Research Collection
Educational Material
Pharmazeutische Chemie Sommersemester 2001
Author(s):
Bader, Reto; Lerch, Mirjam; Zerbe, Oliver Publication Date:
2001
Permanent Link:
https://doi.org/10.3929/ethz-a-004337686
Rights / License:
In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use.
P ROTEINSTRUKTUREN UND NMR S PEKTROSKOPIE Reto Bader
Mirjam Lerch Dr.Oliver Zerbe
Department Angewandte Biowissenschaften Institut für Pharmazeutische Wissenschaften
Sommersemester 2001
1 Peptid und Protein Strukturen ... 2
1.1 Einleitung ... 2
1.2 Die Aminosäuren:... 2
1.3 Gliederung der Protein Strukturen: ... 3
1.4 Gefaltete vs. denaturierte Proteine: ... 4
1.5 Die Sekundärstruktur:... 4
1.6 Helices: ... 6
1.7 b-Blätter:... 8
1.8 Reverse Turns:... 9
1.9 Seitenkettenkonformationen:... 11
1.10 Supersekundärstrukturen: ... 12
1.11 Kräfte zwischen Atomen in Proteinen: ... 13
1.12 Elektrostatische Wechselwirkungen: ... 15
1.13 Wechselwirkungen mit Dipolen:... 15
1.14 Wasserstoffbrücken: ... 17
1.15 Methoden zur Strukturbestimmung:... 18
1.16 Dynamik von Proteinen:... 21
1.17 Informationen über Proteine auf dem Web (aus Branden and Tooze):... 22
1.18 Literatur: ... 22
2 Einleitung zur Strukturbestimmung mit NMR ... 23
3 Sample-Vorbereitung ... 23
4 Spektren-Aufnahme ... 25
4.1 [1H,1H]-COSY:[1H,1H]-Correlated Spectroscopy ... 28
4.2 [1H,1H]-TOCSY: Total Correlation Spectroscopy ... 31
4.3 [1H,1H]-NOESY: Nuclear Overhauser Effect Spectroscopy ... 32
5 Strategie zur Resonanzfrequenz-Zuordnung für kleine, nicht-markierte Proteine ... 35
5.1 Beurteilung der Spektrenqualität... 35
5.2 Spinsystem-Identifikation: NH-aH-bH-... ... 35
5.3 Sequenz-spezifische Zuordnung... 38
6 Strukturrechnung mit NMR-Daten ... 40
6.1 Chemische Shifts ... 40
6.2 3JHN-aH-Kopplungskonstanten ... 40
7 Qualtitätsbeurteilung der berechneten Struktur ... 47
8 Literatur ... 49
9 Anhang: Bezeichnung der Protonen der 20 natürlichen Aminosäuren ... 49
1. P
EPTIDUNDP
ROTEINS
TRUKTUREN1.1 Einleitung:
Die drei-dimensionale Struktur von Peptiden und Proteinen ist sehr eng mit ihrer biolo- gischen Aktivität verknüpft. Um den Wirkmechanismus von Enzymen zu verstehen ist eine genaue Kenntnis der 3D Anordnung der Atome der Polypeptidkette nötig. Ferner ist im Bereich der Molekularen Modellierung von Wirkstoffen strukturelle Information unentbehrlich. Leider ist es bisher noch nicht gelungen, die Primärsequenz von Protein- en in die 3D Struktur zu übersetzen (das sogenannte Faltungsproblem). Das erscheint auf den ersten Blick erstaunlich, sind doch die Kräfte, die zwischen den Atomen wirken, seit langem bekannt und berechenbar. Wenn man aber bedenkt, dass gefaltete Proteine nur um 5-10 kcal pro mol gegenüber dem ungefalteten Zustand stabilisiert sind, ein En- ergiebetrag, der ungefähr einer einzigen Wasserstoffbrücke entspricht, wird klar, dass es einer enormen Präzession bedarf, um die relative Stabilität von Konformeren abzus- chätzen. Die geringe Stabilisierung des gefalteten gegenüber dem ungefalteten Zustand hat im wesentlichen 3 Gründe: a) die Funktion der Protein erfordert eine Flexibilität (z.B. für induced-fit Wechselwirkungen). b) es sollten während der Proteinfaltung keine falsch-gefalteten Intermediate mit niedriger Energie entstehen können. c) Eine Entfal- tung des Proteins ist für die Translokation durch die Membran nötig. Die erfolgreichsten Strukturvorhersagen sind derzeit dann möglich, wenn eine hohe Sequenzhomologie (>40%) zu einem bekannten Protein vorliegt. Dazu muss aber auch aber auch das Hy- drophobizitätsprofil, d.h. die Abfolge von hydrophilen und hydrophoben Aminosäuren, sehr ähnlich sein.
1.2 Die Aminosäuren:
Es gibt 20 proteinogene Aminosäuren, die in von Eukaryonten exprimierten Proteinen vorkommen. Diese unterscheiden sich in der Seitenkette. Die Aminosäuren werden auf-
TABELLE 1.
AS Hydropath AS Hydropath AS Hydropath AS Hydropath
Ile 4.5 Met 1.9 Trp -0.9 Gln -3.5
Val 4.2 Ala 1.8 Tyr -1.3 Asp -3.5
Leu 3.8 Gly -0.4 Pro -1.6 Asn -3.5
Phe 2.8 Thr -0.7 His -3.2 Lys -3.9
Cys 2.5 Ser -0.8 Glu -3.5 Arg -4.5
grund der Hydrophilie/Lipophilie ihrer Seitenketten grob klassifiziert. Es ist in diesem Zusammenhang aber von Bedeutung zu erkennen, das die Hydrophilie einer Seitenkette stark von ihrem Ladungszustand abhängt, wobei die ionisierten Seitenketten sehr viel hydrophiler sind. Peptide/Proteine am pH in der Nähe ihres pI sind daher am schlechtes- ten löslich. Eine Übersicht über die kovalente Struktur und den pK der proteinogenen Aminosäuren befindet sich im Anhang.1
Aminosäuren sind chiral am Cα. Der Cahn-Ingold-Prelog Nomenklatur folgend liegen alle proteinogenen Aminosäuren in der (S) Form vor, mit Ausnahme von Cys (R). Gly und Pro haben eine Sonderstellung. Gly hat keine Seitenkette (2 α-H Atome) und Pro kein Amidproton (ringförmige Seitenkette). Somit ist für Gly der Bereich erlaubter Seitenkettenkonformationen größer und für Pro ist deren Bereich sehr viel eingeschränkter als bei den anderen Resten.
Die Peptidbindung hat einen partiellen Doppelbindungscharakter, weswegen der Dihe- dralwinkel um die Amidbindung 0 oder 180° ist. Für alle Peptidbindungen Xxx-Aaa mit Aaa ungleich Pro ist die trans Konformation sehr viel günstiger (weniger sterische Prob- leme). In unstrukturierten Peptiden wird für die Xxx-Pro Amidbindung dagegen häufig ein gewisser Anteil cis (bis zu 30%) beobachtet. In globulär gefalteten Proteine kommen dagegen durchaus, wenn auch relativ selten, cis Amidbindungen vor.
1.3 Gliederung der Protein Strukturen:
Man unterscheidet bei der Beschreibung der Struktur die
• Primärstruktur, d.h. die Abfolge der Aminosäuren
• die Sekundärstruktur, d.h. die Nahordnung der Polypeptidkette (z.B. Helix, β-Blatt)
• die Tertiärstruktur, die beschreibt, wie die Sekundärstrukturelemente zueinander angeordnet sind und damit einzelne Domänen bilden. Eine Domäne ist ein Teil der Proteinkette, der sich unabhängig vom Rest des Proteins faltet (d.h. eine wohl-defin- ierte Struktur einnimmt). Häufig kodieren Exons für Domänen von Proteinen. Eine Domäne ist auch häufig von der Funktion autark, d.h. eine biologische Funktion wird ausschließlich von einer bestimmten Domäne ausgeübt während eine andere z.B. für die Bindung an einem bestimmten Rezeptor verantwortlich ist.
• Häufig sind Proteine als Multimere aufgebaut. Die Packung der Monomereinheiten zueinander wird durch die Quartärstruktur beschrieben.
1. Die pK Werte der einzelnen Aminosäuren hängen stark von der Umgebung im Protein ab (sog.
pK Shifts)
1.4 Gefaltete vs. denaturierte Proteine:
Korrekt gefaltete Proteine zeichnen sich dadurch aus, dass die Polypeptidkette eine wohl-definierte Struktur einnimmt. Speziell die Backbone Dihedralwinkel regulärer Sekundärstrukturelemente nehmen ausgezeichnete Werte ein. Korrekt gefaltete Pro- teine zeichnen sich in der Regel durch einen relativ scharf definierten Temperaturbere- ich aus, im dem die Struktur verloren geht. Dies lässt sich relativ gut im CD Spektrum verfolgen. Einen Übergang zwischen völlig ungefalteten Polypeptiden und globulären Proteinen bilden die sogenannten “Molten Globules”. Die “Molten Globules” werden als Zwischenstufe bei der Proteinfaltung interpretiert. Sie zeichen sich durch ein Nebe- neinander von gefalteten und ungefalteten Bereichen aus. “Molten Globules” lassen sich aus globulären Proteinen durch Zugabe von denaturierenden Stoffen teilweise erzeugen.
Die globulären Proteine zeichen sich durch eine Reihe von Eigenschaften aus:
• die hydrophoben Reste befinden sich meist im Inneren (dem sogenannten “Core”)
• hydrophile Reste sind eher an der Oberfläche anzutreffen
• die Seitenketten im Core sind sehr dicht gepackt
• φ,ψ Dihedralwinkel sind in den Ramachandran-erlaubten Bereichen.
• Der hydrodynamische Radius ist kleiner als bei ungefalteten Proteinen gleicher Größe
Wird ein globuläres Protein durch Zugabe von Guanidiniumhydrochlorid oder durch Erhitzen denaturiert, so werden hydrophobe Reste dem Wasser exponiert, weswegen denaturierte Proteine in reinem Wasser meist unlöslich sind. Proteine falten sich in Abwesenheit anderer Proteine. Kürzlich wurden aber Hilfsmoleküle, sogenannte Chap- erone, entdeckt, die bei der Faltung helfen, indem sie helfen, dass falsch-gefaltete Inter- mediate sich wieder entfalten. Bislang gilt auch immer noch das Dogma, das es eine eindeutige Faltung gibt, d.h. das ein und das gleiche Protein in einer bestimmten Lösungsumgebung immer in einer einzigen (der gleichen) Konformation vorliegt.
1.5 Die Sekundärstruktur:
Die Sekundärstruktur wird durch die Faltung des Proteinrückgrades (Backbone), welch- es durch den Amidstickstoff, das C-alpha und den Carbonyl-Kohlenstoff gebildet wird, beschrieben.Es werden die folgenden Elemente in Proteinstrukturen angetroffen:
• Helices
• β-Stränge/Blätter
• Turns
daneben gibt es längere Loops, die nicht gut-definierte Backbone Konformationen be- sitzen und auch häufig flexibler sind. Disulfidbrücken, die zwischen Cystein Resten ge- bildet werden, stabilisieren Sekundärstrukturelemente, haben aber auch einen grossen Einfluss auf die Tertiärstruktur, da sie die Fernordnung beeinflussen.
Diese Sekundärstrukturen lassen sich über die entsprechenden Dihedralwinkel bes- chrieben. Sie sind nach IUPAC-IUB Regeln wie folgt definiert :
Die Sekundärstrukturelemente lassen sich über die entsprechenden φ,ψ Dihedralwinkel wie folgt definieren:
FIGUR 2. Definition der Backbone Diederwinkel nach IUPAC
φ ψ ω Anzahl
Reste/
Turn
Ausdehnung der Kette/
Rest (Å) antiparalle-
les β-Blatt
-139° 135° -178° 2.0 3.4
paralleles β- Blatt
-119° 113° 180° 2.0 3.2
TABELLE 3. Definitionen der Dihedralwinkel in Sekundärstrukturelementen ω
ψ φ χ
α α
α α
α β γ
i i i i i
i i i i i
i i i i i
i i i i i
for C C cis to N C for C N trans to C O for C C trans to N H for C N cis to C O
= − −
= − −
= − −
= − −
+ +
0 0 0
0
1 1
1
'
' '
1.6 Helices:
Die α-Helix hat 3.6 Reste per Helix Windung. Korrespondierende Atome benachbarter Helixwindungen sind 5.4 Å entfernt. Die α-Helix wird durch Wasserstoffbrücken, die zwischen dem Carbonylatom des Restes i und dem Amidproton des Restes i+4 gebildet wird, stabilisiert.
Die planare Anordnung der Amidbindung bedingt ein Dipolmoment. Da in der α-Helix alle Wasserstoffbrücken gleichgerichtet sind, addieren sich alle Dipolmoment einer ein- zelnen Helix, so dass das Gesamtdipolmoment für n Reste n*3.5 Debye beträgt. Dies entspricht in etwa 0.5-0.7 positiven bzw. negativen Ladungen am N-oder C-Terminus,
α-Helix -57° -47° 180° 3.6 1.5
310-Helix -49° -26° 180° 3.0 2.0
π-Helix -57° -70° 180° 4.4 1.15
PolyprolineI -83° 158° 0° 3.3 1.9
Polyproli- neII
-78° 149° 180° 3.0 3.12
Polyproli- neIII
-80° 150° 180° 3.0 3.1
FIGUR 4. Fig: Links: Schematische Darstellung der Helix. Mitte: Backbone Darstellung mit Angabe der Richtung des Dipolmomentes. Rechts: Struktur des 434 Repressors.
φ ψ ω Anzahl
Reste/
Turn
Ausdehnung der Kette/
Rest (Å)
TABELLE 3. Definitionen der Dihedralwinkel in Sekundärstrukturelementen
respektive. Deshalb sind negative geladene Aminosäure häufig vor einer Helix und pos- itiv geladene häufig hinter einer Helix anzutreffen. Da der Ladungszustand der Ami- nosäuren vom pH abhängt, ist die Stabilität der Helix mit dem pH korreliert.
Normalerweise ist der Helixdrehsinn rechtsgängig. Viele Helices sind amphiphatisch, d.h. die Reste sind derart angeordnet, dass die Seitenketten der polaren und unpolaren Gruppen zu verschiedenen Seiten der Helix zeigen. Die verschiedenen Aminosäuren ha- ben unterschiedliche Neigungen, bestimmte Sekundärstrukturelemente auszubilden.
Dies wurde statistisch zuerst von Chou und Fasman untersucht (“Chou-Fasman Regeln”). Für α-Helices sind folgende Werte kürzlich bestimmt worden:
Es gibt noch zwei weitere Formen der Helix: die 310- Helix und die π-Helix. Die 310-Helix hat 3 Reste pro Windung, die π-Helix 4.4.
Wasserstoffbrücken werden in diesen beiden Formen zwischen Carbonyl-sauerst- offen der Reste i und Amid- protonen der Reste i+3 bzw.
i+5 gebildet. Damit ist der Durchmesser der 310-Helix kleiner und der der π-Helix größer als in der α-Helix AS
Relative Stabilisierung
(kcal/mol)
AS
Relative Stabilisierun g (kcal/mol)
AS
Relative Stabilisierung
(kcal/mol)
Ala -0.77 Ser -0.35 Val -0.14
Arg -0.68 Gln -0.33 Thr -0.11
Lys -0.65 Glu -0.27 Asn -0.07
Leu -0.62 Cys -0.23 His -0.06
Met -0.50 Ile -0.23s Gly 0
Trp -0.45 Tyr -0.17 Pro 3
Phe -0.41 Asp -0.15
TABELLE 5. Relative Stabilisierungsenergien der einzelnen Aminosäuren in Helices
FIGUR 6. Oben VerschiedeneWasserstoffbrückenpositionen.
Unten: Abhängigkeit des Durchmessers vom Helix Typ.
Die ersten 3 Amidprotonen bzw. die letzten 3 Carbo- nyl Sauerstoffe einer Helix haben keine H-Brücken Partner. Man findet deshalb in Helices häufig soge- nannte N- bzw- C- Capping Reste. Diese bilden über ihre Seitenkettenfunktionen (z.B. über die β-Hydrox- ylgruppe von Ser) Wasserstoffbrücken zu den entspre- chenden Atomen und stabilisieren so die Helix. Beim Proteindesign hilft das Einfügung von geeigneten C- bzw. N-Caps, die Helix zu stabilisieren.
Wie bereits erwähnt, hat die Helix ein Dipolmoment, das am N-Terminus in etwa einer halben positiven und am C-Terminus einer halben negativen Ladung entspricht. Das Einfügen geladener Aminosäuren am C- oder N- Cap hilft ebenfalls, die Helix zu stabilisier- en.
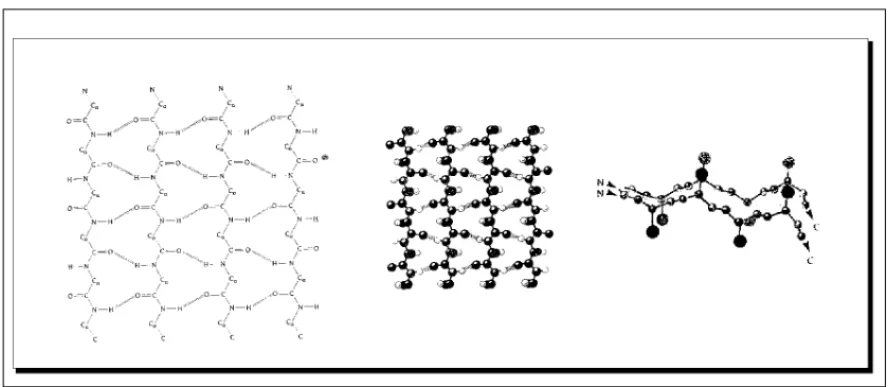
1.7 β-Blätter:
Grundlegende Einheit der β-Blätter ist der β-Strang, der eine gestreckte Konformation der Polypeptidkette darstellt. Der isolierte β-Strang ist nicht stabil, wird aber durch die Aneinanderreihung benachbarter β-Stränge zum sogenannten β-Faltblatt stabilisiert.
Dabei gibt es zwei verschiedene Anordnungen der β-Stränge zueinander:
• die Kopf-Schwanz Anordnung, die zum antiparallelen Faltblatt führt:
FIGUR 8. Fig: Links: Antiparalleles Faltblatt. Mitte: Ball-and-Stick Darstellung. Rechts:
Seitenansicht.
FIGUR 7. Helix N-Cap. Das OH von Ser bildet H-Brücke .
• die Kopf-Kopf Anordnung, die zum parallelen Faltblatt führt:
In β-Faltblättern sind die Seitenketten abwechselnd oberhalb und unterhalb der Ebene des Faltblattes angeordnet. Dabei kommen sich die Seitenketten gegenüberliegender Stränge relativ nahe. Die meisten der in natürlichen Polypeptiden beobachteten β-Falt- blätter sind nicht vollständig planar sondern weisen eine rechtsgängige Verdrillung auf.
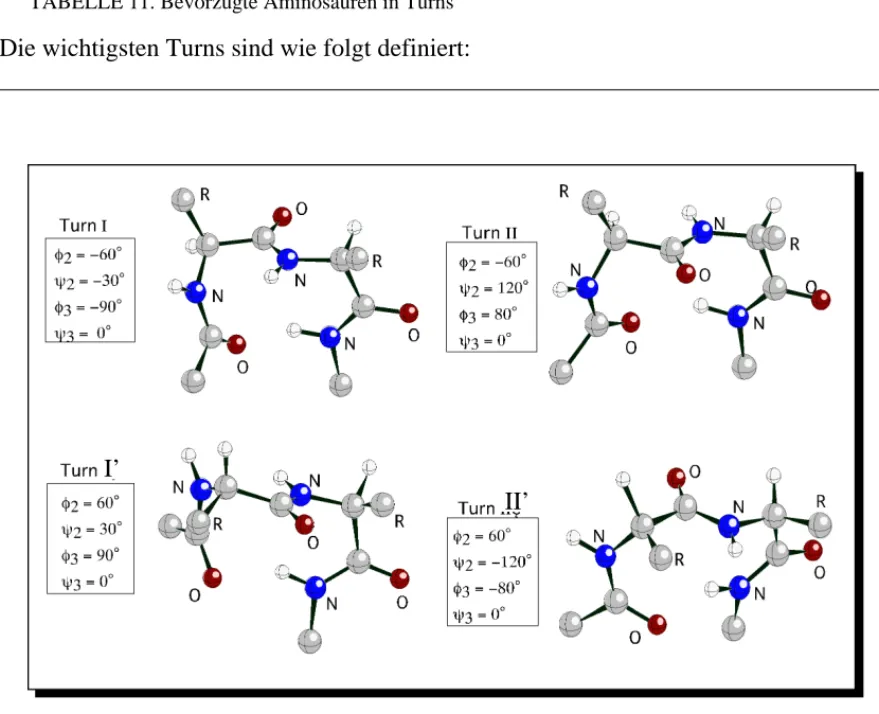
1.8 Reverse Turns:
Reverse Turns spielen eine wichtige Rolle in Protein- strukturen. Sie bewirken, dass die Polypeptidkette ihre Richtung ändert und sind deshalb bevorzugt an der Oberfläche von Proteinen anzutreffen. Turns werden ebenfalls über Wasserstoffbrücken stabilisiert.In soge- nannten γ-turns werden H-Brücken zwischen dem Carbonylsauerstoff des Restes i und dem Amidproton des Restes i+2 gebildet.
β-Turns zeichen sich durch Wasserstoffbrücken zwis- chen dem Carbonylsauerstoff des Restes i und dem Amidproton des Restes i+3 aus. Es gibt eine Vielzahl verschiedener β-Turns, von denen diejenigen vom Typ I und II am häufigsten angetroffen werden.
Eine wichtige Eigneschaft von Turns ist, dass immer wieder ähnliche Reste an bestimmten Positionen der Turns angetroffen werden. Gly ist besonders deshalb
FIGUR 9. Fig: Links:Paralleles Faltblatt. Mitte: Ball-and-Stick Darstellung. Rechts: Seitenansicht.
FIGUR 10. :Oben: γ-Turn.
Unten: inverser γ-Turn.
prädestiniert, weil es wegen der fehlenden Seitenkette ungewöhnliche φ,ψ Kombina- tionen einnehmen kann. Ebenfalls häufig angetroffen wird Pro oder Asn
Die wichtigsten Turns sind wie folgt definiert:
Die oben bereits erwähnten Chou Fasman Regeln erlauben eine Abschätzung der Wahr- scheinlichkeiten, dass eine bestimmte Sequenz eine der Sekundärstrukturen einnimmt.
Folgende Kriterien gelten als Hinweise auf helikale Strukturen:
• innerhalb eines Hexapeptides 4 Reste mit P(Helix)>1.12
• der Durchschnitt für alle Reste <P(Helix)> 1.03
• <P(α)> > <P(β)>
• und für β-Blätter
• Pentapeptid mit mindestens drei Reste mit P(b-Strang) >1.19
Position Typ I Typ I’ Typ II Typ II’
i Asp,Asn,Ser,Cy s
Asp,Asn,Ser,Cy s
Asp,Asn,Ser,Cy s
Asp,Asn,Ser,Cy s
i+1 Pro Gly Pro Gly
i+2 kein Pro Gly Gly, Asn
i+3 Gly Gly
TABELLE 11. Bevorzugte Aminosäuren in Turns
FIGUR 12. : Turns mit den dazugehörigen Backbonedihedralwinkeln.
II’
I’
• der Durchschnitt für alle Reste <P(β-Blatt)> 1.05
• <P(β)> > <P(α)>
Das betreffende Sekundärelement läuft um jeweils umn einen Rest weiter, solange der Durchschnitt der Wahrscheinlichkeit des terminalen Tetrapeptides nicht unter 1.0 fällt. :
1.9 Seitenkettenkonformationen:
Die Seitenkettenkonformationen werden über die Dihedralwinkel χ1 (Cα-Cβ), χ2 (Cβ- Cγ) etc. beschrieben. Aus sterischen Gründen nehmen die Seitenketten bevorzugt ge- staffelte anstatt ekliptische Konformationen ein. Globuläre Proteine zeichen sich auch dadurch aus, dass die Seitenketten definierte Winkel einnehmen, obwohl es in den flex- ibleren Loops teilweise auch Übergänge zwischen verschiedenen Rotameren geben kann.
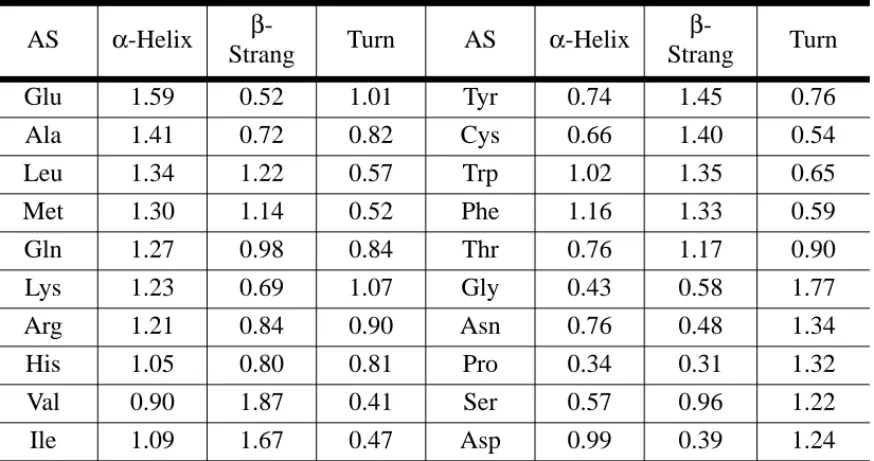
AS α-Helix β-
Strang Turn AS α-Helix β-
Strang Turn
Glu 1.59 0.52 1.01 Tyr 0.74 1.45 0.76
Ala 1.41 0.72 0.82 Cys 0.66 1.40 0.54
Leu 1.34 1.22 0.57 Trp 1.02 1.35 0.65
Met 1.30 1.14 0.52 Phe 1.16 1.33 0.59
Gln 1.27 0.98 0.84 Thr 0.76 1.17 0.90
Lys 1.23 0.69 1.07 Gly 0.43 0.58 1.77
Arg 1.21 0.84 0.90 Asn 0.76 0.48 1.34
His 1.05 0.80 0.81 Pro 0.34 0.31 1.32
Val 0.90 1.87 0.41 Ser 0.57 0.96 1.22
Ile 1.09 1.67 0.47 Asp 0.99 0.39 1.24
TABELLE 13. Propensitäten der einzelnen Aminosäuren für Sekundärstrukturelemente.
FIGUR 14. Darstellungen der Seitenkettenkonformationen. Links: Ekliptisch. Rechts: Gestaffelt.
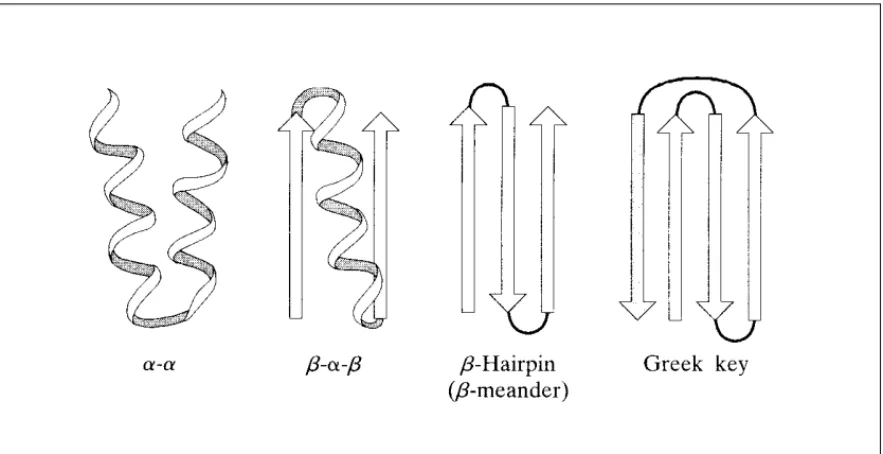
1.10 Supersekundärstrukturen:
Es gibt eine Anzahl von Anordnungen, in denen Sekundärstrukturelemente gegenein- ander gepackt sein können:
Ein bekanntes Motiv ist das Helix-Turn-Helix Motiv. Es wird z.B. bei den DNA bin- denden Proteinen (434 Repressor) oder bei Calcium-bindenden Proteinen (Calbindin) angetroffen. Wie der Name besagt, werden darin zwei Helices über einen Loop verknüpft.
Das β-Hairpin Motiv ist eine einfache Anordnung, um 2 antiparallele β-Stränge über einen kurzen Turn miteinander zu verknüpfen.Überhaupt besitzen Proteine die starke Tendenz, β-Stränge so miteinander zu verknüpfen, dass solche Stränge miteinander verknüpft werden, deren Aminosäuren in der Primärsequenz benachbart sind.
Im β−α−β Motiv werden 2 parallele β-Stränge über einen kurzen Loop, dann eine He- lix, und dann wieder einen kurzen Loop miteinander verknüpft.
Im Greek-Key Motiv werden 4 antiparallele β-Stränge in charakteristischer Art und Weise über Loops miteinander verknüpft. Dieses Motiv hat seinen Namen von den Verzierungen, die auf antiken griechischen Vasen gefunden worden.
FIGUR 15. Supersekundärstrukturelemente
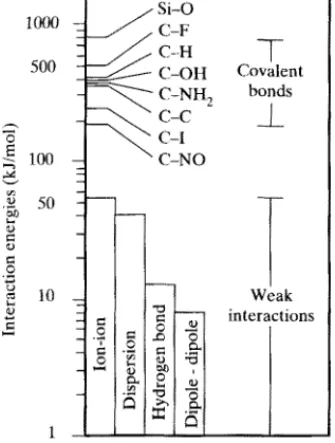
1.11 Kräfte zwischen Atomen in Proteinen:
Es gibt eine Reihe von interatomaren Kräften, die die Faltung des Proteins beein- flussen. Derartige Kräfte können anzie- hender oder abstossender Natur sein. Vom Mechanismus her können sie elektronischer Natur wie z.B. die Wechselwirkung zwis- chen geladenen Atomen oder sterischer Na- tur sein. Man unterscheidet die starken Wechselwirkungen, wie z.B. die kovalenten Bindungen sowie die schwachen Wechsel- wirkungen:
Die schwachen Wechselwirkungen haben eine unterschiedliche Abhängigkeit vom Abstand der wechselwirkenden Atome:
Die sterischen Wechselwirkungen treten immer dann auf, wenn der Abstand zweier Atome d kleiner als die Summe der van-der-Waals Radien (ra/2+rb/2) ist
Typ der Wechselwirkung Abstandsabhängigkeit
Ladung-Ladung 1/r
Ladung-Dipol 1/r2
perm. Dipol - perm. Dipol 1/r3
Ladung - induz. Dipol 1/r4
induz. Dipol - induz. Dipol 1/r6
TABELLE 17. Abstandsabhängigkeit der Wechselwirkungen
FIGUR 18.
FIGUR 16. Grösse wichtiger
Der van-der-Waals Radius für die am häufigsten angetroffenen Atome ist in der fol- genden Tabelle angegeben:
Atome, die van der Waals Anziehungskräfte erfahren, sind ungefähr 0.8 Å weiter vo- neinander entfernt, als wenn sie kovalent gebunden wären.
Die sterische Abstossung zwischen Atomen schränkt die Kombination der ψ,φ Bind- ungswinkel stark ein. Dies wurde von Ramachandran berechnet (“Hard-Sphere” Mod- el):
Wie aus dem Ramachandran Plot ersichtlich ist, sind nur bestimmte Kombinationen sterisch erlaubt. Zudem sind die verschiedenen Sekundärstrukturelemente auf bestim- mte Ramachandran Regionen beschränkt. Glycin Reste, die keine Seitenkette haben, können noch andere Kombinationen einnehmen.
Atom beobachteter Bereich Radius bei
Einfachbindungen
Wasserstoff 1.00-1.54 Å 1.17 Å
Sauerstoff 1.40-1.70 Å 1.40 Å
Stickstoff 1.55-1.60 Å 1.55 Å
Kohlenstoff 1.70-1.78 Å 1.75 Å
Schwefel 1.75-1.80 Å 1.80 Å
TABELLE 19.
FIGUR 20. Fig: Ramachandran Plot
1.12 Elektrostatische Wechselwirkungen:
Die elektrostatischen Wechselwirkungen sind stark und weitreichend. Sie werden nach dem Coulombschen Gesetz beschrieben durch:
In Lösung ist die Wechselwirkung um die Dielektrische Konstante D herabgesetzt:
Solche Wechselwirkung treten in Proteinen zwischen geladenen Gruppen auf. Ein Beispiel ist z.B. die anziehende Wechselwirkung zwischen einer Aspartat CO2- Gruppe und einer NH3+ Lysin Seitenkettenfunktion, die zu einer sogenannten Salzbrücke führt.
Bei der Berechnung der stabilisierenden Wirkung ist allerdings zu bedenken, dass solche Gruppen im ungefalteten Zustand solvatisiert vorliegen.
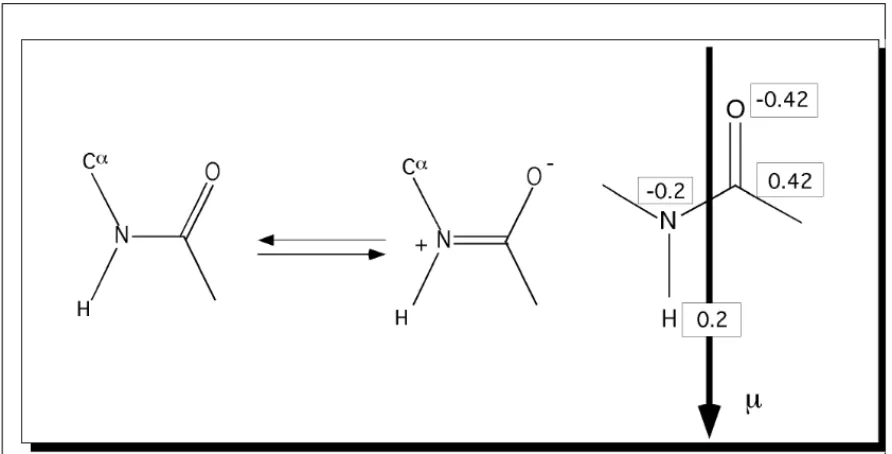
1.13 Wechselwirkungen mit Dipolen:
Die Amidbindung hat Doppelbindungscharakter. Dies ist der Grund, warum der Dihe- dralwinkel ω auf 0° (cis) oder 180° (trans) beschränkt ist. Zudem ist aus den mesomeren Grenzstrukturen erkenntlich, dass es ein Dipolmoment geben sollte:
Das Dipolmoment der Amidbindung ist 3.5 Debye (Wasser hat 1.85 zum Vergleich).
FIGUR 21. Fig: Links: Resonanzformel der Peptidbindung. Rechts: Partialladungen.
∆E ZAZB
ε
2rAB ---
=
∆E ZAZB
ε
2DrAB ---
=
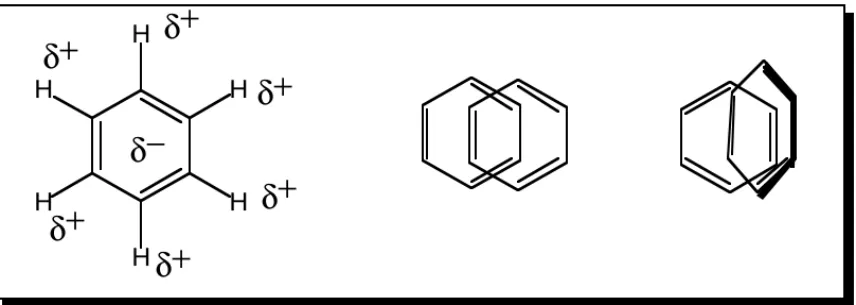
Aromatische Ringe haben ebenfalls ein Dipolmoment, weswegen sie zueinander bevor- zugte Konformationen einnehmen:
Dipolmomente können permanent oder induziert sein. Die Wechselwirkungsenergie zwischen permanenten Dipolen ist proportional r-3.
Die Wechselwirkungen mit induzierten Dipolmomenten werden van-der Waals Wech- selwirkungen genannt und sind proportional r-6. Bei hydrophoben Gruppen spielen die sogenannten London Dispersionskräfte eine wichtige Rolle.
Sie entstehen zwischen 2 Gruppen, die beide kein permanentes Dipolmoment ha- ben, und sind auf die zeitlich asymmetrische Ladungsverteilung der Elektronen zurück- zuführen. Van-der Waals Kräfte werden häufig in Form des Lennard-Jones Poten- tiales ausgedrückt:
Cn und C6 sind Konstanten, wobei der erste Term der Gleichung die Abstossung, die aus dem Verbot der Überlappung der Elek- tronenorbitale resultiert, beschreibt. Meist verwendet man für diese Term n=12. Der zweite Term ist anziehend und wird durch die induzierten Dipolmomente hervorg- erufen.
FIGUR 22. Partialladungen in aromatischen Systemen und deren relative Orientierung
H
H
H H
H
H
δ+
δ+
δ+ δ+
δ+
δ+
δ−
FIGUR 23. Lenard-Jones Potential
E d( ) Cn ---dn C6
---d6 –
=
1.14 Wasserstoffbrücken:
Wasserstoffbrücken spielen eine herausragende Rolle in der Aus- formung von Sekundärstruk- turelementen. Sie treten immer dann auf, wenn zwei elektroneg- ative Atome um die Bindung zu einem Wasserstoffatom konkur- rieren. α-Helices werden durch die Ausbildung von Wasserstoffbrücken zwischen Amidprotonen der Reste i und Carbonlysauerstoffen der Reste i+3 stabilisiert. β-Blätter weisen ebenfalls Wasserstoffbrücken zwischen Amidprotonen und Carbonylsauerstoffen benach- barter Stränge auf. In diesen Wasserstoffbrücken ist der O..H Abstand 1.9-2.0 Å. Normaler- weise ist das Proton kovalent an ein anderes Atom gebunden, den sogenannten Donor.
Der Abstand zum Akzeptor, mit dem es ebenfalls wechselwirkt, ist aber kleiner als die Summe der van der Waals Radien, was auf einen partiellen Bindungscharakter hindeu- tet. Je stärker die Wasserstoffbrücke, desto kleiner ist der Atomabstand zum Akzeptor.
In den stärksten H-Brücken, den symmetrischen H-Brücken, ist der Abstand zum Akzeptor gleich groß wie der Abstand zum Donor. Die Länge und damit die Stärke der Wasserstoffbrücken hängt von der relativen Elektronegativität von Donor und Akzeptor ab. Besonders starke H-Brücken werden zu geladenen Atomen gebildet. Sauerstoff- atome können zwei Wasserstoffbrücken gleichzeitig ausbilden. Wasserstoffbrücken stabilisieren das Protein um 2-10 kcal/mol pro gebildeter Brücke. Man muss allerdings bedenken, dass im denaturierten Protein meistens Wasserstoffbrücken zum Lösungsmittel Wasser ausgebildet werden, so dass der stabilisierende Einfluss auf die Proteinfaltung sehr viel geringer ist.
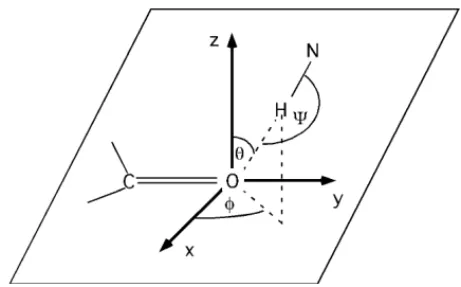
FIGUR 24. Fig.: Abstände in Wasserstoffbrücken.
Während der N-H...O Bindung- swinkel Ψ meist um 180° beträgt, bilden die Carbonylbindung und die Amidbindung nicht eine Achse. Die Winkel Φ und θ nehmen einen weit- eren Bereich ein, dessen häufigste Werte 60° bzw. 0° sind, respektive:
1.15 Methoden zur Strukturbestimmung:
Bei den Methoden zur Untersuchung der 3-dimensionalen Struktur von Proteinen unter- scheidet man zwischen solchen Methoden, die lediglich eine Abschätzung des Gehaltes an Sekundärstrukturen liefern und solchen, die das Studium der Struktur mit atomarer Auflösung erlauben. Zu den ersteren gehören die IR und vor allen Dingen die CD Spe- ktroskopie und zu den letzteren die NMR Spektroskopie und die Einkristall-Rönt- genbeugung (“X-Ray”).
Die Circulardichroismus (CD) Methode hilft zu erkennen, ob das Protein ungefaltet, eher helikal oder eher β-Blatt artig aufgebaut ist. Die Methode beruht darauf, dass die chromophoren Gruppen in Proteinen mit den beiden Ko- mponenten des zirkular polarisi- erten Licht unterschiedlich wechselwirken und daher unter- schiedlich absorbiert werden.
Dies ist immer genau dann der Fall, wenn optisch-aktive Verbin- dungen vorliegen. Diese Absorp- tion ist abhängig von der Wellenlänge. In regelmässigen Sekundärstrukturelementen kom- mt es wegen der definierten Anor-
FIGUR 25. Definition der Winkel in H-Brücke
FIGUR 26. Typische CD Kurven für Helices (α), β-Blatt
(β) und Zufallsknäul(r)
dnung der Chromophore zueinander zur gegenseitigen Wechselwirkung (“Exciton Splitting”), die das Ausmass der Absorption und deren Frequenz beeinflusst. CD Spek- tren brauchen wenig Material und sind schnell aufgenommen und interpretiert. Sie repräsentieren die Summe aller Sekundärelemente und erlauben keine direkte räumliche Auflösung. CD Spektren eignen sich sehr gut, um folgende Punkte schnell abzuklären:
• Ist das Protein überhaupt gefaltet?
• Wenn ja, was ist das dominierende Sekundärstrukturelement?
• Bei welcher Temperatur denaturiert das Protein und ist dieser Vorgang reversibel?
• Findet eine konformationelle Aenderung z.B. bei einer Ligandbindung statt?
Die CD Spektren der verschiedenen Sekundärstrukturelemente haben ein charakteristis- ches Aussehen. Im Prinzip lässt sich ein CD Spektrum mathematisch in die Komponent- en von α-Helix, β-Blatt und Random Coil zerlegen und so deren anteiligen Gehalt bestimmen (Deconvolution).
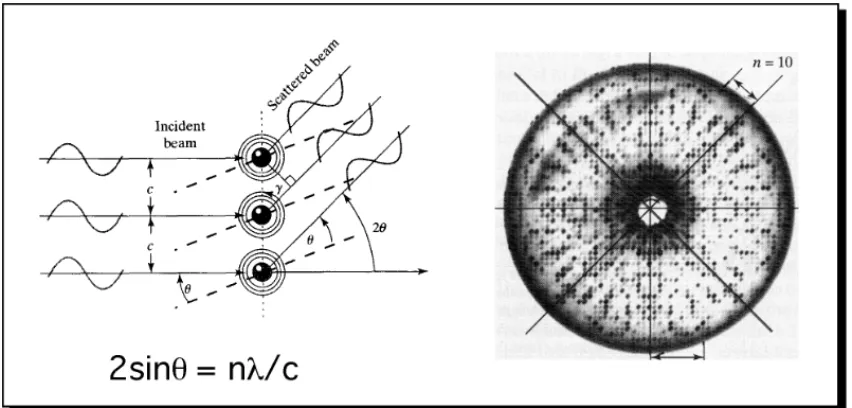
Die wichtigste Methode zur Bestimmung von Proteinen im Festkörper (neben der jetzt langsam an Bedeutung gewinnenden Cryo-Elektronenmikroskopie) ist die Rönt- genbeugung am Einkristall. Die (Schwer)atome des Kristallgitters beugen den (mono- chromatischen) Röntgenstrahl in charakteristischer Weise:
Zwischen den abgelenkten Strahlen kommt es zur konstruktiven und destruktiven Inter- ferenz, so dass am Ende ein charakteristisches Beugungsmuster entsteht. Mit dieser In-
FIGUR 27. Fig.: Links: Brechnung des Röntgenstrahls gemäss Braggs Gesetz. Rechts:Resultierendes Brechungsmuster.
formation kann man dann indirekt eine Rekonstruktion der dreidimensionalen Elektronendichte vornehmen. Der abgelenkte Strahl wird z.B. auf einer photographis- chen Platte detektiert, wobei leider die Phaseninformation des Strahls verloren geht.
Dies führt zu einer erheblichen Komplizierung der Interpretation der Daten. In der Pro- tein Kristallographie werden im wesentlichen zwei Methoden angewandt, um trotzdem die Elektronendichten berechnen zu können:
• bei homologen Strukturen wird das sogenannte Molecular Replacement benutzt, bei dem ein Model der Struktur, das aus einem Homologen erzeugt wurde, benutzt wird, um die Elektronendichteverteilung zu berechnen
• es wird eine Reihe von Schweratomderivaten erzeugt (Isomorphous Replacement)
• (Man arbeitet mit Synchrotonstrahlung verschiedener Wellenlänge (Multiwave- length Anomalous Diffraction))
Insbesondere die erstere Methode führt sehr schnell zu gut-aufgelösten Strukturen, lässt sich aber nur dann anwenden, wenn bereits die Struktur eines Proteins mit hoher Se- quenzhomologie bekannt ist. Die Mehrzahl der Proteinstrukturen, die in der pdB Daten- bank enthalten sind, sind Kristallstrukturdaten. Die X-Ray Methode misst die Struktur des Proteins im Festkörper. Obwohl ein Vergleich von NMR und X-Ray Daten in fast allen Fällen sehr gute Übereinstimmung geliefert hat, kann es an den Kontaktflächen der Moleküle zu sogenannten Packungsartefakten kommen, d.h. der Verlauf der Polypeptidkette ist dort durch Packungseffekte bestimmt. Das grösste Problem in der Röntgenkristallmethode ist es, einen Kristall ausreichender Güte, d.h. sehr hoher Ord- nung, zu erhalten. Im Prinzip werden solche Kristalle aus übersättigten Lösungen hoch aufgereinigter Proteine (>97%) gezüchtet. Ob und wie ein Protein kristallisiert ist aber immer noch sehr schlecht verstanden. Wenn sehr gute Kristalle erhalten wurden, lassen sich heute Auflösungen im Bereich von 1Å erhalten, die auch die Lokalisation von Wasserstoffatomen (sonst nur Schweratome) sowie von Wassermolekülen im Kristall erlaubt. Ein unbestrittener Vorteil der X-Ray Methode gegenüber der NMR Spektrosk- opie ist, dass sie auf Proteine beinahe beliebiger Grösse anwendbar ist, sofern sich diese kristallisieren lassen.
1.16 Dynamik von Proteinen:
Die Polypeptidkette in Proteinen ist nicht überall gleich gut definiert. Besonders an den C- und N- Termini der Kette wird in der Regel eine grosse Flexibilität beobachtet. Diese Flexibilität führt dazu, dass die Elektronendichten für diese Reste zu gering sind und diese sich daher in der Röntgenstrukturanalyse nicht interpretieren lassen (diese Reste bleiben dann “unsichtbar”). In der NMR Spektroskopie äussert sich die Flexibilität darin, dass das Strukturbündel, das aus den Strukturrechnungen resultiert, für diese Reste sehr breit aufgefächert ist (hoher RMSD). In den X-Ray Untersuchungen geben die sogenannten B-Faktoren einen Hinweis auf die thermische Beweglichkeit der ein- zelnen Atome, d.h. hohe B-Faktoren bedeuten, dass diese Atome schlechter definiert sind. Die NMR Spektroskopie erlaubt über Relaxationsmessungen (z.B. 15N Relax- ation) direkt das Ausmass der Beweglichkeit sowie, im Gegensatz zur X-Ray, auch die Zeitskala der relevanten Bewegungsvorgänge zu messen.
Die Dynamik der Proteine ist für ihre Funktion von essentieller Bedeutung. Katalytische Vorgänge gehen einher mit der Umgruppierung von an dem katalytischen Schritt beteiligten Atomen und erfordern daher eine gewisse Flexibilität. In der Regel sind längere Loops in Proteinen flexibel. Häufig wird auch eine Bewegungen von Domänen zueinander (sog. “Hinge-Motion”) beobachtet. Aromatische Ringe führen häufig sog.
Ringflips mit hoher Frequenz durch. Ferner diffundiert Wasser relativ schnell durch den Core von Proteinen.
Die Dynamik von Proteinen auf der Picosekunden/Nanosekunden Zeitskala lässt heutzutage relativ gut mit Moleküldynamik (MD) Methoden berechnen.
1.17 Informationen über Proteine auf dem Web (aus Branden and Tooze):
Es gibt eine Reihe von relevanten Adressen im Internet, um Struktur- und Sequenzin- formationen zu erhalten:
• die Homepage der Brookhaven Datenbank (pdb):
http://www.rcsb.org/pdb
• die Homepage des schweizer Institutes für Bioinformatik (ExPASy), die die autom- atische Sequenzdatenbank SWISSPROT enthält:
http://www.expasy.ch
• für Proteintopologien CATH
http://www.biochem.ucl.ac.uk/bsm/cath
• oder TOPS, mit dem sich nach Topologien suchen lässt http://www3.ebi.ac.uk/tops/
• oder FSSP:
http://www2.ebi.ac.uk/dali/fssp
• DALI vergleicht die Koordinaten mit bekannten Koordinaten, um Ähnlichkeiten mit bekannten Proteinen aufzudecken.
• für Sequenzalignments:
Pfram
http://www.sanger.ac.uk/Pfam oder
ProDom:
http://www.protein.toulouse.inra.fr/prodom.html
1.18 Literatur:
• Carl Branden, John Tooze: Introduction to Protein Structure, Garland Publisher 1999.
• Thomas E. Creighton, Proteins; Structure and Molecular Properties, Freeman, 1993.
• Georg E. Schultz, R.H. Schirmer, Principles of Protein Structure, Springer 1985.
• K.E. van Holde, W.C. Johnson, P.S. Ho, Physical Biochemistry, Prentice Hall 1998.
• Alan Fersht, Structure and Mechanism in Protein Science, Freeman 1999.
P EPTID NMR
2. E
INLEITUNGZURS
TRUKTURBESTIMMUNGMITNMR
NMR Spektroskopie ist die Methode zur Strukturaufklärung von Proteinen in Lösung.
Im Gegensatz zur Röntgeneinkristalldiffraktometrie erlaubt sie die Untersuchung der Struktur unter annähernd physiologischen Bedingungen. Leider ist die Routineanwendung der NMR Methode derzeit noch auf Proteine unter 30 kDa beschränkt.
Die Strukturaufklärung mittels NMR beruht im wesentlichen auf der Ausnutzung des
“Nuklear Overhauser Effektes” (NOE), der sich im NOESY zwischen Protonen, die weniger als 5 Å voneinander entfernt sind, beobachten lässt1:
(GL 1)
Mit Hilfe der Information über die Nachbarschaftsbeziehungen aller Protonen zueinandern lässt sich dann die Struktur berechnen. Die Strukturaufklärung ist in folgende Schritte unterteilt:
• Etablierung von geeigneten Messbedingungen (Probe ist monomer , stabil etc.)
• Messung der 2D oder 3D NMR Spektren
• Sequentielle Zuordnung aller Protonenfrequenzen (von jedem einzelnen Proton muss dessen Frequenz bekannt sein)
• Zuordnung der Kreuzpeaks im NOESY zu den Protonen in der Primärsequenz
• Integration des NOESY und Umsetzung der Intensitäten in Distanzein-schränkun- gen
• Bestimmung von Kopplungskonstanten für Diederwinkeleinschränkungen
• Strukturrechnung
• Refinement
• Ueberprüfung der Qualität der Struktur
3. S
AMPLE-V
ORBEREITUNGGute Messbedingungen sind Voraussetzung für qualitativ gute Spektren, ohne die eine gute Auflösung der Struktur nicht zu erreichen ist, da diese mit Hilfe dieser Spektren gerechnet wird. Folgende Kriterien sollten bei der Probenvorbereitung beachtet werden:
1. unter τc versteht man die Korrelationszeit, d.h. die Zeit, die das Molekül für eine 360° Drehung benötigt
NOE∼r–6⋅ f( )τc
1. Das Protein muss in nativer und funktioneller Konformation vorliegen:
• Globuläre Faltung (nicht nur kurze Peptide sind oft sehr flexibel, nur eine Subpopu- lation ist gefaltet, CD checken!)
• geeigneten pH wählen (möglichst physiologisch, falsche Ladungszustände können zu Aggregation führen, aber pH Abhängigkeit des H,D Austausch für Amidprotonen beachten! (vergl. 6.))
• Temperatur variieren, bis Qualität und Signaldispersion optimal ist (oft: schärfere Linien bei erhöhter Temperatur)
• Ionenstärke beeinflusst das Aggregationsverhalten (Hofmeister Serie)
• Screening geeigneter Bedingungen z.B. mit CD-Spektroskopie
• Löslichkeit muss beim pH nach Wahl hoch genug sein (pI!). Die Konzentration sollte mindestens 1mM betragen (in 0.5ml). Bei halber Konzentration muss doppelt solange gemessen werden, um das gleiche Signal/Rausch-Verhältnis zu erhalten!
Evtl. Zusatz von Detergentien/Salz.
2. Keine Kontaminationen, genügend hohe Reinheit des Proteins (>95%, genügende Anzahl Reinigungsschritte, Check via HPLC/MS/SDS-PAGE etc.).
3. In Abhängigkeit vom Molekulargewicht:
• Anzahl Signale im Protonenspektrum: Jedes Proton gibt mindestens einen Peak im Spektrum (Ausnahme: langsamer Austausch), d.h. je grösser das Molekulargewicht, desto komplexer das Spektrum (Signalüberlappung!)
Je grösser das Molekül, desto länger seine Korrelationszeit τc (η: Viskosität, r:
hydrodynamischer Radius, T: Temperatur in Kelvin, k: Boltzmann-Konstante)
(GL 2)
Lange Korrelationszeiten führen zu breiten Linien im Spektrum: Geringe Signalintensität und mehr Signalüberlagerungen!
• Zuordnung der Resonanzen mit homonuklearen Protonenspektren oft einfach bis zu ca 50AS, bei 50-100 AS empfiehlt sich 15N-Labelling, bei > 100 AS (bis gegen 200 AS) zusätzlich 13C und evtl. Deuterium-Labelling.
4. Aggregationszustand:
• Monodisperse Verteilung anstreben, da verschiedene Aggregatszustände evtl. zu verschiedenen Signalsätzen führen können.
• Wenn möglich in Monomerform messen (Check via Dynamic Light Scattering):
kleine Moleküle geben schärfere Signale
5. Stabilität unter den gewählten Bedingungen prüfen (CD, FPLC) τc 4πηr3
---3kT
=
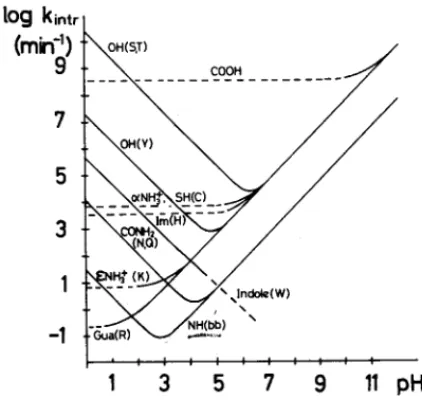
Hydrophile Peptide misst man meist in 90% H20/D2O, damit die labilen Protonen (NH) nicht durch Deuterium ausgetauscht sind. Der Backbone-NH Austausch von freien Amidprotonen ist bei einem pH um 3.0 im Minimum, aus Stabilitätsgründen misst man oft bei einem pH zwischen 4 und 5. (Fig. 1) Sind die Amidprotonen in Wasserstoffbrücken involviert (z.B. in einer Helix), oder befinden sie sich im Core und sind nicht Lösungsmittel-zugänglich, tauschen sie sehr viel langsamer aus und können auch bei höherem pH gut beobachtet werden.
4. S
PEKTREN-A
UFNAHMEDie Strukturbestimmung von Peptiden via NMR basiert auf der Beobachtung des sogenannten Nuklear Overhauser Effektes (NOEs) zwischen Protonen (siehe oben).
Um die NOEs den Protonen zuordnen zu können, müssen zunächst deren Resonanzfrequenzen ermittelt werden. Aufgrund der grossen Protonenzahl (3-13 pro Aminosäure) und der daraus resultierenden Signalüberlappung ist es bereits in kleinen Peptiden undenkbar, die Signale in einem normalen 1D Protonenspektrum zuzuordnen.
Eine schematische Verteilung der verschiedenen Protonen ist in der folgenden Figur gezeigt:
FIGUR 28. Logarithmische Darstellung der intrinsischen Austauschraten kintr versus pH für Lösungsmittel-zugängliche, labile Protonen in Wasser bei 25˚C.
Elegant lassen sich die Zuordnungsprobleme allerdings mit Hilfe der zweidimensionalen NMR-Spektroskopie lösen. Grundprinzip aller 2D-Experimente ist die Korrelation zweier Frequenzen miteinander. Die Korrelationsintensitäten entsprechen der dritten Dimension (Darstellung wie von Höhenlinien auf Landkarten).
Werden auf den beiden Achsen chemische Verschiebungen aufgetragen, spricht man von zweidimensionaler Verschiebungs-korrelierter Spektroskopie. Die horizontal liegende Frequenzachse (Abszisse) wird als F2-, die vertikale als F1-Achse bezeichnet (Ordinate). Welche Eigenschaften ein Protonenpaar (A, B) haben muss, damit es in einem 2D-Spektrum korreliert erscheint, d.h. durch zwei Kreuzpeaks (entspricht einem Berg auf der Landkarte) bei den beiden zugehörigen Protonenfrequenzen (νA, νB) bzw.
(νB, νA) in F2 und F1 verbunden ist, wird durch das NMR-Experiment genau definiert.
In allen homonuklearen Spektren wird jede Protonenfrequenz auch mit sich selber korreliert (νA, νA). Dies führt zu den sogenannten Diagonalpeaks, oft die intensivsten
FIGUR 29. Chemische Shift-Bereiche für die verschiedenen Typen von 1H-Resonanzen in Ubiquitin (aus Cavanagh et al.: Protein NMR Spectroscopy).
Signale im 2D-Spektrum. Alle Diagonalpeaks zusammen bilden die Diagonale des Spektrums und entsprechen einem Standard 1D Protonenspektrum.
Eine grosse Fülle an 2D Experimenten wurde entwickelt, so dass viele komplexe Fragestellungen durch sequentielle Applikation der geeigneten Experimente beantwortet werden können. Zur Resonanzfrequenzzuordnung von Protonenspektren eines Peptids werden standardmässig 3 verschiedene zweidimensionale homonukleare [1H,1H]-korrelierte Experimente gemacht: das COSY, das TOCSY und das NOESY.
Diese liefern 2D-Spektren, bei denen auf beiden Frequenzachsen 1H-chemische Verschiebungen miteinander korreliert sind (siehe Abschnitt 4.1 auf Seite 29,Abschnitt 4.2 auf Seite 32, Abschnitt 4.3 auf Seite 33).
Eine kurze grundsätzliche Bemerkung zur Aufnahme von Spektren mit Wasser als Lösungsmittel: Das bei weitem intensivste Signal in einem normalen 1D- Protonenspektrum liegt selbstverständlich bei der Wasserfrequenz (um 4.7ppm bei 37˚C). Da in diesem Frequenzbereich auch die meisten Cα-H liegen, muss das Wassersignal unterdrückt werden, z.B. durch vorheriges Einstrahlen auf die Wasserfrequenz werden die Wasserprotonen gesättigt, was die Spins der
FIGUR 30. Schema eines 2D Spektrums mit Angabe der Kreuz- und Diagonalpeaks sowie der externen Projektionen
ν1
ν1
F1
F2
Gegendiagonale: ν1 = -ν1 Diagonale: ν1 = ν1
ν2
ν2
(ν1, ν1) (ν1, ν2)
(ν2, ν2) (ν2, ν1)
Wasserprotonen für das nachfolgende Experiment quasi unsichtbar macht. Ein schmales Wasserband ist jedoch auch in wasserunterdrückten Spektren gut sichtbar.
4.1 [1H,1H]-COSY:[1H,1H]-Correlated Spectroscopy
[1H,1H]-Korrelation aufgrund skalarer [1H,1H]-Kopplungen mit Kopplungskonstanten J > 2Hz
Pro Memoria: In Molekülen treten benachbarte magnetische Kerndipole miteinander in Wechselwirkung. Solche Spin-Spin-Kopplungen beeinflusen die Energien und somit die Frequenzen der beobachteten Kerne. Es kommt zu Aufspaltungen der Signale in Dubletts, Tripletts etc. Bei der skalaren (indirekten) Kopplung erfolgt die Wechselwirkung über die Bindungen, d.h. indirekt, während bei der direkten (dipolaren) Kopplung eine direkte Wechselwirkung durch den Raum stattfindet, die sich allerdings in Lösung nicht als Aufspaltung zeigt.
Die Effizienz des COSY Experimentes nimmt mit zunehmender Linienbreite, d.h.
zunehmendem Molekulargewicht, drastisch ab. Dies ist ein Grund dafür, dass für grössere Moleküle Experiemente mit isotopenmarkierten Verbindungen eingesetzt werden müssen.
Die Peaks in einem COSY lassen sich an Hand ihrer Position im Spektrum wie folgt grob klassifizieren:
a. alle nichtlabilen, nichtaromatischen Seitenkettenprotonen ausser βH - γCH3 von Thr, δH-δH von Pro und βH-βH von Ser.
b. αH-βCH3 von Ala und βH-γCH3 von Thr.
c. αH-βH von Val, Ile, Leu, Glu, Gln, Met, Pro, Arg, und Lys.
d. αH-βH von Cys, Asp, Asn, Phe, Tyr, His und Trp.
e. αH-αH von Gly, αH-βH von Thr, δH-δH von Pro, αH-βH und βH-βH von Ser.
FIGUR 31. Skalare (links) und dipolare (rechts) Kopplungen
.
f. aromatische Ringprotonen, inklusive 2H-4H von His, sowie Seitenketten- Amidprotonen von Asn und Gln.
g. Hauptketten (Backbone) NH-αH.
h. δCH2-εNH von Arg.
Von spezieller Bedeutung ist die Backbone NH-αH-Region (g) von Spektren, die in Wasser aufgenommen wurden (in D2O tauschen die Backbone NH-Gruppen gegen D aus, ND gibt kein Signal). Sie wird Fingerprint-Region der Aminosäure-Sequenz genannt und kann als erstes Kriterium zur Qualitätsbeurteilung des Samples sowie der Spektren dienen. Jede L-Aminosäure (Ausnahme:Pro) führt zu einem einzelnen NH- αH Kreuzpeak, und jedes Gly gibt entweder 2 Kreuzpeaks (wenn die beiden αH verschiedene chemische Verschiebungen haben) oder einen einzelnen Peak mit einer charakteristischen Multiplett-Feinstruktur. Die N-terminale AS kann wegen des raschen Austauschs der α-Aminoprotonen mit Wasser nicht beobachtet werden (Abbildung 28 auf Seite 26).
FIGUR 32. [1H,1H]-COSY von Neuropeptid Y (NPY)
Die skalaren Kopplungen werden überwiegend zwischen Protonen, die durch 3 Bindungen separiert sind (sog. vicinalen Protonen) beobachtet. Mehrere Gründe können dafür verantwortlich sein, dass nicht die maximal mögliche Anzahl an Kreuzpeaks in der Fingerprint Region sichtbar sind: Signalüberlagerungen (Spektren werden erneut bei leicht erhöhter oder erniedrigter Temperatur aufgenommen, wodurch die Signal meist auf verschiedene Weise schieben), sehr kleine JNH-αΗ- Kopplungskonstanten (in stabilen α-Helices ist sie < 4 Hz), die aus Dihedralwinkeln nahe 90° resultieren (s. Abbildung 44 auf Seite 41), oder schneller Austausch mit dem Wasser.
Die COSY Peaks zeigen eine Multiplet-Feinstruktur. Diese resultiert aus den aktiven und passiven Kopplungen der beiden Protonen, deren skalare Wechselwirkung den Peak verursacht. Unter der aktiven Kopplung versteht man die Kopplung der beiden zu dem Peak gehörenden Protonen zueinander. Die passiven Kopplungen sind die Kopplungen der beiden Protonen zu weiteren Protonen. Aktive Kopplungen sind in anti-Phase (die beiden Komponenten haben unterschiedliches Vorzeichen) passive Kopplungen sind in-Phase:
FIGUR 33. COSY Kreuzpeakpattern für ein Spinsystem
ppm
2.00 ppm
4.00 F1 J(A,B)
J(A,B)
F2 J(A,C)
C
A B pinsystem:
4.2 [1H,1H]-TOCSY: Total Correlation Spectroscopy
Das TCOSY Experiment liefert [1H,1H]-Korrelation von Signalen aller Protonen eines Spinsystems. Ein Spinsystem setzt sich zusammen aus allen Spins, die zum gleichen (skalaren) Kopplungsnetzwerk gehören. Protonen verschiedener Aminosäuren gehören immer zu verscheidenen Spinsystemen (es gibt keine skalare Kopplung über die Amidbindung). In manchen Aminosäuren sind alle Protonen Bestandteil eines Spinsystemes (z.B. Ile), andere bestehen aus 2 Spinsystemen (z.B. Phe):
Das Erkennen der Spinsysteme ist ein wichtiger, erster Schritt im Verlauf der Resonanzzuordnung, und das TOCSY Experiment spielt bei Peptiden hier eine herausragende Rolle (siehe Abschnitt 5.2 auf Seite 36).
Durch Variation der sogenannten Mischzeit des Experimentes kann man bestimmen, wie weitreichend die Korrelationen innerhalb der beobachteten Spinsysteme sein werden. Wählt man z.B. eine kurze Mischzeit von 12ms, so beobachet man im 2D Spektrum ausschliesslich Kreuzpeaks zwischen geminal und vicinal koppelnden Protonen. Wählt man eine lange Mischzeit von ca 80ms, so erwartet man Kreuzpeaks für alle Protonen des Spinsystems. Um die Spinsysteme gut zu erkennen, ist es sinnvoll in einen Bereich des Spektrum zu schauen, der eine möglichst gute Signalseparation aufweist. Das ist normalerweise der Bereich der Amidprotonen (ca. 7-10 ppm), ev.
Ueberlappungsprobleme lassen sich häufig durch die Aufnahme der Spektren bei zwei verschiedenen Temperaturen beseitigen (der Shift der Amid Protonen ist stark temperaturabhängig).
Ein Vergleich der Aussagekraft von COSY und TOCSY ist in der folgenden Figur gut zu erkennen:
FIGUR 34. Spinsysteme von Tyr (J) und Arg(X) im TOCSY Experiment
.
4.3 [1H,1H]-NOESY: Nuclear Overhauser Effect Spectroscopy
Im NOESY-Experiment sind auf beiden Frequenzachsen 1H-chemische Verschiebungen aufgetragen. Kreuzpeaks resultieren aufgrund von 1H, 1H- Wechselwirkungen durch den Raum (dipolare, direkte Kopplung), sind also nur vom
FIGUR 35. Vergleich der Region der Amidprotonen (F2), aliphatischen Protonen (F1) für COSY (A) und TOCSY (B) sowie der Kopplungen aliphatischer Protonen untereinander im COSY (C) und TOCSY (D).
Abstand der Kerne voneinander abhängig, während die Anzahl der zwischen den Atomen liegenden Bindungen keinen Einfluss hat.
(GL 3)
Dipolare Kopplungen lassen sich in der hochauflösenden NMR Spektroskopie, d.h. in flüssiger (isotroper) Phase, nicht direkt aus dem Spektrum herauslesen, weil die Kopplungen aufgrund der raschen Reorientierung der Moleküle in Lösung zeitlich herausgemittelt werden. Die dipolaren Kopplungen sind aber die Ursache des Kern- Overhauser Effekts (nuclear Overhauser effect, NOE) - vgl. mit Gleichung 1, auf Seite 24 - und können somit aufgrund der Distanzabhängigkeit der Intensität zur Strukturbestimmung von Proteinen herangezogen werden.
Die Signalphase der Kreuzpeaks im NOESY-Experiment ist abhängig vom Vorzeichen des NOEs. Als Faustregel kann man sich merken, dass der NOE für kleine Moleküle .
TABELLE 36. Besonderheiten des NOESY-Experiments:
Signalphase Diagonalpeak
Signalphase Kreuzpeak kleine Moleküle,
niedervisköse Lösungsmittel
positiv negativ
mittlere Moleküle positiv sehr schwache Signale (positiv oder negativ)
grosse Moleküle positiv positiv
FIGUR 37. Abhängigkeit des homonuklearen Kern-Overhauser-Effekts zwischen Protonen vom Produkt aus Resonanzfrequenz und molekularer Korrelationszeit.(Abb. aus Günther: NMR-Spektr.)
D≈r–6
positiv und für grosse Moleküle negativ ist. Der NOE für mittlere Moleküle befindet sich gerade im Bereich des Vorzeichenwechsels und ist deshalb klein oder nullAuch im NOESY lassen sich aus der Lage der Peaks schon Rückschlüsse über die Art der involvierten Protonen erhalten:
a. NH; aromatische - NH; aromatische.
b. NH; aromatische - αH; δH von Pro; βH von Ser und Thr.
c. NH; aromatische - aliphatische Seitenketten.
d. αH; δH von Pro; βH von Ser und Thr - αH; δH von Pro; βH von Ser und Thr.
e. αH; δH von Pro; βH von Ser und Thr - aliphatische Seitenketten.
f. Aliphatische Seitenketten - aliphatische Seitenketten.
Nicht nur liefert das NOESY als einziges der 3 erwähnten Experimente Informationen über Protonendistanzen innerhalb des Peptids - es ist auch das Schlüsselexperiment bei der sequentiellen Zuordnung der AS-Spinsysteme zur Aminosäuresequenz. Man bedenke, dass 2 AS-Reste immer zu verschiedenen Spinsystemen gehören und deshalb nie durch Kreuzpeaks in einem COSY oder TOCSY-Experiment verbunden sind! Nur mit Hilfe des NOESY lassen sich die Spinsysteme über die Amidbindungen miteinander verknüpfen (s. 4.3).
FIGUR 38. (H,H)-NOESY von ProteinD (Phagen Hüllprotein)
5. S
TRATEGIEZURR
ESONANZFREQUENZ-Z
UORDNUNG FÜRKLEINE,
NICHT
-
MARKIERTEP
ROTEINE5.1 Beurteilung der Spektrenqualität
Anhand der Fingerprint-Region im COSY-Spektrum wird die Anzahl der beobachtbaren Spinsysteme bestimmt. Fehlen mehr als 10% der maximal möglichen Zahl an Resten, sollte der Grund für die Abwesenheit der Peaks bestimmt und die Spektrenaufnahme optimiert werden (Temperatur, pH, Salz etc.)
In einem zweiten Schritt müssen alle Spektren gleich kalibriert werden. Als interner Standard wird dabei oft die Wasserfrequenz verwendet (T=Temperatur in [K]):
(GL 4)
Durch Übereinanderlegen von TOCSY und COSY, bzw. NOESY und COSY kann nun auch die Qualität der Fingerprint-Region von TOCSY und NOESY abgeschätzt werden. Hierbei ist zu beachten, dass im NOESY eine grössere Anzahl an zusätzlichen (interresiduellen, meist sequentiellen NHi-Hαi-1) Kreuzpeaks vorhanden sein können.
Es lohnt sich ausserdem meist, Spektren bei zwei verschiedenen Temperaturen aufzunehmen, um in Fällen von Signalüberlappung eindeutige Interpretationen zu erhalten (siehe Abschnitt 5.3 auf Seite 39).
5.2 Spinsystem-Identifikation: NH-αΗ−βΗ−...
Im ersten Schritt versucht man, die Spinsysteme von der Hauptkette aus (Spins, die zum gleichen Spinsystem wie das betrachtete Amidproton gehören) zu identifizieren.
Es gelingt meist nicht, die Aminosäuren direkt zu indentifizieren (Ausnahmen sind Thr, Ser, Gly). Die Klassifizierung nach Typ des Spinsystemes erlaubt aber später unter Zuhilfenahme der sequentiellen Information aus dem NOESY für Tri/
Tetrapeptidsegmente eine eindeutige Zuordnung.
Kriterien für die Klassifizierung hierzu sind z.B.
• die Länge der Spinsysteme
es gibt kurze Spinsysteme (Typ J, z.B. Ser), lange Spinsysteme (z.B. Lys)
• bestimmte Peak-“Patterns“ (siehe Anhang):
Man sucht sich einen Bereich, in dem die Spinsysteme möglichst gut separiert sind.
Dies ist meist in der NH Region der Fall. Bei kurzen Peptiden ist die Analyse des NH (F2)/H-aliphatisch (F1) Bereiches im TOCSY sehr nützlich. Manche Ami-
H2O
( )
δ 7 83, T
96 9, --- ppm –
=
nosäuren lassen sich schon anhand der chemischen Verschiebungen ziemlich ein- deutig bestimmen:
Ser: β−Protonen sehr tief, keine Methylgruppe
Thr: β−Protonen sehr tief, plus Methylgruppe um 1 ppm Ala: keine β-Protonen, jedoch Mehtylgruppe um 1 ppm
Gly gibt als einzige Aminosäure ein Triplett für die Amidresonanz. Pro hat kein Amidproton, zeigt aber ein charakteristisches Spinsystem im TOCSY.
Methylgruppen tragen neben Ala und Thr nur noch Val, Leu und Ile. Val, Leu und Ile können aufgrund der unterschiedlichen Verknüpfungen von Methyl- und Methylengruppen im COSY unterschieden werden. Kurze Spinsysteme (nur NH, α- und β-Protonen) besitzen neben Ser nur Cys, Asp, Asn, Phe, His, Trp und Tyr (sog.
Typ J-Spinsysteme). Die Verschiebungsbereiche der Protonen sind schemtisch in der folgenden Abbildung gegeben
FIGUR 39. 1H-chemische Verschiebungen von aliphatischen Protonen. Mittelwert und Standardabweichung wurden aus Daten von 13 Proteinen bestimmt.
(Abbildung aus Cavanagh et al.: Protein NMR Spectroscopy)
Die sogenannten “Random-Coil” Verschiebungen (Shifts in unstrukturierten Tripeptiden) sind in der nachfolgenden Tabelle dargestellt:
Die Mitglieder der letzten Gruppe von AS-Resten, Lys, Arg, Met, Gln, Glu und Pro, besitzen alle 2 γ-Protonen, gekoppelt an die β−Protonen. Weil die shifts der β−
Protonen in dieser Gruppe höher als 2.2 ppmliegen, werden diese Reste auch Typ-U (upfield) Spinsysteme genannt. Met, Gln und Glu haben ihre γH-Resonanzen tiefer als jene der β-Protonen und vice versa für Arg, Lys, Pro und Leu.
Für die aliphatischen und aromatischen Protonen der Aminosäuren gibt es charakteristische Pattern in der COSY und TOCSY Spektren, die im Anhang schematisch zusammengefasst sind.
TABELLE 40. Random coil chemische Verschiebungen für die 20 natürlichen Aminosäuren im Dipeptid mit nachfolgendem Alanin
Rest NH Hα Hβ Andere
Ala 8.24 4.32 1.39
Cys(red) 8.32 4.55 2.93, 2.93
Cys(ox) 8.43 4.71 3.25, 2.99
Asp 8.34 4.64 2.72, 2.65
Glu 8.42 4.35 2.06, 1.96 γCH2 2.31, 2.31
Phe 8.30 4.62 3.14, 3.04 2,6H 7.28; 3,5H 7.38; 4H 7.32
Gly 8.33 3.96
His 8.42 4.73 3.29, 3.16 2H 8.58; 4H 7.29
Ile 8.00 4.17 1.87 γCH2 1.45, 1.16; γCH3 0.91; δCH3 0.86
Lys 8.29 4.32 1.84, 1.75 γCH2 1.44, 1.44; δCH2 1.68, 1.68;
εCH2 2.99, 2.99; εNH3+ 7.81 Leu 8.16 4.34 1.62, 1.62 γCH 1.59; δCH3 0.92, 0.87 Met 8.28 4.48 2.11, 2.01 γCH2 2.60, 2.54; εCH3 2.10 Asn 8.40 4.74 2.83, 2.75 γNH2 7.59, 6.91
Pro - 4.42 2.29, 1.94 γCH2 2.02, 2.02; δCH2 3.63, 3.63 Gln 8.32 4.34 2.12, 1.99 γCH2 2.36, 2.36; δNH2 7.52, 6.85 Arg 8.23 4.34 1.86, 1.76 γCH2 1.63, 1.63; δCH2 3.20, 3.20;
εNH 8.07
Ser 8.31 4.47 3.89, 3.87
Thr 8.15 4.35 4.24 γCH3 1.21
Val 8.03 4.12 2.08 γCH3 0.94, 0.93
Trp 8.25 4.66 3.29, 3.27 2H 7.27; 4H 7.65; 5H 7.18; 6H 7.25;
7H 7.50
Tyr 8.12 4.55 3.03, 2.98 2,6H 7.14; 3,5H 6.84
5.3 Sequenz-spezifische Zuordnung
Hat man die Spinsysteme charakterisiert, so versucht man sie über NOEs aneinander zu hängen. Hierzu gibt es charakteristische, kurze sequentielle Abstände, die je nach Sekundärstruktur variieren.
.
Natürlich werden auch nicht-sequentielle kurze Abstände beobachtet, daher sollte man nur starke und mittelstarke NOEs für die sequentielle Zuordnung benutzen. In gestreckten Konformationen wie in β−Faltblättern oder im Zufallsknäul (random-coil)
FIGUR 41. NOESY-Diagonal- und Kreuzpeaks in der NH (F2)/ NH (F1)- Region. Starke
(sequentielle) dNN-Kreuzpeaks werden v.a. in helikalen Segmenten, augrund der kurzen (sequentiellen) NHi-NHi+1 Abstände beobachtet und erlauben es, die sequentielle Anordnung der Spinsysteme zu bestimmen durch Zuordnung der NH-shifts (sog. NOESY-walk in der NH/NH-Region)
FIGUR 42. NOESY-Kreuzpeaks in der NH(F2)/αH (F1)-Region. Starke (sequentielle) dαΝ- Kreuzpeaks werden v.a. in gestreckten (β−Blättern und random coil) Konformationen aufgrund der kurzen NHi+1/αHi-Abstände beobachtet, oft gleichzeitig in Kombination mit dβN-Kreuzpeaks.
7 ppm
987
ppm 4
31 245
9 8 7 pm
4.543.5 ppm 3
31 245
N H
Cα C
N αααα
C O
α O
i+1
-1
werden kurze sequentielle NHi+1-αHi beobachtet. In helikalen Abschnitten sind die NHi-NHi+1 Abstände kurz. Durch Vergleich von NOESY und COSY lassen sich die intraresiduellen von den sequentiellen (interresiuduellen) NH-αΗ Peaks unterscheiden. Mit geeigneten Programmen wie XEASY können identische Frequenzausschnitte aus COSY/TOCSY und NOESY gleichzeitig nebeneinander angezeigt werden und intraresiduelle von interresiduellen NOEs schnell unterschieden werden. Der Vergleich von COSY und NOESY eignet sich für die sequentielle Aneinandereihung der Spinsysteme:
Die kurzen sequentiellen Abstände, bzw. die daraus resultierenden NOEs ermöglichen es nun, kurze Segmente zusammenzuhängen, und man kann dann meist unter Zuhilfenahme der Information über die Art der Spinsysteme entscheiden, um welche Aminosäure es sich handelt.
FIGUR 43. : Fingerprint Region im COSY (links) und NOESY (rechts) Spektrum.