HEINRICH-HEINE-UNIVERSITÄT DÜSSELDORF Mathematisch-Naturwissenschaftliche Fakultät
Institut für Theoretische Chemie und Computerchemie
Quantenchemische Charakterisierung von Diarylethen-Malachitgrün- Chromophoren im elektronischen Grundzustand
2016
Bachelor-Arbeit zur Erlangung des akademischen Grades eines Bachelor of Science (B. Sc.)
im Studiengang Wirtschaftschemie Vorgelegt von:
Alexander Becker aus Schwerte
März 2016
Erstgutachterin: Frau Prof. Dr. Christel M. Marian Zweitgutachter: Herr PD Dr. Klaus Schaper
I
Ich versichere an Eides statt, dass ich meine vorliegende Bachelorarbeit selbständig ohne Hilfe Dritter und ohne Benutzung anderer als der angegebenen Quellen und Hilfsmittel angefertigt und die den benutzten Quellen wörtlich oder inhaltlich entnommenen Stellen als solche kenntlich gemacht habe. Diese Arbeit hat in gleicher oder ähnlicher Form noch keiner Prüfungsbehörde vorgelegen.
Düsseldorf den 10.März 2016
________________________________
Alexander Becker
II
Danksagung
Ich bedanke mich zuallererst bei Frau Prof. Dr. Christel Marian für die Möglichkeit, diese Bachelorarbeit an ihrem Lehrstuhl anzufertigen und für ihren Rat und ihre Unterstützung während der Arbeit. Weiterer Dank gilt auch meinem stets ansprechbaren und sehr geduldigem Betreuer Dr. Oliver Weingart. Natürlich bedanke ich mich beim gesamten Lehrstuhl für die nette Aufnahme und die stete Hilfestellung bei großen und kleinen Problemen im Laufe dieser Arbeit. Abschließend danke ich allen Personen die das vergangene Studium zu einer unvergesslichen Zeit gemacht haben und meiner Familie für alles, was sie mir gegeben und ermöglicht hat.
III
Kurzzusammenfassung
In dieser Arbeit werden Diarylethen-Malachitgrün-Chromophore mit quantenchemischen Verfahren hinsichtlich Geometrie, Infrarot-Spektrum und UV-Spektrum untersucht. Im Rahmen der Untersuchung des UV-Spektrums erfolgt ein Vergleich der Ergebnisqualität zwischen gängigen TDDFT-Rechnungen und DFT/MRCI-Rechnungen mit spezieller Parametrisierung. Untersuchungsgegenstand ist ein Diarylethen-Malachitgrün-Chromophor, der durch Kombination des gängigen organischen Photoschalters Diarylethen mit dem Triphenyl-Farbstoff Malachitgrün entsteht. Der Chromophor zeichnet sich durch seine reversible Schaltbarkeit zwischen sechs verschiedenen Zuständen aus, die durch Kombination der photochromen Eigenschaften des Diarylethens mit dem halochromen Charakter des Malachitgrüns entstehen. Basierend auf der Röntgenstruktur wurden erfolgreich die stationären Geometrien verschiedener Diarylethen-Malachitgrün-Chromophore bestimmt. Es zeigte sich, dass vorhandene Rotationsachsen relativ hohe Barrieren aufweisen. Der Einfluss von Acetonitril auf die Geometrie erwies sich als gering, er steigt geringfügig mit der Ladung an.
Das geöffnete Isomer ist energetisch günstiger, die doppelt geladene Spezies zeigte die niedrigste Reaktionsenthalpie. Die C-C Bindung zwischen den beiden Einzelkomponenten verkürzt sich mit zunehmender Ladung und bei Ringschluss, was auf verbesserte Konjugation zwischen beiden Komponenten zurückzuführen ist. Das Infrarot-Spektrum wird im aromatischen Bereich durch dominante Valenzschwingungsbanden charakterisiert, deren Intensität mit der Ladung des Moleküls erheblich zunimmt. Die UV-Spektren sind durch intensive, im sichtbaren Bereich des Lichtes liegende Banden der S1-Anregung charakterisiert und zeigen meist einen Charge-Transfer-Charakter. TDDFT-Rechnungen mit dem CAM- B3LYP Funktional ergeben keine guten Ergebnisse für diese Spektren, da die intensiven Banden stark blauverschoben wurden. DFT/MRCI-Rechnungen mit Tight-Parametrisierung nach Lyskov eignen sich deutlich besser zur Berechnung der Spektren dieser Chromophore, versagen aber auch bei einem der Chromophore.
IV
Inhalt
Abbildungsverzeichnis ... V Tabellenverzeichnis ... VI Abkürzungsverzeichnis ... VII
1. Einleitung ... 1
2. Quantenchemische Grundlagen ... 3
2.1. Die Schrödinger-Gleichung ... 3
2.2. Die Born-Oppenheimer-Näherung ... 3
2.3. Potentialhyperflächen und Geometrieoptimierungen ... 4
2.4. Mehrelektronensysteme und der Hartree-Fock-Formalismus ... 5
2.5. Elektronenkorrelation und Multikonfigurationsansätze ... 6
2.6. Dichtefunktionaltheorie ... 7
2.7. Zeitabhängige Dichtefunktionaltheorie ... 9
2.8. DFT/MRCI-Methoden ... 10
2.9. Infrarot-Spektroskopie ... 11
2.10. Solvens-Effekte ... 11
3. Diarylethene ... 12
4. Malachitgrün ... 14
5. Verwendete Programme und Methoden ... 14
6. Ergebnisse ... 15
6.1. Abweichungen zur Röntgenstruktur ... 15
6.2. Rotationsbarrieren in Diarylethen-Malachitgrün ... 17
6.3. Valenzstrukturen von 1C und 1D ... 18
6.4. Lösungsmitteleinflüsse ... 19
6.5. Die Verbindung von Diarylethen und Malachitgrün ... 20
6.6. Relative Energien der Isomere ... 20
7. Infrarot-Spektren und Schwingungsanalyse ... 22
7.1. Infrarot-Spektrum der Struktur 1A ... 22
7.2. Infrarot-Spektrum der Struktur 1B ... 24
7.3. Infrarot-Spektrum der Struktur 1C ... 25
7.4. Infrarot-Spektrum der Struktur 1D ... 26
7.5. Infrarot-Spektrum der Struktur 1E ... 27
7.6. Infrarotspektrum der Struktur 1F ... 28
7.7. Zusammenfassung ... 29
V
8. UV-Spektren ... 30
8.1. UV-Spektren von Diarylethen ... 30
8.2. UV-Spektren von Malachitgrün ... 32
8.3. UV-Spektrum von Struktur 1A ... 34
8.4. UV-Spektrum von Struktur 1B ... 36
8.5. UV-Spektrum von Struktur 1C ... 37
8.6. UV-Spektrum von Struktur 1D ... 39
8.7. UV-Spektrum von Struktur 1E ... 40
8.8. UV-Spektrum von Struktur 1F ... 42
8.9. Vergleich der Strukturen 1A bis 1F hinsichtlich des S1-Zustandes ... 44
8.10. Vergleich von TDDFT und DFT/MRCI-Methoden ... 45
8.11. Zusammenfassung ... 49
Anhang ... 51
Literaturverzeichnis ... 51
Geometrien ... 53
Abbildungsverzeichnis Abbildung 1: Diarylethen im geöffneten (l.) und geschlossenem Zustand (r.) ... 1
Abbildung 2: Malachitgrün (l. Carbinalbase, m. Kation, r. Dikation) ... 1
Abbildung 3: Photo- und halochrome Reaktionen des DAE-Malachitgrün-Chromphores ... 2
Abbildung 4: cis- (links) und trans-Isomere (rechts) des Stilbens ... 12
Abbildung 5: Ringschluss des Stilbens ... 12
Abbildung 6: Diarylethen in geöffneter (l.) und geschlossener Form (r.) ... 13
Abbildung 7: DAE-Schalter nach Irie ... 13
Abbildung 8: DAE-Malachitgrünschalter nach Kasuno et al. ... 13
Abbildung 9: Malachitgrün als Carbinolbase (l.), Kation und Dikation (r.) ... 14
Abbildung 10: Röntgenstruktur (blau) und Vakuumstruktur (rot) der Verbindung 1A ... 15
Abbildung 11: Abweichung der Bindungslängen zwischen berechneter Geometrie und Röntgenstruktur ... 16
Abbildung 12: Bindungswinkelabweichungen zwischen berechneter Geometrie und Röntgenstruktur ... 16
Abbildung 13: Diederwinkelabweichung zwischen der berechneten Geometrie und Röntgenstruktur ... 16
Abbildung 14: Torsionsachse in DAE-MG ... 17
Abbildung 15: Rotationsbarrieren um C-C Einfachbindungen im DAE-MG ... 17
Abbildung 16: Valenzstruktur von Struktur 1E und 1F nach Kasunu et al. ... 18
Abbildung 17: Valenzstruktur von Struktur 1E und 1F basierend auf Berechnung ... 18
Abbildung 18: Lösungsmitteleinfluss auf die Geometrie von Verbindung 1F ... 19
Abbildung 19: Verbindung von DAE und MG ... 20
Abbildung 20: Infrarot Spektrum (Struktur 1A) ... 22
VI
Abbildung 21: Ringe der Struktur 1A ... 23
Abbildung 22: Infrarot Spektrum (Struktur 1B) ... 24
Abbildung 23: IR-Spektrum der Struktur 1C (geänderte Skalierung) ... 25
Abbildung 24: IR-Spektrum der Struktur 1D ... 26
Abbildung 25: IR-Spektrum der Struktur 1E ... 27
Abbildung 26: IR-Spektrum der Struktur 1F (geänderte Skalierung) ... 28
Abbildung 27: Mesomere Strukturen von Struktur 1C ... 29
Abbildung 28: Struktur 1E in Valenzschreibweise ... 29
Abbildung 29: Diarylethenisomere mit geöffneter und geschlossener Ringstruktur ... 31
Abbildung 30: Mit DFT/MRCI berechnete Absorptionsspektren von Diarylethenisomeren .. 31
Abbildung 31: HOMO -> LUMO Übergänge von Diarylethen-Isomeren ... 32
Abbildung 32: Mit DFT/MRCI berechnete Absorptionsspektren von Malachitgrün in AN ... 33
Abbildung 33: Berechnetes und experimentelles Absorptionsspektrum in AN (Struktur 1A) 34 Abbildung 34: Molekülorbitale der Struktur 1A ... 35
Abbildung 35: Berechnetes und experimentelles Absorptionsspektrum in AN (Struktur 1B) 36 Abbildung 36: HOMO und LUMO der Struktur 1B ... 37
Abbildung 37: Berechnetes und experimentelles Absorptionsspektrum (Struktur 1C) ... 37
Abbildung 38: Molekülorbitale der Struktur 1C ... 38
Abbildung 39: Berechnetes und experimentelles Absorptionsspektrum (Struktur 1D) ... 39
Abbildung 40: Molekülorbitale der Struktur 1D ... 40
Abbildung 41: Berechnetes und experimentelles Absorptionsspektrum (Struktur 1E) ... 40
Abbildung 42: Molekülorbitale der Struktur 1E ... 41
Abbildung 43: Absorptionswellenlängen und Anregungen von Struktur 1F ... 42
Abbildung 44: Molekülorbitale der Struktur 1F ... 43
Abbildung 45: Absorptionswellenlängen der HOMOLUMO Übergänge ... 44
Abbildung 46: Vergleich von TDDFT und DFT/MRCI-Methode für ein Malachitgrünkation ... 45
Abbildung 47: Vergleich von TDDFT- und DFT/MRCI Methode (Struktur 1A) ... 46
Abbildung 48: Vergleich von TDDFT- und DFT/MRCI-Methode (Struktur 1B) ... 46
Abbildung 49: Vergleich von TDDFT- und DFT/MRCI-Methode (Struktur 1C) ... 47
Abbildung 50: Vergleich von TDDFT- und DFT/MRCI-Methode (Struktur 1D) ... 47
Abbildung 51: Vergleich von TDDFT- und DFT/MRCI-Methode (Struktur 1E) ... 48
Abbildung 52: Vergleich von TDDFT- und DFT/MRCI-Methode (Struktur 1F) ... 48
Tabellenverzeichnis Tabelle 1: Abweichung zwischen Vakuum- und LM-Geometrie (Acetonitril) ... 19
Tabelle 2: Bindungslängen und Diederwinkel der Verbindung von DAE und MG ... 20
Tabelle 3: Relative Stabilität der Isomere ... 21
Tabelle 4: Charakteristische Banden und Schwingungen (Struktur 1A) ... 23
Tabelle 5: Charakteristische Banden und Schwingungen (Struktur 1B) ... 24
Tabelle 6: Charakteristische Banden und Schwingungen (Struktur 1C) ... 25
Tabelle 7: Charakteristische Banden und Schwingungen (Struktur 1D) ... 26
Tabelle 8: Charakteristische Banden und Schwingungen (Struktur 1E) ... 27
Tabelle 9: Charakteristische Banden und Schwingungen (Struktur 1F) ... 28
Tabelle 10: Absorptionswellenlängen und Anregungen von DAE (geöffnet) ... 31
VII
Tabelle 11: Absorptionswellenlängen und Anregungen von DAE (geschlossen) ... 31
Tabelle 12: Halochromatizität von Malachitgrün (λm aus DFT/MRCI Rechnung) ... 32
Tabelle 13: Absorptionswellenlängen und Anregungen der Carbinolbase des Malachtigrüns 33 Tabelle 14: Absorptionswellenlängen und Anregungen des Malachitgrünkations ... 34
Tabelle 15: Absorptionswellenlängen und Anregungen des Malachitgründikations ... 34
Tabelle 16: Absorptionswellenlängen und Anregungen von Struktur 1A ... 35
Tabelle 17: Absorptionswellenlängen und Anregungen von Struktur 1B ... 36
Tabelle 18: Absorptionswellenlängen und Anregungen von Struktur 1C ... 38
Tabelle 19: Absorptionswellenlängen und Anregungen von Struktur 1D ... 39
Tabelle 20: Absorptionswellenlängen und Anregungen von Struktur 1E ... 41
Tabelle 21: Absorptionswellenlängen und Anregungen von Struktur 1F ... 42
Tabelle 22: Vergleich der energetischen Lage der S1-Anregung ... 44
Abkürzungsverzeichnis
BL Bindungslänge
BR Brückenring
BW Bindungswinkel
CI Configuration Interaction
CT Charge-Transfer
DAE Diarylethen
DFT Dichtefunktionaltheorie
DW Diederwinkel
HF Hartree-Fock
HOMO Highest Occupied Molecular Orbital
IR Infrarot
LM Lösungsmittel
LUMO Lowest Unoccupied Molecular Orbital
MG Malachitgrün
MO Molekülorbital
SG Schrödinger-Gleichung
SCRF Self-Consistent Reaction-Field
TDDFT Time-dependent density functional theory
UV Ultraviolett
1 1. Einleitung
Im Bestreben, elektronische Komponenten immer kleiner und effizienter zu machen, wurden in den letzten Jahren große Fortschritte in der Molekularelektronik gemacht. Hierbei bietet die monomolekulare Elektronik Möglichkeiten, Systeme im Nanometerbereich zu entwickeln. Die in dieser Arbeit charakterisierten Diarylethen-Malachitgrün-Chromophore sind eine Kombination aus einem Diarylethen (DAE, Abbildung 1) und Malachitgrün (MG, Abbildung 2). Diarylethene sind photochrome Substanzen, deren Umwandlung bzw. Schaltung lichtinduziert erfolgt. Durch Licht wird bei DAE eine Ringöffnung bzw. ein Ringschluss induziert, es ist somit möglich, zwischen zwei Zuständen reversibel zu wechseln1.
Abbildung 1: Diarylethen im geöffneten (l.) und geschlossenem Zustand (r.)
In einer Studie von Kasunu und Uchida et al.2 wird ein DAE-Schalter mit Malachitgrün kombiniert. Malachitgrün ändert seine Farbe mit seinem Ladungszustand bzw. dem pH-Wert.
Abbildung 2: Malachitgrün (l. Carbinalbase, m. Kation, r. Dikation)
So entsteht ein molekularelektronisches System, das auf verschiedene Arten „schaltbar“ ist; es ist möglich, die Umwandlung verschiedener Isomere untereinander gezielt zu induzieren und somit zu steuern. Halochrom, also abhängig vom pH-Wert der Lösung, jedoch unabhängig von der photochromen Schaltung sind so drei weitere Zustände, insgesamt also 6 Zustände möglich.
1 (Irie 2010)
2 (M. Kasuno, K. Uchida, Y. Tatsumi et al. 2014)
2
Abbildung 3: Photo- und halochrome Reaktionen des DAE-Malachitgrün-Chromphores3 Ziel der Arbeit ist es, die Strukturen des Ausgangsmaterials und der in Abbildung 3 dargestellten Verbindungen mit quantenchemischen Verfahren möglichst genau zu charakterisieren und diese Ergebnisse mit experimentellen Befunden zu vergleichen. Es werden geometrische Eigenschaften dokumentiert und mit der Röntgenstruktur von 1A verglichen. Des Weiteren werden die Infrarot-Spektren bestimmt und mit den experimentellen Daten (für Struktur 1A und 1B vorhanden) verglichen. Abschließend werden die UV-Spektren berechnet und mit den experimentellen Spektren verglichen. Im Rahmen dessen wird auch die Leistungsfähigkeit von DFT/MRCI-Rechnungen mit einer speziellen Parametrisierung nach Lyskov4 gegenüber TDDFT-Rechnungen als Benchmark dokumentiert und bewertet.
3 (M. Kasuno, K. Uchida, Y. Tatsumi et al. 2014)
4 (Lyskov et al. 2016)
3 2. Quantenchemische Grundlagen
2.1. Die Schrödinger-Gleichung
Die auf Erwin Schrödinger zurückgehende Schrödinger-Gleichung (SG) ist die Gleichung deren Lösung die Wellenfunktion ψ eines Systems ergibt5. Die SG ist eine partielle Differentialgleichung 1. Ordnung bezüglich der Zeit und 2. Ordnung bezüglich des Ortes.
Entsprechend lautet die zeitabhängige SG mit dem Hamilton-Operator 𝐻̂: 𝐻̂𝜓 = 𝑖ħ𝜕𝜓
𝜕𝑡 [2.1]
Durch Separierung von zeitabhängigen und ortsabhängigen Komponenten ergibt sich die zeitunabhängige Schrödinger-Gleichung, die für zeitunabhängige Hamilton-Operatoren gilt6:
𝐻̂𝜓 = 𝐸𝜓 [2.2]
wobei E als Energie des betrachteten Systems postuliert wird7. 2.2. Die Born-Oppenheimer-Näherung
Die in dieser Arbeit betrachteten Systeme bestehen sowohl aus Elektronen als auch aus Atomkernen. Eine genaue Lösung für solche Vielteilchensysteme zu ermitteln, ist problematisch. Zur Vereinfachung der Lösung solcher Probleme bei chemischen Systemen wird angenommen, dass sich die Elektronen aufgrund ihrer den Kernen gegenüber deutlich geringeren Masse und erhöhten Geschwindigkeit der Anordnung der Kerne direkt anpassen.
Unter dieser Prämisse lassen sich Kernbewegungen und Elektronenbewegungen trennen. Der Hamilton-Operator 𝐻̂ wird zerlegt in die Summe der Kernanteile 𝐻̂𝐾 und des elektronischen Hamilton-Operators 𝐻̂𝑒.
𝐻̂𝐺𝑒𝑠 = 𝐻̂𝐾+ 𝐻̂𝑒 [2.3]
Die Wechselwirkung zwischen Elektronen und Kernen ist hierbei im elektronischen Hamilton- Operator enthalten. Auch die Wellenfunktion des Systems ψ wird genähert in Anteile von Kernen und Elektronen zerlegt:
ψ(𝑟⃗, 𝑅̅) ≈ ψ𝑒(𝑟⃗, 𝑅̅) ψ𝐾(𝑅̅) [2.4]
5 (Atkins 1993), S.289
6 (Atkins 1993), S.290
7 (Levine 2014), S.13
4
wobei die Elektronenkoordinaten 𝑟⃗ nur Variablen der elektronischen Zustandsfunktion sind und beide Funktionen parametrisch von den nicht variablen, da räumlich fixierten, Kernkoordinaten 𝑅̅ abhängen. Die Lösung von Gleichung 2.2. für den elektronischen Teil ergibt die elektronische Schrödinger-Gleichung:
𝐻̂𝑒ψ𝑒(𝑟⃗,𝑅̅) = E𝑒(𝑅̅)ψ𝑒(𝑟⃗,𝑅̅) [2.5]
Addiert man zur elektronischen Energie E𝑒(𝑅̅) noch die potentielle Energie 𝑉𝐾𝐾(𝑅̅), resultierend aus der Abstoßung der Kerne, erhält man die Gesamtenergie in Abhängigkeit von 𝑅̅8:
E(𝑅̅) = E𝑒(𝑅̅) + 𝑉𝐾𝐾(𝑅̅) [2.6]
2.3. Potentialhyperflächen und Geometrieoptimierungen
Alle Energien E(𝑅⃗⃗) eines Zustandes ergeben zusammen betrachtet die Potentialhyperfläche des Zustandes. Bei gegebenen drei Raumrichtungen ergeben sich bei N Kernen für eine Bewegung auf dieser Potentialhyperfläche 3N Möglichkeiten. Drei dieser Möglichkeiten beschreiben eine gleichgerichtete Bewegung aller Kerne (Translation), weitere drei Möglichkeiten entfallen auf Rotationsbewegungen. Insgesamt 3N-6 entfallen bei nicht-linearen Strukturen auf Schwingungen bei denen sich die räumliche Anordnung der Kerne zueinander ändert.
Kernanordnungen (Konformationen), die mit einem Minimum auf der Potentialhyperfläche korrespondieren, sind Konformere der Verbindung9. Für ein Minimum auf der Potentialhyperfläche ist es notwendige Bedingung, dass die erste Ableitung der Energiefunktion bezüglich der Kernkoordinaten gleich Null ist. Ein Satz aller 3N-6 ersten Ableitungen in einem Punkt wird als dessen Gradient bezeichnet10. Ein Punkt, für den der Gradient gleich Null ist, wird als stationärer Punkt bezeichnet. Neben Minima genügen auch Sattelpunkte diesem Kriterium, wobei die Unterscheidung bei Betrachtung der zweiten Ableitung erfolgt. Klassisch werden diese zweiten Ableitungen in der Hesse-Matrix dargestellt.
Sind alle Eigenwerte der Hesse-Matrix für einen Punkt positiv, ist dieser ein Minimum. Ein Maximum in einer einzigen Richtung entspricht einem Übergangszustand. Das Finden von Minima und den korrespondierenden Geometrien ist Ziel der Geometrieoptimierung, hierfür können verschiedene Algorithmen verwendet werden. Es ist zu unterscheiden, ob es sich bei einem gefundenen Minimum um ein lokales oder globales Minimum handelt.
8 (Reinhold 2015), S.276 ff.
9 (Levine 2014), S.481
10 (Levine 2014), S.483
5
2.4.Mehrelektronensysteme und der Hartree-Fock-Formalismus
Für Wasserstoff-ähnliche Systeme mit einem einzigen Elektron ist die exakte Wellenfunktion bekannt, die Schrödinger-Gleichung ist hier als Einelektronen-SG exakt lösbar. Für Systeme mit mehr als einem Elektron muss prinzipiell eine Mehrelektronen-SG gelöst werden, deren Hamilton-Operator lautet11:
𝐻̂ = − ħ2
2𝑚𝑒∑𝑛𝑖=1∇𝑖2− ∑ ∑ 𝑍𝛼𝑒2
4𝜋𝜀0𝑟𝑖+ ∑ ∑ 𝑒2
4𝜋𝜀0𝑟𝑖𝑗 𝑛𝑗=𝑖+1 𝑛−1𝑖=1
𝑛𝑖=1
𝐾𝛼=1 [2.7]
Der erste Summenterm stellt den Operator der kinetischen Energie für n Elektronen dar, der zweite Summenterm die potentielle Energie in Abhängigkeit von Ladungen 𝑍𝛼𝑒 und Entfernung 𝑟𝑖, der dritte Summenterm die Elektron-Elektron Wechselwirkungen in Abhängigkeit ihres Abstandes 𝑟𝑖𝑗. Durch die Indizes der Doppelsumme werden hierbei Doppelzählung und Selbstwechselwirkung verhindert.
Aus der Ununterscheidbarkeit der Elektronen im System folgt die Bedingung, dass sie als identische Teilchen symmetrisch oder antisymmetrisch gegenüber ihrer Vertauschung (Austausch) sein müssen, wobei Elektronen als Fermionen ferner antisymmetrisch gegenüber einem Austausch sind12. Um dieser Bedingung Rechnung zu tragen, kann die Zustandsfunktion als Slaterdeterminante in der allgemeinen Form13 dargestellt werden:
𝜓 = 1
√𝑁![
ф1(1) ⋯ ф1(𝑁)
⋮ ⋱ ⋮
ф𝑁(1) ⋯ ф𝑁(𝑁)
] [2.8]
wobei 1
√𝑁! der Normierung dient.
Grundsätzlich ist es möglich, Mehrelektronen-SG mittels Separation in lösbare Einelektronen- SG zu überführen, sofern Elektron-Elektron Wechselwirkungen vernachlässigt werden14. Um diese genähert zu berücksichtigen, wird ein Potential 𝑉𝑒𝑓𝑓 eingeführt – ein Elektron wechselwirkt nicht mehr mit allen anderen Elektronen einzeln, sondern vielmehr mit deren gemitteltem Feld. Hierdurch kann Gleichung 2.7 überführt werden in:
𝐻̂ = − ħ2
2𝑚𝑒∑𝑛𝑖=1∇𝑖2− ∑ 𝑍𝑒2
4𝜋𝜀0𝑟𝑖
𝑛𝑖=1 + ∑𝑛𝑖=1𝑉𝑖𝑒𝑓𝑓(𝑟𝑖) [2.9]
11 (Levine 2014), S.289
12 (Reinhold 2015), S.227f
13 (Reinhold 2015), S.229
14 (Reinhold 2015), S.233
6
Im Rahmen des Hartree-Fock-Formalismus (HF) wird nun die Wechselwirkung eines Elektrons mit dem gemittelten Feld gemäß Gleichung 2.9 und 2.8 durch zwei Integrale15 beschrieben:
𝑗̂𝑙(𝑖)ф𝑘(𝑖) = [∫𝑒2ф𝑙∗(𝑗)ф𝑟 𝑙(𝑗)
𝑖𝑗 𝑑𝑟⃗⃗⃗𝑑𝜎𝑗 𝑗] ф𝑘(𝑖) [2.10]
𝐾̂𝑙(𝑖)ф𝑘(𝑖) = [∫𝑒2ф𝑙∗(𝑗)ф𝑟 𝑘(𝑗)
𝑖𝑗 𝑑𝑟⃗⃗⃗𝑑𝜎𝑗 𝑗] ф𝑙(𝑖) [2.11]
Integriert wird über die drei Raumrichtungen sowie die Spinkoordinate. 𝑗̂𝑙 wird als Coulomb- Operator, 𝐾̂𝑙 als Austauschoperator bezeichnet. Werden Operator für kinetische und potentielle Energie in Gleichung 2.9 zusammengefasst zu ℎ̂𝑖 (Core-Hamiltonian), ergibt sich der Fock- Operator (oder effektiver Ein-Elektronen-Operator) als Summe aus diesem und dem Coulomb- wie auch dem Austauschoperator16:
𝑓̂𝑖 = ℎ̂𝑖 + ∑𝑛𝑙=1(𝑗̂𝑙(𝑖) − 𝐾̂𝑙(𝑖))
𝑙≠𝑘
[2.12]
bzw. für Gleichung 2.2 genähert mit 2.12:
𝑓̂𝑖ф𝑘(𝑖) = 𝜖𝑘ф𝑘(𝑖) [2.13]
2.5. Elektronenkorrelation und Multikonfigurationsansätze
Aufgrund der Näherungen des effektiven Potentials in Gleichung 2.9 sind nur Teile der Elektron-Elektron-Wechselwirkungen im Hartree-Fock-Formalismus berücksichtigt.
Elektronen bewegen sich nicht unabhängig von den anderen Elektronen im System, sie sind vielmehr korreliert. Teile der Korrelation sind implizit bereits in dem Hartree-Fock- Formalismus durch die Antisymmetrisierung enthalten17. Der Energieunterschied zwischen der theoretisch korrekten (nicht relativistischen) Energie und der in Hartree-Fock bestimmten Energie wird als Korrelationsenergie bezeichnet:
𝐸𝐾𝑜𝑟𝑟𝑒𝑙𝑎𝑡𝑖𝑜𝑛 = 𝐸𝑛𝑖𝑐ℎ𝑡−𝑟𝑒𝑙𝑎𝑡𝑖𝑣𝑖𝑠𝑡𝑖𝑠𝑐ℎ− 𝐸𝐻𝐹 [2.14]
Ein Teil der fehlenden Korrelation resultiert aus der direkten (instantanen) Korrelation der Bewegung von Elektronen, dies wird als dynamische Korrelation bezeichnet. Des Weiteren entstehen durch die Verwendung einer einzigen Slater-Determinante als Wellenfunktion in HF Ungenauigkeiten bei der Beschreibung von Dissoziationen (z.B. des H2-Moleküls in zwei H- Atome) und Verbindungen mit niedrigen energetischen Unterschieden zwischen besetzten und
15 (Reinhold 2015), S.237 und S.240
16 (Reinhold 2015), S.241
17 (Levine 2014), S.298
7
unbesetzten Orbitalen (Ozon-Molekül)18. Um die Korrelationsenergie mit zu erfassen, gilt es, Beiträge von Konfigurationswechselwirkungen zu berücksichtigen. (engl. Configuration Interaction (CI)). Für den Grundzustand ist der wichtigste Beitrag der der Grundkonfiguration, in der nur die energetisch niedrigsten Spinorbitale besetzt sind (𝜓0). Hinzu kommen im Rahmen der CI die verschieden substituierten Konfigurationen (einfach substituiert (𝜓𝑆), zweifach substituiert (𝜓𝐷), dreifach (𝜓𝑇), …), in denen besetzte Orbitale mit virtuellen Orbitalen vertauscht sind. Diese können mit entsprechenden Koeffizienten versehen werden, die Koeffizienten selbst können mittels linearer Variation bestimmt werden:
𝜓0+ 𝐶𝑆𝜓𝑆+ 𝐶𝐷𝜓𝐷+ 𝐶𝑇𝜓𝑇+ ⋯ [2.15]
Das Prinzip von Gleichung 2.15 entspricht einer vollständigen CI-Entwicklung (Full-CI), in der Realität muss eine Beschränkung auf die wichtigsten (beitragsstärksten) Konfigurationen erfolgen. Für den Grundzustand sind besonders Zweifach-Substitutionen von Bedeutung (CID), aufgrund der verhältnismäßig geringen Anzahl werden Einfach-Substitutionen miteinbezogen (CISD)19. Eine Form des CI-Verfahrens ist das Multi-Referenz-CI-Verfahren (MRCI). Als Referenz dient beim MRCI-Verfahren nicht die die reine Grundzustandsdeterminante, sondern es wird ein bereits aus mehreren Konfigurationen gebildeter Referenzraum genutzt.
2.6. Dichtefunktionaltheorie
Hohenberg und Kohn gelang 1964 der Beweis, dass alle molekularelektronischen Eigenschaften des Grundzustandes durch dessen Einelektronendichte 𝜌0 festgelegt werden (Hohenberg-Kohn-Theorem)20. Analog zum Mehrteilchensystem des Hartree-Fock- Formalismus verwendeten Kohn und Sham eine Slater-Determinante aus Ein- Elektronenfunktionen zur Beschreibung der Grundzustandsfunktion 𝜓𝑘 eines Systems aus N nicht wechselwirkenden Elektronen21(Kohn-Sham-Orbitale). Für die Elektronendichte des Grundzustandes gilt dann:
𝜌(𝑟⃗) = ∑𝑁𝑘=1𝑒|𝜓𝑘(𝑟⃗)|2 [2.16]
Der Zusammenhang zwischen Energie 𝐸 und der Elektronendichte 𝜌(𝑟⃗) stellt das Energiefunktional 𝐸[𝜌] her22:
18 (Levine 2014), S.526
19 (Reinhold 2015), S.260
20 (Levine 2014), S.552
21 (Reinhold 2015), S.270
22 (Reinhold 2015), S.266
8
𝐸 = 𝐸[𝜌] [2.17]
Dieses kann aufgeteilt werden in Beiträge der kinetischen Energie 𝑇[𝜌], Elektron- Elektronwechselwirkungen 𝑉𝑒𝑒[𝜌] und einem systemabhängigen externen Potential 𝑉𝑒𝑥𝑡(𝑟⃗)23.
𝐸[𝜌] = 𝑇[𝜌] + 𝑉𝑒𝑒[𝜌] + ∫ 𝑉𝑒𝑥𝑡(𝑟⃗)𝜌(𝑟⃗) 𝑑𝑟⃗ [2.18]
Nach Kohn und Sham wird 𝑇[𝜌] genähert durch 𝑇𝑆[𝜌] mit Hilfe der Kohn-Sham-Orbitale beschrieben:
𝑇𝑆[𝜌] = ∑ ∫ 𝜓𝑘∗(𝑟⃗𝑖) [− ħ2
2𝑚𝑒∆𝑖]
𝑁𝑘=1 𝜓𝑘(𝑟⃗𝑖)𝑑𝑟⃗𝑖 [2.19]
Der Anteil der Coulombwechselwirkungen an 𝑉𝑒𝑒[𝜌] wird analog durch 𝐽[𝜌] dagestellt:
𝐽[𝜌] =1
2∑ ∑ ∫ ∫ 𝜓𝑘∗(𝑟⃗𝑖)𝜓𝑙∗(𝑟⃗𝑗)𝑒2
𝑟𝑖𝑗 𝑁𝑙=1
𝑙≠𝑘
𝑁𝑘=1 𝜓𝑘(𝑟⃗𝑖)𝜓𝑙(𝑟⃗𝑗)𝑑𝑟⃗𝑖𝑑𝑟⃗𝑗 [2.20]
Noch nicht in 𝐽[𝜌] enthaltene Austauschwechselwirkungen und Korrelationen sowie durch die Näherung in Gleichung 2.19 entstandene Fehler werden im Austausch-Korrelations-Funktional 𝐸𝑋𝐶[𝜌] zusammengefasst:
𝐸𝑋𝐶[𝜌] = (𝑇[𝜌] − 𝑇𝑆[𝜌]) + (𝑉𝑒𝑒[𝜌] − 𝐽[𝜌]) [2.21]
Gemäß Gleichung 2.17 bis 2.20 gilt also24:
𝐸[𝜌] = 𝑇𝑆[𝜌] + 𝐽[𝜌] + 𝐸𝑋𝐶[𝜌] + ∫ 𝑉𝑒𝑥𝑡(𝑟⃗)𝜌(𝑟⃗) 𝑑𝑟⃗ [2.22]
Die DFT umfasst im Gegensatz zu HF die Elektronenkorrelation und ist prinzipiell exakt, sofern alle Funktionale in Gleichung 2.22 bekannt sind. Das Austausch-Korrelations-Funktional ist leider nicht bekannt, vielmehr gibt es diverse Ansätze, dieses zu nähern. Das verwendete BH- LYP Funktional25 besteht aus einem Austauschteil, der zur Hälfte durch das B88-Funktional und zur Hälfte durch HF-Austausch beschrieben wird (BH), und dem Korrelationsfunktional von Lee, Yang und Parr (LYP). Das in dieser Arbeit u.a. verwendete B3-LYP Funktional beinhaltet Slater-, HF- und Becke-Austauschfunktionale sowie Korrelationsfunktionale von Lee, Yang und Parr (LYP) sowie Vosko, Wilk und Nusair (VWN)26 der Form:
𝐸𝑋𝐶𝐵3𝐿𝑌𝑃 = 0,8𝐸𝑋𝑆𝑙+ 0,2𝐸𝑋𝐻𝐹+ 0,72𝐸𝑋𝐵88+ 0,19𝐸𝐶𝑉𝑊𝑁 + 0,81𝐸𝐶𝐿𝑌𝑃 [2.23]
23 (Reinhold 2015), S.269
24 (Reinhold 2015), S.270f
25 (Becke 1988)
26 (Reinhold 2015), S.273; (Levine 2014) S.566
9
Im Rahmen der Coulomb-Attenuated-Methode (CAM, bspw. CAM-B3LYP27) wird der Austauschterm für langreichweitige Wechselwirkungen korrigiert. Hierzu werden langreichweitige Austauschwechselwirkungen durch HF-Austauschwechselwirkungen beschrieben28. Dies soll Ergebnisse für langkettige konjugierte Systeme verbessern und die Beschreibung von Charge-Transfer-Anregungen mithilfe der folgenden zeitabhängigen Dichtefunktionaltheorie verbessern.
2.7. Zeitabhängige Dichtefunktionaltheorie
Für die zeitabhängige Dichtefunktionaltheorie (engl. time-dependent DFT (TDDFT)) wird ausgehend von der Dichtefunktionaltheorie des elektronischen Grundzustandes die Auswirkung einer Störung durch ein schwaches, oszillierendes, zeitabhängiges und räumlich einheitliches elektrisches Feld untersucht29. Nach dem Runge-Gross-Theorem ist bei einem gegebenen Anfangszustand die zeitabhängige Elektronendichte durch das ebenfalls zeitabhängige externe Potential bestimmt30. Der Einfluss dieses zeitabhängigen Potentials wird ausgehend vom Grundzustand störungstheoretisch betrachtet. Für das externe Potential gilt31
𝑉𝑒𝑥𝑡(𝑟⃗, 𝑡) = 𝑉(0)(𝑟⃗) + 𝑉(1)(𝑟⃗, 𝑡) [2.24]
mit dem (nicht zeitabhängigen) Coulomb-Potential 𝑉(0)(𝑟⃗) der Form 𝑉(0)(𝑟⃗) = − ∑ 𝑍𝑎𝑒2
𝑟𝑎𝑖
𝐾𝑎=1 [2.25]
Und der Störung durch die z-Komponente des äußeren elektrischen Feldes 𝐸⃗⃗
𝑉(1)(𝑟⃗, 𝑡) = 𝑒|𝐸⃗⃗|𝑧 cos (𝜔𝑡) [2.26]
Die zeitliche Entwicklung der Elektronendichte kann störungstheoretisch beschrieben werden, eine gängige Vereinfachung ist die Beschränkung auf die erste Ordnung (Linear-Response).
Durch Fourier-Transformation kann eine frequenzabhängige Darstellung der linearen Antwortfunktion mit einem vollständigen Satz von Mehrelektronenfunktionen 𝜓𝑚 und deren Energien 𝐸𝑚 abgeleitet werden32:
𝜒(𝑟⃗, 𝑟⃗´, 𝜔) = lim
𝜂→0+∑ {〈𝜓0,𝜌̂(𝑟⃗)𝜓𝑚〉〈𝜓𝑚,𝜌̂(𝑟⃗´)𝜓0〉
ħ𝜔−(𝐸𝑚−𝐸0)+𝑖𝜂 −〈𝜓0,𝜌̂(𝑟⃗)𝜓𝑚〉〈𝜓𝑚,𝜌̂(𝑟⃗´)𝜓0〉
ħ𝜔+(𝐸𝑚−𝐸0)+𝑖𝜂 }
𝑚 , [2.27]
27 (Yanai, Tew und Handy 2004)
28 (Peach et al. 2006)
29 (Levine 2014), S. 570
30 (Reinhold 2015), S.273
31 (Reinhold 2015), S.274
32 (Reinhold 2015), S.275
10 wobei 𝜌̂(𝑟⃗) der Teilchendichteoperator ist. Für
𝐸𝑚− 𝐸0 = ħ𝜔 [2.28]
wird der Realteil des Nenners im ersten Term Null, die Funktion hat für diese Frequenz eine Polstelle. Unter Verwendung von Kohn-Sham-Orbitalen können so auch Beziehungen zwischen den Anregungsenergien und den Intensitäten hergestellt werden33, so ist es möglich, Absorptionsspektren für vertikale Anregungen aus dem Grundzustand zu berechnen. Durch Variation der Geometrien des Grundzustandes ist ferner auch die Potentialhyperfläche des angeregten Zustandes berechenbar. LR-TDDFT Rechnungen können gute Ergebnisse liefern, jedoch grundsätzlich nur für Anregungen eines einzelnen Valenzelektrons34.
2.8. DFT/MRCI-Methoden
Zur korrekten Beschreibung von angeregten Zuständen und der richtigen Berechnung der Absorptionsspektren müssen verschiedene Konfigurationen im Rahmen einer CI-Rechnung berücksichtigt werden. CI-Entwicklungen ergeben gute Korrelationsenergien, sind jedoch aufwändig und für große Moleküle nicht praktikabel. Dichtefunktional-Rechnungen sind indes auch für größere Moleküle anwendbar und beispielsweise bei der Geometrieoptimierung Standard. Für die Beschreibung von angeregten Zuständen und Absorptionsspektren reichen DFT-Rechnungen alleine nicht aus. Die Idee hinter der DFT/MRCI-Methode35 ist es, Teile der Korrelation durch MRCI-Rechnungen zu erhalten und Teile aus DFT-Rechnungen. Die dynamische Korrelation wird durch die Dichtefunktionaltheorie gut beschrieben und folglich wird die dynamische Korrelation auch mittels DFT berechnet. Die statische Korrelation wird mittels einer modifizierten CI-Rechnung nach Grimme und Waletzke berechnet, wobei Kohn- Sham-Orbitale genutzt werden. Durch Parametrisierung der Hamilton-Matrix kann eine doppelte Berücksichtigung von dynamischen Korrelationsbeiträgen verhindert werden, hierzu werden Außerdiagonalterme für Konfigurations-Zustandsfunktionen mit großem Energieunterschied gegen Null skaliert. Die in dieser Arbeit verwendete Parametrisierung nach Lyskov36 sieht Parameter für die Skalierung ergänzender Coulomb-Anteile (begründet durch den unterschiedlichen Orbitalenergieabstand von KS und HF), Austauschwechselwirkungen, sowie zwei Parameter der Dämpfungsfunktion von Außerdiagonalelementen vor. Zur Anwendung bei größeren Molekülen sind diese Parameter auch für eine engere Auswahlgrenze bei einer Energiedifferenz zwischen den Konfigurationen von 0,8 Eh optimiert, diese wurden
33 (Reinhold 2015), S.276
34 (Levine 2014), S.570
35 (Grimme und Waletzke 1999)
36 (Lyskov et al. 2016)
11
auch in der folgenden Arbeit verwendet. Die neue Parametrisierung verbessert die Beschreibung von Doppelanregungen mit vier offenen Schalen.
2.9. Infrarot-Spektroskopie
Infrarot-Spektroskopie wird als analytische Methode verwendet, um Information über Identität und Struktur von Substanzen zu erhalten. Es wird experimentell die Transmission von infrarotem Licht durch eine Probe in Abhängigkeit von der Wellenzahl gemessen.
Infrarotes Licht regt bei Absorption Schwingungen an, die annähernd in Normalschwingungen zergliedert werden können. Wie in Kapitel 2.3 beschrieben gibt es bei einem nicht-linearen Molekül aus N-Atomen 3N-6 mögliche unabhängige Koordinaten, die nicht auf Rotation oder Translation entfallen. Normalschwingungen sind Linearkombinationen von solchen Koordinaten die für einen quadratischen Potentialverlauf dynamisch unabhängig sind37. In Annahme einer quadratischen bzw. harmonischen Näherung (harmonischer Oszillator) wird die Schwingungsenergie beschrieben durch38:
𝐸𝑣𝑖𝑏 = ∑ (𝜈𝑘+1
2) ℎ𝑓𝑘
3𝑁−6𝑘=1 [2.27]
Mit 𝑓𝑘 als Frequenz der k-ten Mode und 𝜈𝑘 als Schwingungsquantenzahl (mit den Werten 0,1,2,3,…). In harmonischer Näherung können aus den Eigenwerten der Hesse-Matrix die Kraftkonstanten und aus den Eigenvektoren die Normalschwingungen bestimmt werden. Durch die Änderung des Dipolmomentes kann die Intensität berechnet werden.
Schwingungen entlang der Bindungsachse werden als Valenzschwingungen, Schwingungen unter Veränderung des Bindungswinkels als Deformationsschwingungen bezeichnet.
Valenzschwingungen können, symmetrisch unter Erhalt der Symmetrie oder asymetrisch unter Verlust derer stattfinden. Deformationsschwingungen können innerhalb der Ebene als Dreh- und als Scherschwingung, aus der Ebene hinaus als Wipp- und als Drehschwingung stattfinden.
2.10. Solvens-Effekte
Die bisherigen Beschreibungen quantenchemischer Grundlagen bezogen sich auf ein einzelnes isoliertes Molekül im Vakuum. Die experimentellen Werte und die daraus ableitbaren Eigenschaften der untersuchten Strukturen werden jedoch auch durch ihre Umgebung beeinflusst. Um dem Effekt von Lösungsmitteln auf quantenchemische Eigenschaften Rechnung zu tragen, kann eine Umgebung aus einzelnen Lösungsmittelmolekülen
37 (Atkins 1993), S.228
38 (Levine 2014), S.498
12
berücksichtigt werden (explizite Solvatation). Alternativ kann eine Lösung als kontinuierliches Dielektrikum angesehen werden, das wiederum eine Kavität umschließt, welche das gelöste Molekül beinhaltet39. Das Dielektrikum wird durch seine relative Permittivität 𝜀𝑟 charakterisiert. Die Solvatation wird hier implizit beschrieben. Verschiedene Methoden existieren, die sich im Wesentlichen durch Form und Größe der Kavität und den Term, der die Wechselwirkungen zwischen Molekül und Umgebung beschreibt, unterscheiden40. In dieser Arbeit werden das Polarizable Continuum Model (PCM) mit einer parametrisierten, durch vdW-Radien beschriebenen Kavität und das Conductor-like Screening Model (COSMO), das eine Kavität auf Basis der Molekülform nutzt, verwendet.
3. Diarylethene
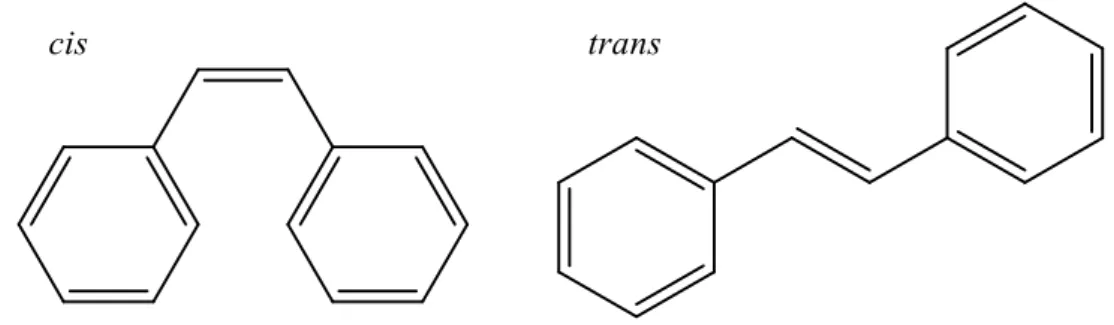
Diarylethene sind Derivate des Stilbens. Stilben kann als cis- wie auch als trans-Isomer vorliegen:
Abbildung 4: cis- (links) und trans-Isomere (rechts) des Stilbens
Lichtinduziert kann aus der cis-Konfiguration ein Ringschluss erfolgen, nach Woodward- Hoffmann verläuft der photoinduzierte Ringschluss hierbei konrotatorisch41.
Abbildung 5: Ringschluss des Stilbens
Die Reaktion des Stilbens ist thermisch reversibel, in Folge dessen ist das geschlossene Isomer thermisch nicht stabil (T-Typ-Photochrom). Durch Substitution der aromatischen Ringe durch
39 (Levine 2014), S.511
40 (Levine 2014), S.512
41 (Irie 2010), S.473
13
Heterocyclen (bspw. Thiophene oder Furane) entsteht ein photoschaltbares System dessen beide Isomere thermodynamisch stabil sind. Die Reaktionen sind nicht thermisch reversibel, jedoch photochemisch reversibel (P-Typ-Photochrom)42.
Abbildung 6: Diarylethen in geöffneter (l.) und geschlossener Form (r.)
Der Ringschluss steht in Konkurrenz zur cis/trans-Isomerisierung, was der photochromen Ausbeute schadet. Durch die Substitution der Ethen-Einheit kann dieses verhindert werden43. Des Weiteren kann durch Einführung von Alkylgruppen an der 2- und 6-Position eine oxidative Zersetzung verhindert werden44. Ein früher Diarylethen-Schalter der diese Merkmale aufweist, wurde von Irie et al. entwickelt45. Dieses 1,2-bis(2,5-dimethyl-3-theinyl)perfluorocyclopenten (Abbildung 7) ist auch Basis des in dieser Arbeit untersuchten Photoschalters von Kasuno et al.46 (Abbildung 8).
Abbildung 7: DAE-Schalter nach Irie
Abbildung 8: DAE-Malachitgrünschalter nach Kasuno et al.
42 (Irie 2010), S.473
43 (Barrois 2014), S.69
44 (Barrois 2014), S.69
45 (Irie 2010)
46(M. Kasuno, K. Uchida, Y. Tatsumi et al. 2014)
14 4. Malachitgrün
Malachitgrün ist ein N-methylierter Triphenyl-Farbstoff. Aufgrund seiner intensiven blau- grünen Farbe wird MG heute noch zum Färben von Seide, Baumwolle, Leder und Papier verwendet47. Des Weiteren wird es auch als Fungizid und Bakterizid zur Behandlung von Fischen verwendet, MG ist als gentoxisch und kanzerogen eingestuft48. Aufgrund seiner halochromatischen Eigenschaften ist es als pH-Indikator verwendbar, die Umschlagpunkte liegen bei pH 0,2 bis 1,8 sowie 11,5 bis 13,249.
Abbildung 9: Malachitgrün als Carbinolbase (l.), Kation und Dikation (r.) 5. Verwendete Programme und Methoden
Die Geometrieoptimierung und Frequenzanalyse wurde in Gaussian0950 durchgeführt. Beides erfolgte auf DFT-Basis mit dem Funktional B3LYP, es wurde ein feineres DFT-Grid genutzt (Ultrafine) um sicherzustellen, dass das globale Minimum gefunden wurde. Basissatz für alle Rechnungen war SVP51. Der SVP-Basissatz beschreibt die Rumpforbitale mit einer kontrahierten Gaußfunktion aus 6 primitiven Gaußfunktionen und die Valenzorbitale mit einer aus drei primitiven Gaußfunktionen bestehenden kontrahierten Gaußfunktion und einer primitiven Gaußfunktion. Hinzukommen Polarisationsfunktionen (p-Funktionen für Wasserstoff und d-Funktionen für Elemente der 2.Periode). Die DFT-MRCI Rechnungen erfolgten in Turbomole52, verwendet wurde das BHLYP Funktional. Es wurde die Parametrisierung nach Lyskov53 mit Tight-Parametersatz verwendet. Als Energy-Cutoff wurde 0,8 Eh gewählt. Als Referenzfunktionen dienten maximal doppelt angeregte Konfigurationen mit 8 Elektronen in 8 Orbitalen. Mit Ausnahme von Struktur 1C, für die 50 Zustände bestimmt wurden, wurden 10 Zustände berechnet. TDDFT-Rechnungen erfolgten in Gaussian09.
Aufgrund des langen konjugierten Systems in einigen Verbindungen und des Charge-Transfer-
47 (Spektrum Akademischer Verlag, Heidelberg 1998)
48 (Bundesinstitut für Risikoberwertung 2014)
49 (Bruneau 2012)
50 (Gaussian Inc. 2009)
51 (Hariharan und Pople 1973)
52 (TURBOMOLE GmbH 2011)
53 (Lyskov et al. 2016)
15
Charakters einiger Übergänge wurde mit CAM-B3LYP54 ein Funktional mit Coulomb- Attenuating-Methode gewählt, um langreichweitige Wechselwirkungen besser darzustellen55. Es wurden 30 Zustände (bzw. 50 Zustände für Struktur 1A) berechnet. Zur Darstellung der Geometrien wurden für diese Arbeit Jmol56 und VMD57 genutzt. Berechnete Spektren wurden in xmgrace und Gabedit bearbeitet und erstellt. Die Frequenzen der IR-Spektren wurden mit einem Faktor von 0,958 skaliert58. Sowohl für IR- wie auch für UV-Spektren wurde eine Lorentzfunktion für die Erstellung der Spektren gewählt. Lösungsmitteleffekte wurden bei Gaussian09 durch das Polarizable-Continuum-Modell (PCM), bei Turbomole durch das Conductor-like Screening Model (COSMO) beschrieben.
6. Ergebnisse
6.1.Abweichungen zur Röntgenstruktur
In Abbildung 10 sind die berechnete Geometrie (rot) und die röntgenspektroskopisch bestimmte Struktur59 (blau) gezeigt. Gegenüber der Röntgenstruktur ist eine Torsion im Bereich des Malachitgrüns zu beobachten.
Abbildung 10: Röntgenstruktur (blau) und Vakuumstruktur (rot) der Verbindung 1A
Die Änderung der Bindungslängen gegenüber der Röntgenstruktur sind in Abbildung 11 dargestellt und unterscheiden sich kaum von der Kristallstruktur, durchschnittlich weichen die Bindungslängen um 0,7% ab, maximal um 1,68%.
54 (Yanai, Tew und Handy 2004)
55 (Adamo und Jacquemin 2013)
56 (Jmol: an open-source Java viewer for chemical structures in 3D)
57 (Humphrey, W., Dalke, A. und Schulten, K 1996)
58 (National Institute of Standards and Technology 2015)
59 (M. Kasuno, K. Uchida, Y. Tatsumi et al. 2014)
16
Abbildung 11: Abweichung der Bindungslängen zwischen berechneter Geometrie und Röntgenstruktur
Die Änderung der Bindungswinkel sind in Abbildung 12 dargestellt, die Dimethylaminogruppen (grün markiert) sowie die Bindungswinkel um das Zentralatom des Malachitgrüns (rot markiert) zeigen größere Abweichungen, die maximale prozentuale Abweichung liegt bei 3,22%, die Standardabweichung liegt bei einem Grad.
Abbildung 12: Bindungswinkelabweichungen zwischen berechneter Geometrie und Röntgenstruktur
Analog zeigen auch die Diederwinkel die größten Abweichungen zwischen den beiden Strukturen im Bereich der Dimethylaminogruppen (grün) und des Malachitgrüntetraeders (rot markiert). Die Abweichungen betragen bis zu 30˚, die Standardabweichung liegt bei 9,2˚.
Abbildung 13: Diederwinkelabweichung zwischen der berechneten Geometrie und Röntgenstruktur
-0,04 -0,02 0 0,02 0,04
Å
Δ Bindungslängen
-4 -2 0 2 4 6
ΔWinkel in Grad
Δ Bindungswinkel
-40 -20 0 20 40
ΔWinkel in Grad
Δ Diederwinkel
17
Die Röntgenstruktur von Diarylethen-Malachitgrün-Chromophoren unterscheidet sich von DAE-MG in Vakuum besonders bezüglich Bindungswinkel und Diederwinkel der zwei Diaminogruppen (Vgl. Abbildung 13) und der Torsion um die C-C Einfachbindung, dargestellt in Abbildung 14, im Malachitgrün um ca. 20˚ (Vgl. Abbildung 10):
Abbildung 14: Torsionsachse in DAE-MG
Vermutlich sind Packungseffekte Ursache für diese Veränderungen.
6.2. Rotationsbarrieren in Diarylethen-Malachitgrün
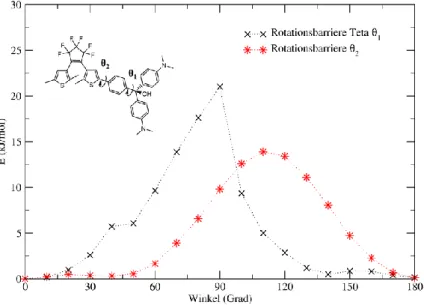
Um energetisch günstigere Rotamere auszuschließen und weitere Erkenntnisse über den geometrischen Charakter zu erhalten, wurden für Struktur 1A die Rotationsbarrieren um zwei C-C Bindungen berechnet:
Abbildung 15: Rotationsbarrieren um C-C Einfachbindungen im DAE-MG
Die Rotationsbarrieren um die beiden Achsen liegen bei ca. 15 bis 23 kJ/mol. Während der Rotation um die θ1-Achse kommt es zu sterischer Hinderung der aromatischen Ringe, in Folge dessen sich die Ringe verdrehen. Durch die daraus bedingten Geometrieänderungen kommt es
18
zu Sprüngen zwischen Schritt 5 bzw. 6 und 10 und 11, die mit einer beginnenden Drehung um die eigene Achse (zw. 5 und 6) und der weiteren Drehung beim Passieren des Aromatischen Ringes (zw. 10 und 11) zusammenhängen. Die Rotationen um C-C Einfachbindungen sind in Folge sterischer Hinderung mit energetisch hohen Barrieren versehen, es ist daher nicht von einer freien Rotation bei Raumtemperatur auszugehen. Die Ausgangsposition entsprach bezüglich dieser beiden Koordinaten dem Minimum, so dass ein energetisch günstigeres Rotamer ausgeschlossen werden kann. Daher wurden diese Parameter für die Optimierung der restlichen Strukturen verwendet.
6.3. Valenzstrukturen von 1C und 1D
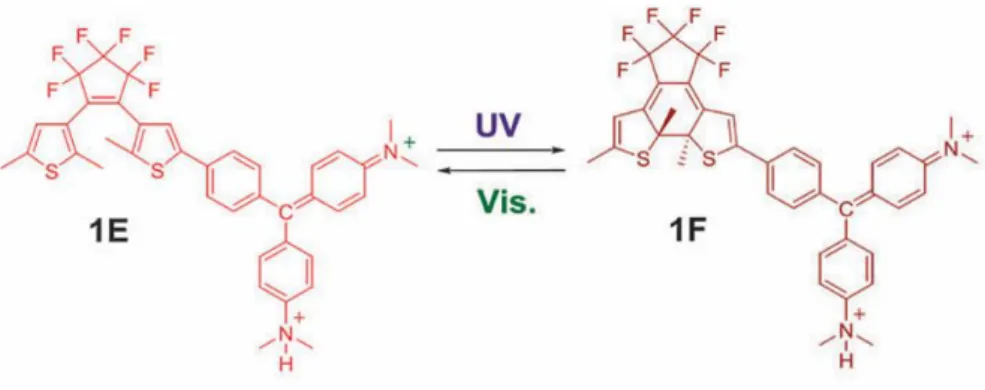
Bei der Untersuchung von Struktur 1E und 1F stellte sich heraus, dass die von Kasuno et al.
vorgeschlagene Valenzschreibweise
Abbildung 16: Valenzstruktur von Struktur 1E und 1F nach Kasunu et al.60
nicht dem Befund entspricht. Die Orientierung des protonierten Ringes zum Diarylethen ist anders und entspricht in Valenzschreibweise vielmehr
Abbildung 17: Valenzstruktur von Struktur 1E und 1F basierend auf Berechnung
60 (M. Kasuno, K. Uchida, Y. Tatsumi et al. 2014)
19 6.4.Lösungsmitteleinflüsse
Als Lösungsmittel in der Studie von Kasuno et al. wurde mit Acetonitril ein polares aprotisches Lösungsmittel verwendet. Die Einflüsse auf die Geometrie wurden durch eine Geometrieoptimerung mit implizitem Lösungsmitteleinfluss untersucht.
Tabelle 1: Abweichung zwischen Vakuum- und LM-Geometrie (Acetonitril)
Struktur σΔBindungslänge Δ BLmax σΔBindungswinkel Δ BWmax σΔDiederwinkel ΔDWmax
1A 0,0017 Å 0,0057 Å 0,1316 ˚ 0,3977 ˚ 1,2536 ˚ 5,2338 ˚ 1B 0,0019 Å 0,0054 Å 0,1663 ˚ 0,6209 ˚ 1,7879 ˚ 7,1366 ˚ 1C 0,0027 Å 0,0086 Å 0,2201˚ 0,7584 ˚ 1,2551 ˚ 3,7159 ˚ 1D 0,0040 Å 0,0123 Å 0,2924 ˚ 0,9923 ˚ 1,1281 ˚ 4,7525 ˚ 1E 0,0062 Å 0,0190 Å 0,3720 ˚ 1,1068 ˚ 1,8549 ˚ 5,1231 ˚ 1F 0,0109 Å 0,0283 Å 0,6239 ˚ 1,8562 ˚ 2,2712 ˚ 9,7688 ˚
Die Geometrieveränderung ist für Bindungslängen und –winkel gering. Die größte Auswirkung des Lösungsmittels auf die Geometrie ist bei den kationischen Strukturen 1E und 1F zu beobachten. In Abbildung 18 werden Vakuumgeometrie (rot) und die Struktur in Acetonitril (grün) dargestellt, es ist gut zu erkennen, dass besonders die Geometrie des Malachitgrüns beeinflusst wird. Der Einfluss eines polaren Lösungsmittels auf die Geometrie ist relativ gering und nimmt mit zunehmender Ladung zu (Vgl. Tabelle 1). Dies erscheint nachvollziehbar, da die zunehmend polaren Strukturen verstärkt mit dem polaren Lösungsmittel Acetonitril wechselwirken. Für die Berechnung der UV-Spektren wurden diese Geometrien im Lösungsmittel genutzt, für die IR-Spektren erfolgten die Berechnungen auf Basis der Vakuumstrukturen.
Abbildung 18: Lösungsmitteleinfluss auf die Geometrie von Verbindung 1F
20
6.5. Die Verbindung von Diarylethen und Malachitgrün
Beide Komponenten sind durch die in Abbildung 19 rot markierte C-C Einfachbindung zwischen zwei aromatischen Ringen verbunden.
Abbildung 19: Verbindung von DAE und MG
Deren Bindungslänge nimmt von 1A nach 1F ab. In Tabelle 2 sind die Diederwinkel für Struktur zwischen Malachitgrün und Diarylethen zu finden. Der Diederwinkel ist i.d.R. gering bis nahezu planar. Ausnahme bildet die Struktur 1A, der Grund ist vermutlich die verbesserte Konjugation des Systems für die Strukturen 1B bis 1F (durch Ringschluss zu 1B und der Erweiterung des konjugierten Systems auf die aromatischen Ringe nach Abspaltung der OH- Gruppe). Hierzu passen auch die verkürzten Bindungslängen der Bindung in Folge besserer Konjugation, verbunden mit zunehmendem Doppelbindungscharakter der Bindung.
Tabelle 2: Bindungslängen und Diederwinkel der Verbindung von DAE und MG Struktur Bindungslänge [Å] Diederwinkel1,2,3,4
1A 1,470 21,9˚
1B 1,465 4,6˚
1C 1,457 5,0˚
1D 1,455 0,02˚
1E 1,44 1,9˚
1F 1,436 1,0˚
6.6. Relative Energien der Isomere
Die absoluten Energien der einzelnen Strukturen sind nur bedingt interpretierbar, aus der relativen energetischen Lage der Isomere lassen sich aber Näherungen bezüglich der Reaktionsenthalpie und der freien Enthalpie berechnen. Aus Tabelle 3 sind Enthalpie und freie Enthalpie des Ringschlusses zu entnehmen, hierzu wurden die berechneten Energien und
21
thermischen Korrekturen für 298,15 Kelvin beider Isomere berechnet. Die Differenz der Enthalpie bzw. freien Enthalpie beider Isomere entspricht deren Reaktionsenthalpie bzw. freien Reaktionsenthalpie. Für alle Strukturen ist die geöffnete Form energetisch niedriger, die niedrigste Reaktionsenthalpie ist bei der doppelt geladenen Spezies zu beobachten. Die thermodynamischen Energien des Ringschlusses reichen in der vorliegenden Form nicht aus, um konkrete Aussagen über den Ringschluss zu machen. Hierfür müsste der Reaktionsverlauf im angeregten Zustand untersucht werden, gerade auch im Hinblick auf Nebenreaktionen, die zur Ermüdung des Photoschalters führen.
Tabelle 3: Relative Stabilität der Isomere
Reaktion ΔH in kJ/mol ΔG in kJ/mol
47,06 63,45
51,08 62,18
56,32 69,61
38,73 51,40
22 7. Infrarot-Spektren und Schwingungsanalyse
7.1. Infrarot-Spektrum der Struktur 1A
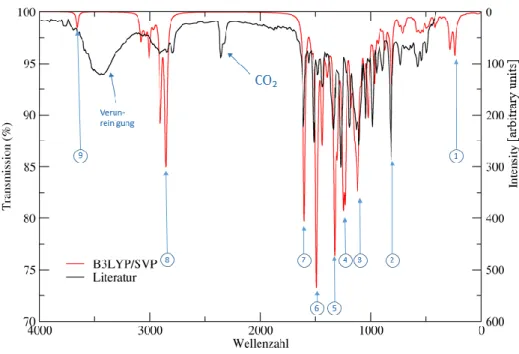
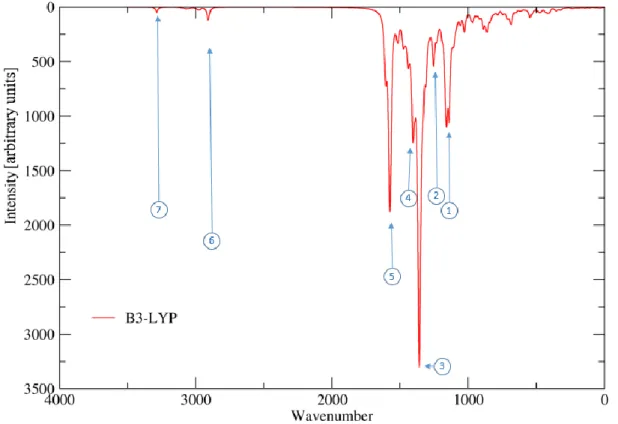
In Abbildung 20 sind das experimentelle IR-Spektrum61 (schwarz), sowie das berechnete Spektrum (rot) dargestellt.
Abbildung 20: Infrarot Spektrum (Struktur 1A)
Das Spektrum deckt sich gut mit den experimentellen Werten, im experimentellen Spektrum sind ferner noch Verunreinigungen bei ca. 3500 Wellenzahlen (evtl. Wasser; Rückstände aus der Synthese) sowie ein Peak bei 2360 Wellenzahlen zu erkennen, der CO2 zuzuordnen ist. Die charakteristischen Banden aus Abbildung 20 sind in Tabelle 4 beschrieben. Im Bereich zwischen ca. 500 bis 1700 Wellenzahlen sind Signale verschiedenster Schwingungen in und zwischen den Ringen des Malachitgrüns (MG1,2,3) und des Diarylethens (DAE1,2) sowie des Brückenringes (BR) zu erkennen.
61 (M. Kasuno, K. Uchida, Y. Tatsumi et al. 2014)
23 Abbildung 21: Ringe der Struktur 1A
In Tabelle 4 sind den charakteristischen (nummerierten) Banden aus Abbildung 20 die jeweiligen Schwingungen zugeordnet.
Tabelle 4: Charakteristische Banden und Schwingungen (Struktur 1A)
Nr. Wellenzahl
[cm-1] Intensität Beschreibung
1 242 70 Wippschwingung der OH-Gruppe
2 813 56 Wippschwingung aller aromatischen Protonen
3 1121 159 Symmetrische Valenzschwingung C-C Einfachbindungen und C-S Bindungen in DAE1, DAE2 und BR
4 1251 246 Symmetrische Valenzschwingung der C-C-Bindungen im BR
5 1330 249 Symmetrische C-N Schwingung
6 1492 332 Symmetrische C-N Schwingung
7 1603 270 Symmetrische C-C Schwingung MG1 und MG2
8 2855 99 Symmetrische Valenzschwingung Protonen der Methylgruppen an den Diaminogruppen
9 3660 30 OH-Valenzschwingung
Besonders intensive Banden entstehen bei Schwingungen unter Beteiligung von Heteroatomen, besonders der Stickstoffe an MG1 und MG2. Im höheren Wellenzahlenbereich liegen charakteristische Signale der Methylgruppen sowie der OH-Gruppe.
24 7.2. Infrarot-Spektrum der Struktur 1B
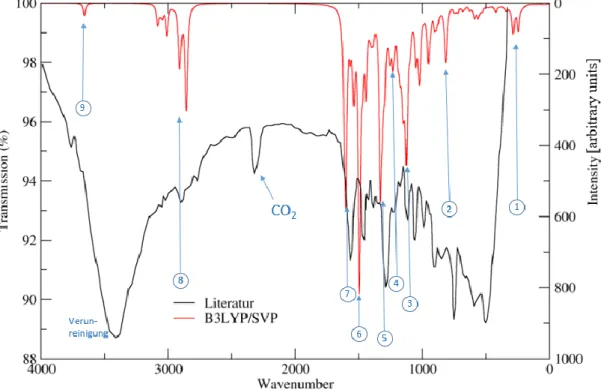
In Abbildung 22 sind für Struktur 1B berechnetes und experimentelles62 Spektrum dargestellt.
Das experimentelle Spektrum zeigt erhebliche Untergrundsignale und scheint stark verunreinigt zu sein.
Abbildung 22: Infrarot Spektrum (Struktur 1B)
Der Ringschluss hat nur geringen Einfluss auf die Lage der Banden. Stärker wird deren Intensität verändert, wobei besonders Banden Nr.3 und 4 in ihrer Intensität verändert werden.
Die Intensität von Bande Nr.3 nimmt ggü. Struktur 1A zu, die Intensität von Nr.4 nimmt ab.
Tabelle 5: Charakteristische Banden und Schwingungen (Struktur 1B) Nr. Wellenzahl
[cm-1] Intensität Beschreibung
1 245 61 Wippschwingung der OH-Gruppe
2 814 61 Wippschwingung aller aromatischen Protonen
3 1125 266
Symmetrische Valenzschwingung C-C Einfachbindungen und C-S Bindungen in DAE1, DAE2 und BR
4 1248 56 Symmetrische Valenzschwingung der C-C-Bindungen im BR 5 1330 263 Symmetrische C-N Valenzschwingung
6 1492 316 Symmetrische C-N Valenzschwingung
7 1603 281 Symmetrische C-C Valenzschwingung MG1 und MG2
8 2856 97
Symmetrische Valenzschwingung Protonen der Methylgruppen an den Diaminogruppen
9 3660 35 OH-Valenzschwingung
62 (M. Kasuno, K. Uchida, Y. Tatsumi et al. 2014)
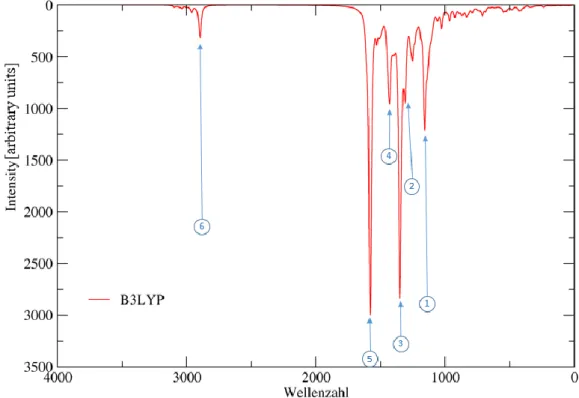
25 7.3. Infrarot-Spektrum der Struktur 1C
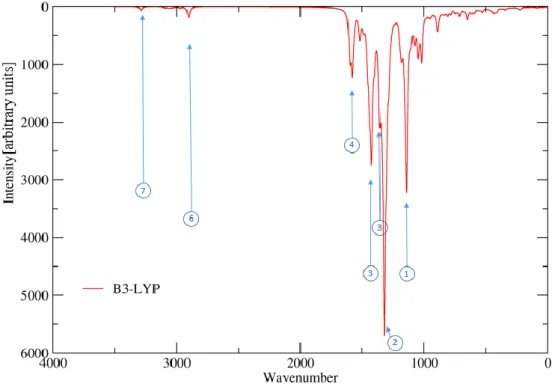
Erwartungsgemäß fehlen im Spektrum für 1C gegenüber den Spektren von 1A und 1B Signale, die auf Schwingungen der OH-Gruppe zurückzuführen sind. Im Vergleich zu den Spektren von 1A und 1B nehmen die Intensitäten der Banden im aromatischen Bereich deutlich zu.
Abbildung 23: IR-Spektrum der Struktur 1C (geänderte Skalierung)
Das Signal der Methylgruppen an den Diaminogruppen ist gegenüber Struktur 1A und 1B um ca. 40 cm-1 blau verschoben, die intensivsten Banden gehören zu Valenzschwingungen in den aromatischen Systemen des Malachitgrüns, besonders zu den Valenzschwingungen der Stickstoffe.
Tabelle 6: Charakteristische Banden und Schwingungen (Struktur 1C) Nr. Wellenzahl
[cm-1] Intensität Beschreibung
1 1157 566 Wippschwingung Protonen MG1 und MG2
2 1309 426 Valenzschwingung C1 zu MG1
3 1350 1902 Valenzschwingung C-C und C-N Bindung MG1 und MG2
4 1427 573
Scherschwingung Protonen der Methylgruppen an den Diaminogruppen
5 1583 1694 Valenzschwingung C-C und C-N Bindung MG2 und MG3
6 2898 160
Symmetrische Valenzschwingung Protonen der Methylgruppen an den Diaminogruppen
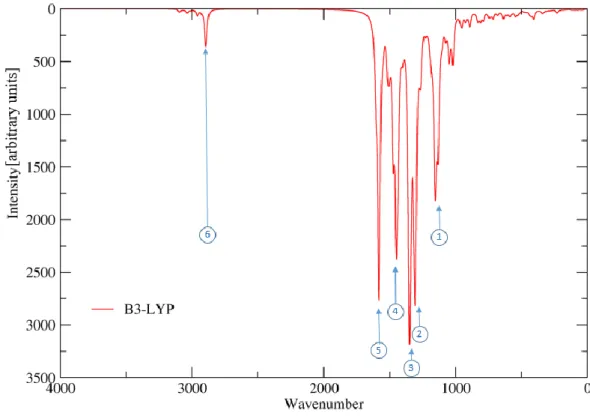
26 7.4. Infrarot-Spektrum der Struktur 1D
Die Lage der einzelnen Signale der Struktur 1D ähnelt denen der Struktur 1C, es bestehen jedoch Unterschiede hinsichtlich deren Intensität. Signal Nr.1, Nr.2 und Nr.4 sind deutlich intensiver.
Abbildung 24: IR-Spektrum der Struktur 1D
Signal Nr.4 gehört zur C-C Valenzschwingung im Aromaten MG1, es überlagert hier die im Spektrum von Struktur 1C noch erkennbaren Signale der Methylgruppen.
Tabelle 7: Charakteristische Banden und Schwingungen (Struktur 1D) Nr. Wellenzahl
[cm-1] Intensität Beschreibung
1 1155 466 Wippschwingung Protonen MG1 und MG2
2 1306 1931 Valenzschwingung C1 zu MG1
3 1350 1503
Symmetrische Valenzschwingung der C-C und C-N Bindungen MG1 und MG2
4 1452 1366 Symmetrische C-C Valenzschwingung MG1
5 1583 1674
Symmetrische Valenzschwingung C-C und C-N Bindung MG2 und MG3
6 2896 165
Symmetrische Valenzschwingung Protonen der Methylgruppen an den Diaminogruppen
27 7.5.Infrarot-Spektrum der Struktur 1E
Das N-H gebundene Proton gibt ein Signal bei ca. 3300 cm-1. Eine Veränderung gegenüber 1C/1D lässt sich im IR-Spektrum an Signal Nr.1 erkennen, das sich der C-N Valenzschwingung zuordnen lässt. Es liegt mit ca. 1160 cm-1 bei einer deutlich niedrigeren Wellenzahl als die C- N-Valenzschwingung der anderen Diaminogruppe (Signal Nr.3, ca. 1360 cm-1).
Abbildung 25: IR-Spektrum der Struktur 1E
Signale, denen Valenzschwingungen im Bereich der Malachitgrüngruppe zugrunde liegen, geben analog zu 1C und 1D sehr intensive Banden.
Tabelle 8: Charakteristische Banden und Schwingungen (Struktur 1E) Nr. Wellenzahl
[cm-1] Intensität Beschreibung
1 1159 704 C-N Valenzschwingung MG2
2 1253 358 Asymmetrische Valenzschwingung MG2 und MG3
3 1359 2493 C-N Valenzschwingung MG3
4 1405 684 Symmetrische C-C Valenzschwingung DAE1
5 1570 1170
Symmetrische C-C Valenzschwingung MG1,MG2 und MG3
6 2910 84
Symmetrische Valenzschwingung Protonen der Methylgruppen an den Diaminogruppen
7 3286 49 Valenzschwingung Proton MG2