Identification of in vivo interactions of insulin receptor substrate 1 in murine liver
132
0
0
Volltext
(3)
(4)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
Abbildung

+7
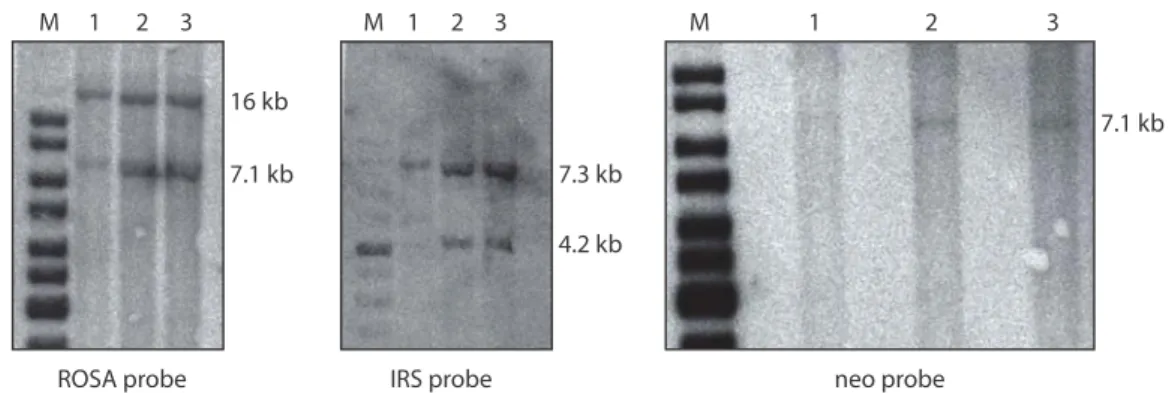
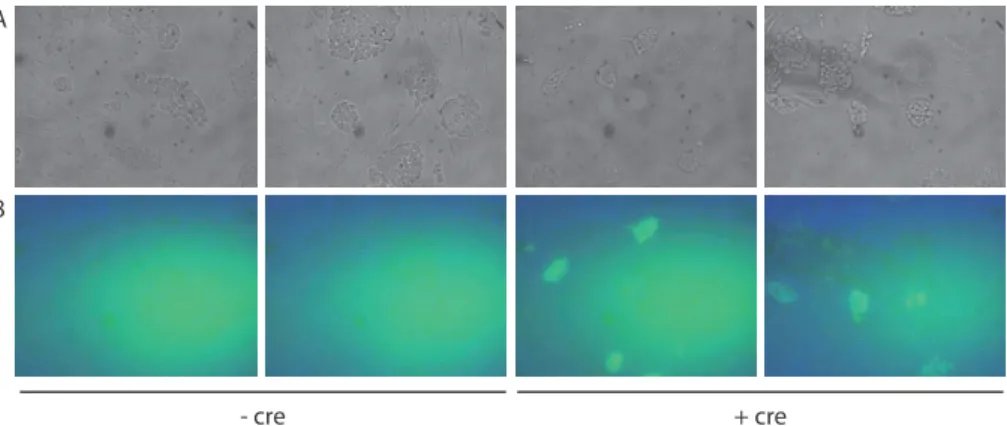
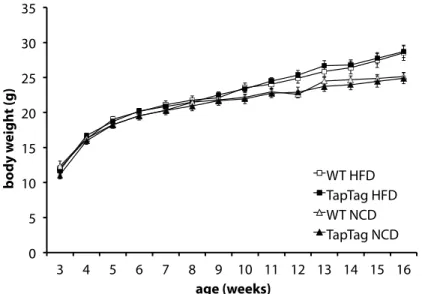
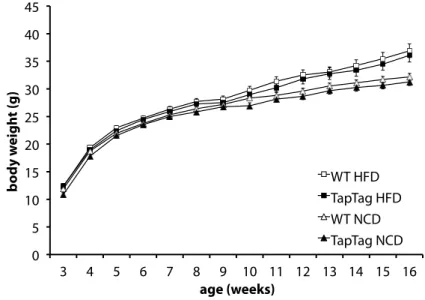
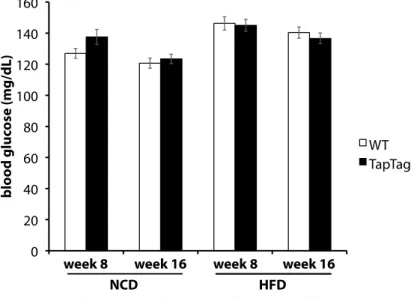
ÄHNLICHE DOKUMENTE
Activation of the JNK Pathway Is Implicated in IRS-1 mRNA Expression—To study the involvement of JNK in the synthesis of IRS-1 mRNA, FAK ⫺/⫺ cells were treated with anisomycin
Der zerebralen Ischämie im Allgemeinen konnten verschiedene ätiopathogenetische Grundlagen zugeordnet werden. Eine grobe Einteilung lässt sich durch die Differenzierung in
Insulin-like growth factor (IGF) binding protein-2, independently of IGF-1, induces GLUT-4 translocation and glucose uptake in 3T3-L1 adipocytes.. OXID MED CELL
Membranes prepared from insect cells expressed the 5-HT 7(a) receptor wild type and different C-terminal cysteine mutants were incubated with [ 35 S]-GTPγS in the presence or
Figure 36 Severe alteration of different signalling pathways in the hippocampus of IRSp53 haploinsufficient and deficient mice following NMDA receptor dependent LTP induction..
Die Immunzytochemie von PIST und Giantin, einem integralen Membranprotein des cis- und medialen Golgi-Apparates, zeigt, dass die Proteine nah bei einander lo- kalisiert sind
Dieses geschieht dadurch, daß nach .Sub- traktion der unspezifischen Bindung (hier nach Sub- traktion des bei der höchsten totalen Insulinkonzen- tration von 409 / Testansatz
As there was a difference observed between the percentage of 1C5-positive cells in the CD4 single-positive and the CD8 single-positive thymocytes it became important to know not
