → LUMO
Tab. 10: Auswahl der Singulett-Anregungen aus der Singulett-Geometrie von fac-Ir(thpy)3 mit 21 Wurzeln, es werden die hellsten (f(L) > 0,10) und die energetisch niedrigsten Anregungen mit einem hohen Beitrag der Übergänge zwischen HOMO-2 bis LUMO+2 gewählt
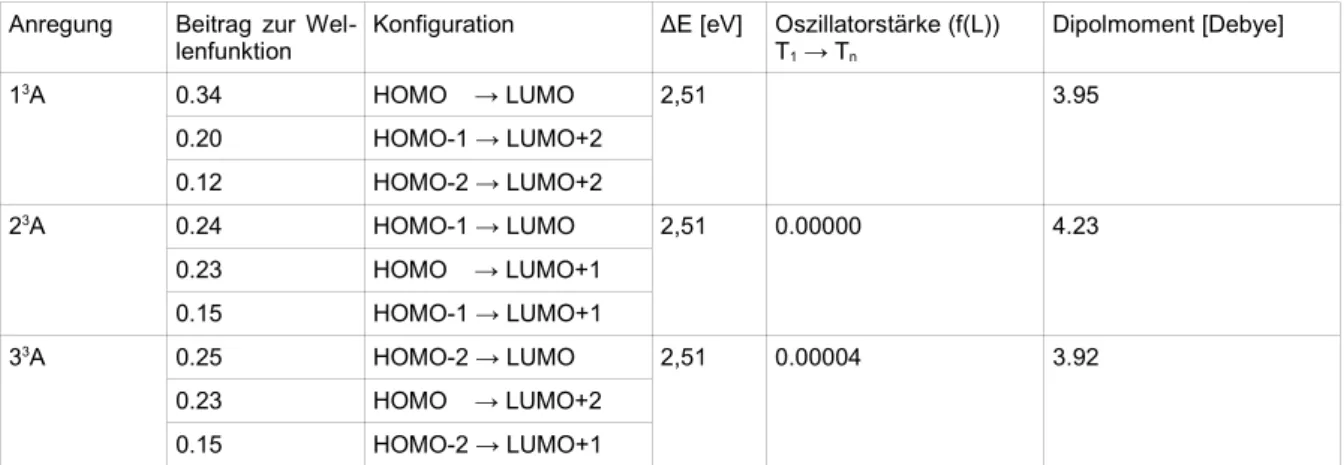
Alle Konfigurationen aus Tabelle 9 finden sich in den ersten drei Triplett-Anregungen wieder.
Anregung Beitrag zur Wel-
lenfunktion Konfiguration ΔE [eV] Oszillatorstärke (f(L))
T1 → Tn Dipolmoment [Debye]
13A 0.34 HOMO → LUMO 2,51 3.95
0.20 HOMO-1 → LUMO+2
0.12 HOMO-2 → LUMO+2
23A 0.24 HOMO-1 → LUMO 2,51 0.00000 4.23
0.23 HOMO → LUMO+1
0.15 HOMO-1 → LUMO+1
33A 0.25 HOMO-2 → LUMO 2,51 0.00004 3.92
0.23 HOMO → LUMO+2
0.15 HOMO-2 → LUMO+1
Tab. 11: Auswahl der Triplett-Anregungen aus der Singulett-Geometrie von fac-Ir(thpy)3 mit 20 Wurzeln, es werden die energetisch niedrigsten Anregungen mit einem hohen Beitrag der Übergänge zwischen HOMO-2 bis LUMO+2 gewählt.
Das berechnete Absorptionsspektrum von fac-Ir(thpy)3 bei 0K im Vakuum wurde durch Gaußfunktionen mit ei- ner Peakverbreiterung von 1000 cm-1 verbreitert (Abb. 10). Es liegt in weiten Teilen im ultravioletten Bereich des elektromagnetischen Spektrums (> 26.000 cm-1), nur eine Bande bei ca. 2,5 x 104 cm-1 befindet sich im sichtbaren Bereich. Beim kristallinen Feststoff ist also eine orange-rote Farbe zu erwarten. Die stärkste Absorp- tionsbande ist dem Übergang 171A mit der vergleichsweise hohen Oszillatorstärke von 0,25602 zuzuordnen.
Hierfür spricht auch die Energie dieses Überganges, die mit 4,11 eV ≈ 33148 cm-1 der Lage der maximalen Ab- sorption bei etwa 3,3 x 104 cm-1 entspricht. Neben dem globalen Absorptionsmaximum wird das Spektrum durch weitere Banden bei ca. 3,05 x 104 cm-1, 2,7 x 104 cm-1 und bei ca. 2,5 x 104 cm-1 geprägt. Die beiden letzteren vereinen sich bei der gewählten Gaußverbreiterung zu einem breiteren Absorptionsbereich. Auch die übrigen Anregungen mit hoher Oszillatorstärke aus Tabelle 10 lassen sich im Anregungsspektrum wiederfinden: Anre- gung 111A bei 30568 cm-1, 121A bei 30616 cm-1. 161A ist bei 33121 cm-1 zu finden. 181A tritt bei 33296 cm-1 auf sowie 191A bei 33311 cm-1.
Abbildung 10 zeigt ferner das experimentelle Anregungsspektrum von fac-Ir(thpy)3 in CH2Cl2 bei Raumtempe- ratur. Auch hier reicht das Spektrum vom Ultravioletten bis zum Sichtbaren, endet aber rotverschoben erst bei etwa 2,0 x 104 cm-1. Charakteristisch sind zwei Absorptionsbereiche: Zum einen die intensivste Bande bei 3,33 x 104 cm-1. Der zweite breitere Bereich wird durch eine Bande bei ca. 3,1 x 104 cm-1 begrenzt und erstreckt sich bis ca. 2,0 x 104 cm-1. Hierin sind zwei Schultern bei ca. 2,9 x 104 cm-1 und bei ungefähr 2,5 x 104 cm-1 zu finden [34].
Vergleicht man die Spektren, so entsprechen sich die Absorptionsmaxima bei 3,33 x 104 cm-1 und auch die Schulter bei ungefähr 2,5 x 104 cm-1 findet sich wieder. Das in der Berechnung vorhandene Maximum bei 3,05 x 104 cm-1 wird im Experiment als einziges nicht gefunden. Insgesamt lässt sich festhalten, dass die Absorptions- Maxima gut übereinstimmen und dass sich die Zweiteilung des Spektrums in zwei charakteristische Bereiche erkennen lässt. Die energetische Untergrenze des Spektrums ist hypsochrom verschoben. Die maximale Bande ist gegenüber den berechneten Ergebnissen verbreitert. Die Unterschiede können durch verschiedene Effekte bedingt sein. Zum einen wird in der Berechnung durch die Anzahl der gewählten Wurzeln ein zu geringer Ener- giebereich abgedeckt und die Berechnung wurde für einen isolierten Komplex im Vakuum durchgeführt. Umge- bungseffekte werden so nicht berücksichtigt. Auch Spin-Bahn-Kopplungseffekte wurden nicht berücksichtigt, was vor allem den niederenergetischen Bereich durch Triplettabsorption bzw. Energieverschiebung beeinflusst.
Ungenauigkeiten können aber auch im Experiment zu finden sein, da die Spektroskopie vor allem bei niedrige- ren Anregungsenergien an ihre Grenzen stößt.
Abb. 10: Normierte Singulett-Absorption von fac-Ir(thpy)3, Vergleich berechnet (rot) zum Experiment (schwarz) [34]
3.3.2 fac-Ir(ppy)3
Die berechneten Anregungsspektren der beiden Komplexe ähneln sich. Es finden sich starke Absorptionsban- den bei 2,8 und 2,6 x 104 cm-1. Die größte Absorption tritt bei ca. 3,4 x 104 cm-1 auf. Zwar ist das Spektrum von fac-Ir(thpy)3 gegenüber fac-Ir(ppy)3 um ca. 0,08 x 104 cm-1 rotverschoben, doch alle vier in 3.3.1 beschriebenen charakteristischen Absorptions-Maxima von Ir(thpy)3 finden sich wieder. Nicht verschoben ist als einziges die Bande bei ca. 3,05 x 104 cm-1, welche einer Schulter des fac-Ir(ppy)3 bei der selben Wellenzahl entspricht. Im Anregungsspektrum des fac-Ir(ppy)3 finden sich darüber hinaus zwei weitere Banden im höherenergetischen Bereich bei 4,05 und 3,75 x 104 cm-1. Beim hier berechneten Anregungsspektrum des fac-Ir(thpy)3 sind keine entsprechenden Anregungen zu finden, da die DFT/MRCI-Rechnungen mit weitaus weniger Wurzeln durchge- führt wurden. Hinzu kommt, dass sich die MOs HOMO-3, -4 und -5 der Komplexe in ihrem d-Charakter stark unterscheiden, was die Anregungen beeinflusst, zu denen sie beitragen. Während alle HOMO(-n) des fac- Ir(thpy)3 gleichermaßen d-Elektronendichte aufweisen, haben die MOs HOMO-3 bis -5 im fac-Ir(ppy)3 fast aus- schließlich Ligand-Charakter.
Abb. 11: Berechnete normierte Singulett-Absorptionen, Vergleich fac-Ir(thpy)3 (rot) zu fac-Ir(ppy)3 (blau)
In fac-Ir(ppy)3 sind an der hellsten Anregung 221A bei 35011 cm-1 die Konfigurationen HOMO-2 → LUMO+4 und HOMO-1 → LUMO+5 beteiligt. Diese Hauptbeiträge liegen über denen bei der hellsten Anregung 171A bei fac-Ir(thpy)3, es kommt zu einer Blauverschiebung. An der zweithellsten Anregung 161A bei 33358 cm-1, welche auch zum Hauptmaximum beiträgt, sind die MOs HOMO-3,-4 und -5 beteiligt. Die energetische Lage von 161A ist im fac-Ir(ppy)3 verschoben, da alle der stark zur Wellenfunktion beitragenden Konfigurationen von denjenigen MOs ausgehen, die sich in ihrer Elektronendichte-Verteilung am stärksten vom fac-Ir(thpy)3 unter- scheiden (HOMO-3,-4 und -5). Das Absorptions-Maximum in fac-Ir(ppy)3 geht also auf eine gemischte LC- und MLCT-Anregung zurück.
Das erste Maximum, das sich im fac-Ir(thpy)3 nicht findet (bei ca. 4,05 x 104 cm-1), geht auf Anregungen aus tiefer liegenden Orbitalen (HOMO-7 und -8) zurück (351A, 361A ). Die Anregungen 231A, 241A und 251A tra- gen zum Maximum bei 3,75 x 104 cm-1 (4,6 eV) bei. An ihnen sind in großem Maß HOMO-3,-4 und -5 betei- ligt, sie haben also anteilig LC-Charakter. Neben der höheren Zahl der Wurzeln, kann also die unterschiedliche Dichteverteilung zur abweichenden Gestalt der Anregungsspektren beitragen.
Anregung Beitrag zur Wellenfunktion Konfiguration ΔE [eV] Oszillatorstärke
(f(L)) Dipolmoment [Debye]
161A 0.14 HOMO-4 → LUMO+1 4.14 0.31981 1.49
0.14 HOMO-3 → LUMO+2
0.12 HOMO-5 → LUMO
181A 0.29 HOMO → LUMO+4 4.19 0.11965 3.10
0.15 HOMO-4 → LUMO
191A 0.29 HOMO → LUMO+5 4.20 0.11419 3.32
0.15 HOMO-3 → LUMO
221A 0.18 HOMO-2 → LUMO+4 4.34 0.38555 0.73
0.17 HOMO-1 → LUMO+5
231A 0.13 HOMO-4 → LUMO+3 4.58 0.02426 2.19
0.11 HOMO-4 → LUMO
0.11 HOMO-3 → LUMO+3
241A 0.13 HOMO-3 → LUMO+3 4.59 0.02425 2.03
0.11 HOMO-3 → LUMO
0.11 HOMO-4 → LUMO+3
251A 0.20 HOMO-5 → LUMO+3 4.60 0.02075 2.07
0.15 HOMO-4 → LUMO+4
0.11 HOMO-3 → LUMO+5
351A 0.07 HOMO-7 → LUMO 4.97 0.01192 5.07
361A 0.07 HOMO-8 → LUMO 4.98 0.01392 5.03
Tab. 12: Auswahl der Singulett-Anregungen aus der Singulett-Geometrie von fac-Ir(ppy)3 mit 40 Wurzeln
3.4 Triplett-Geometrie: Struktur
3.4.1 fac-Ir(thpy)3
Für die Berechnungen in Triplett-Geometrie wird die Besetzung ausgewählt, welche einer Anregung HOMO → LUMO im oben gezeigten MO-Diagramm in Singulett-Geometrie entspricht. Aufgrund der energetischen Nähe wären auch Triplett-Besetzungen unter Einbezug von HOMO-1 und -2 sowie von LUMO+1 und +2 denkbar.
Diese Orbitale sind jeweils nahezu entartet, was die Anwendung von Time Dependent DFT-Methoden nötig
metrie ausgegangen wird, hat das System höchstmögliche Freiheit zu relaxieren. Da UDFT für die Geometrie- optimierung verwendet wird, wird der niedrigste Triplett-Zustand T1 und kein höher liegender erreicht. UDFT optimiert immer den niedrigsten Zustand einer irreduziblen Darstellung. In der hier gewählten C1-Symmetrie mit der irreduziblen Darstellung a für alle Molekülorbitale, entspricht dies dem Zustand 1a. Die nachfolgende Tabelle 13 zeigt selektiert diejenigen Bindungslängen, die sich von der Singulett-Geometrie unterscheiden und die Bindungen zum Zentralatom. Eine Tabelle aller Bindungslängen der Triplett-Geometrie ist im Anhang zu finden.
fac-Ir(thpy)3 Bindungslängeberechnet [Å] Abweichung zur Singulett-Geometrie [Å]
Ir1-N2 2.18 -0.01
Ir1-C39 1.98 -0.03
N2-C4 1.33 -0.01
N2-C5 1.40 0.03
C4-C7 1.40 0.01
C7-C3 1.42 0.02
C3-C6 1.37 -0.01
C5-C6 1.43 0.02
C5-C43 1.40 -0.04
C43-C39 1.48 0.09
C39-C40 1.40 -0.03
C40-C41 1.39 0.01
C41-S42 1.78 0.06
Ir1-N12 2.21 0.02
Ir1-C32 2.01 0.01
Ir1-N22 2.21 0.02
Ir1-C46 2.01 0.00
Tab. 13: fac-Ir(thpy)3 ausgewählte Bindungslängen der Triplett-Geometrie und deren Abweichung gegenüber der Singulett-Geometrie
Die Bindungslängen in Triplett-Geometrie weisen gegenüber der Singulett-Geometrie einige Veränderungen auf. Die Elektronendichte-Verteilungen in den Molekülorbitalen können hierauf erste Hinweise geben. Die Vor- aussagen aus Abschnitt 3.2 lassen sich dabei teilweise bestätigen. Die stärksten Bindungslängen-Veränderungen betreffen denjenigen Liganden, der das Stickstoff-Atom 2 enthält. Die Bindung zwischen Atom 43 und 39 ist um 0,09 Å und zwischen Atom 41 und 42 um 0,06 Å verlängert. Die vorausgesagte Veränderung der Bindungen in den Liganden mit Stickstoff-Atom 12 und 22 ist allerdings nicht zu finden, obwohl an ihnen beim Übergang fast alle Elektronendichte abgezogen wird. Hier finden sich nur vernachlässigbar kleine Änderungen unter 0,01 Å gegenüber der Singulett-Geometrie.
Über die Voraussagen hinaus verkürzt sich die verbrückende Bindung zwischen Atom 5 und 43 um 0,04 Å. Die Liganden in Triplett-Geometrie weisen untereinander Bindungslängen-Unterschiede auf, im Gegensatz zur Sin- gulett-Geometrie. Diese asymmetrischen Bindungslängen lassen sich durch die Jahn-Teller-Verzerrung erklä- ren, wofür auch die leicht unterschiedlichen N-Ir-Bindungslängen sprechen. Die Bindungen zu N12 und N22 verlängern sich um 0,02 Å, was durch die gesunkene Elektronendichte an diesen Liganden erklärbar ist. Dage- gen ist bei Ir-N2 eine Bindungsverkürzung um 0,01 Å zu beobachten. Das Zentralatom weist zwar in Triplett- Geometrie kaum noch d-Elektronendichte auf, die für Wechselwirkungen zur Verfügung stehen könnte. Doch gleichzeitig erhöht sich an diesem Liganden die Elektronendichte, wie oben beschrieben. In Summe mehren sich so die bindenden Wechselwirkungen zwischen Iridium und N2.
![Abb. 5: MO-Schema eines oktaedrisch koordinierten Komplexes, nur Ligandenorbitale mit lokaler σ- σ-Symmetrie werden berücksichtigt (links) [11]; Mögliche Auswirkungen von π-Bindungen auf Δ: (a) bei](https://thumb-eu.123doks.com/thumbv2/1library_info/4531361.1596265/10.892.257.666.141.551/oktaedrisch-koordinierten-komplexes-ligandenorbitale-symmetrie-berücksichtigt-auswirkungen-bindungen.webp)
![Abb. 6: Übergänge in Phosphoreszenz-Emittern unter potentieller Ausnutzung aller gebildeten Exzitonen [14]](https://thumb-eu.123doks.com/thumbv2/1library_info/4531361.1596265/11.892.350.572.604.806/abb-übergänge-phosphoreszenz-emittern-potentieller-ausnutzung-gebildeten-exzitonen.webp)
![Abb. 10: Normierte Singulett-Absorption von fac-Ir(thpy) 3 , Vergleich berechnet (rot) zum Experiment (schwarz) [34]](https://thumb-eu.123doks.com/thumbv2/1library_info/4531361.1596265/23.892.234.703.155.425/abb-normierte-singulett-absorption-vergleich-berechnet-experiment-schwarz.webp)
