Adsorption von SO 2
in modifizierten DMOF-1-Strukturen
Adsorption of SO
2in modified DMOF-1 structures
Institut für Theoretische Chemie und Computerchemie Mathematisch-Naturwissenschaftliche Fakultät
Heinrich Heine Universität Düsseldorf
Abschlussarbeit
zur Erlangung des akademischen Grades Bachelor of Science
vorgelegt von
Felix Schäfer am 01.09.2020
Erstprüfer: PD Dr. Oliver Weingart Zweitprüfer: Prof. Dr. Christoph Janiak
Eidesstattliche Erklärung
Hiermit versichere ich, die vorliegende Abschlussarbeit selbstständig und nur unter Ver- wendung der von mir angegebenen Quellen und Hilfsmittel verfasst zu haben. Sowohl inhaltlich als auch wörtlich entnommene Inhalte wurden als solche kenntlich gemacht. Die Arbeit hat in dieser oder vergleichbarer Form noch keinem anderen Prüfungsgremium vorgelegen.
Datum: Unterschrift:
Danksagungen
Zuerst möchte ich mich bei PD Dr. Oliver Weingart für sein entgegengebrachtes Vertrauen bedanken, an diesem Projekt arbeiten zu dürfen. Vielen Dank für die Betreuung während der Arbeit und die Möglichkeit, neue Programme und Methoden zu erlernen und anzuwenden.
Vielen Dank für die stete Ansprechbarkeit bei Fragen und Problemen. Zusätzlich möchte ich mich für die Erstellung des Erstgutachtens meiner Arbeit bedanken.
Des Weiteren möchte ich mich bei Prof. Dr. Christoph Janiak für die Bereitschaft bedanken, als Zweitgutachter meine Arbeit zu beurteilen.
Dem gesamten Arbeitskreis des Instituts für Theoretische Chemie und Computerchemie spreche ich hiermit meinen Dank für die Unterstützung bei technischen Herausforderungen, bei der Literaturrecherche und für eine angenehme und sichere Arbeitsatmosphäre, trotz der SARS-CoV-2 Pandemie, aus.
Bei Dr. Shang Hua Xing und Simon Hédé möchte ich mich zudem für die Bereitstellung von Daten, Darstellungen und Inputs bedanken.
Abschließend möchte ich mich bei meiner Lebenspartnerin, meiner Familie und meinen Freunden bedanken, die mich während meines Studiums und meiner Bachelorarbeit unterstützt haben.
Zusammenfassung
Anthropogenes Schwefeldioxid entsteht zu großen Teilen bei Verbrennungsprozessen. Es stellt ein gesundheitliches Risiko und eine Gefährdung für die Umwelt dar, weshalb Möglichkeiten gesucht werden, Schwefeldioxid aus dem Kamingas zu entfernen. Da es selbst modernsten Entschwefelungsmethoden nicht gelingt, Schwefeldioxid vollständig zu entfernen, rückt die Stoffklasse der porösen metallorganischen Gerüstverbindungen (metal organic frameworks, MOFs) in das Blickfeld neuester Forschungen.
Diese neuartige Verbindungsklasse besteht aus Metallionen und verknüpfenden Linker- Molekülen und ist in der Lage, Gase zu trennen und zu speichern. In dieser Arbeit werden verschiedene Verbindungen des DMOF-1 betrachtet, welches aus Zink, Terephthalsäure (1,4-benzenedicarboxylic acid, BDC) und Triethylendiamin besteht. Das MOF muss dabei vor der drohenden Hydrolyse und gegen das Schwefeldioxid des feuchten Kamingases geschützt werden. Dafür werden als Linker Varianten mit zusätzlichen Methylgruppen zum Schutz gegen Degradation eingesetzt. Zusätzlich kann Nickel als Metallzentrum verwendet werden, welches als besonders stabil gegenüber dem aggressiven Schwefeldioxid und Wasser gilt. Die Strukturen der DMOFs werden mit Hilfe von Computerprogrammen erstellt und mittels DFT-Verfahren mit einer Basis aus ebenen Wellen optimiert. Anhand dieser optimierten DMOFs wird die Adsorption von Schwefeldioxid molekülmechanisch mit Hilfe von Monte-Carlo-Verfahren simuliert. Des Weiteren werden Bindungsstellen des Schwefeldioxids und ihre Bindungsenergien in den modifizierten DMOF-Strukturen bestimmt. Bei den Untersuchungen wurden insgesamt fünf Bindungspositionen für die DMOF-Strukturen mit Zink identifiziert, für Nickel konnten drei Positionen festgestellt werden. Die Linker mit Methylgruppen zum Schutz vor Degradation, wiesen kleinere Poren in der MOF-Struktur auf. Dadurch ist die maximale Schwefeldioxidaufnahme zwar geringer, jedoch sind durch die zusätzlichen Wechselwirkungen die Bindungsenergien und damit auch die Affinität für Schwefeldioxid größer. Die Simulationen gaben das Schwefeldioxid Adsorptionsverhalten bei niedrigen Drücken qualitativ gut wieder. Vor allem die maximale Aufnahmekapazität der stabilen Ni-DMOF-TM-Verbindung, welche eine Möglichkeit für die zukünftige Kamingasreinigung darstellen könnte, ließ sich in guter Näherung reproduzieren.
Abstract
Anthropogenic sulfur dioxide is largely produced during combustion processes. It represents a health risk and a hazard to the environment. Therefore, methods to remove sulfur dioxide from the flue gas are required. Since even the most modern desulfurization methods are unable to remove sulfur dioxide completely, the substance class of porous metal organic frameworks (MOFs) became the focus of the latest research.
This novel compound class consists of metal ions and linker molecules and is able to separate and store gases. In this thesis, different compounds of DMOF-1 are considered, which consists of zinc, terephthalic acid (1,4-benzenedicarboxylic acid, BDC) and triethyle- nediamine. The DMOF must be protected against hydrolysis and reactions with the sulfur dioxide of the humid flue gas. For this purpose, linker variants with additional methyl groups and nickel as an alternative metal node are used. The latter showed an increased stability with the reactive flue gas. The structures of the DMOFs are created with the help of computer programs and optimized by means of DFT in a plane-wave basis. On the basis of these optimized DMOFs, the adsorption of sulfur dioxide is simulated by molecular mechanics using Monte-Carlo methods. Furthermore, binding sites of the sulfur dioxide and their binding energies in the modified DMOF structures are determined. Five binding positions were found for the DMOF structures with zinc and three for the structures with nickel. The linkers with methyl groups resulted in smaller pores. Due to these smaller MOF pores, the maximum sulfur dioxide uptake is lower, but because of the additional interactions, the binding energies are higher and therefore also the affinity for sulfur dioxide.
The simulations could qualitatively replicate the adsorbtion properties of the DMOFs at low pressures. Especially the adsorbtion capacity of the stable Ni-DMOF-TM, which is a promising candidate for future flue gas purification, was reproduced with good accuracy.
Abkürzungsverzeichnis
ADC 9,10-Anthracendi-carbonsäure (9,10-anthracenedicarboxylic acid) BDC Terephthalsäure (1,4-benzenedicarboxylic acid)
C Kohlenstoff
CCDC Cambridge Crystallographic Data Centre
Co Kobalt
CO2 Kohlenstoffdioxid Carbondioxid
Cu Kupfer
DABCO Triethylendiamin (1,4-diazabicyclo[2.2.2]octane) DFT Dichte-Funktional-Theorie(density functional theory) DMBDC Dimethylterephthalsäure (2,5-dimethyl terephthalic acid)
GGA gradientenkorrigiertes Dichtefunktional (generalized gradient approximation)
H Wasserstoff
LDA lokale Dichtenäherung (local density Approximation)
LSDA lokale spin Dichtenäherung (local spin density Approximation) MOF metallorganische Gerüstverbindung (metal-organic framework)
N Stickstoff
NDC 1,4-Naphthalendicarbonsäure (1,4-naphthalenedicarboxylic acid)
Ni Nickel
NOx Stickoxid
O Sauerstoff
PBE Funktional nach Perdew, Burke und Ernzerhof Pos. Position
PP Pseudopotential
RRKJ Rappe-Rabe-Kaxiras-Joannopoulos
SBU sekundären Baueinheiten (secondary building units) SO2 Schwefeldioxid
TMBDC Tetramethylterephthalsäure (tetramethyl-1,4-benzenedicarboxylic acid) Zn Zink
Inhaltsverzeichnis
Eidesstattliche Erklärung . . . I Danksagungen . . . II Zusammenfassung . . . III Abstract . . . IV Abkürzungsverzeichnis . . . V
1 Einleitung 3
2 Stand der Forschung 5
3 Zielsetzung 10
4 Theorie 11
4.1 Metallorganische Gerüstverbindungen . . . 11
4.2 Dichtefunktionaltheorie . . . 12
4.2.1 Funktionale . . . 13
4.2.2 Dispersionskorrektur . . . 13
4.3 Ebene Wellen . . . 13
4.3.1 Basisfunktion der ebenen Wellen . . . 13
4.3.2 Pseudopotentiale . . . 14
4.3.3 k-Raum . . . 15
4.4 BFGS-Geometrieoptimierung . . . 16
4.5 Magnetisierung . . . 16
4.6 Reales Gas . . . 17
4.6.1 Van der Waals-Gleichung . . . 17
4.6.2 Peng-Robinson-Gleichung . . . 18
4.6.3 Chemisches Potential und Fugazität . . . 18
4.7 Lennard-Jones-Potential . . . 19
4.8 Monte-Carlo-Simulationen . . . 19
4.9 Adsorption und Adsorptionsisotherme . . . 20
5 Durchführung 22 5.1 Erstellung der DMOF-Strukturen . . . 22
5.2 Strukturoptimierung . . . 22
5.3 Bindungsstellen . . . 23
5.4 Berechnung der Ladungen . . . 23
5.5 Monte-Carlo-Simulation . . . 24
6 Ergebnisse 25 6.1 Voruntersuchungen . . . 25
6.1.1 Dispersionskorrektur . . . 25
6.1.2 Konvergenz bei verschiedenen Cutoffs . . . 26
6.1.3 Magnetisierung der Ni-DMOFs . . . 28
6.2 Vergleich der Strukturen . . . 29
6.3 Adsorption . . . 30
6.3.1 Zn-DMOF-Varianten . . . 30
6.3.2 Ni-DMOF-Varianten . . . 33
6.3.3 Ni-DMOF-TM Superzelle . . . 35
6.3.4 Adsorptionsisothermen . . . 37
7 Diskussion 41 8 Ausblick 43 9 Anhang 56 9.1 Stand der Forschung . . . 56
9.2 Ergebnisse . . . 57
9.3 Pseudopotentiale . . . 76
9.4 Kristallstrukturen . . . 76
9.5 Lennard-Jones-Parameter . . . 77
1 Einleitung
Anthropogenes Schwefeldioxid (SO2) entsteht vor allem bei Verbrennungsprozessen großer industrieller Anlagen, wie z.B. bei der Müllverbrennung oder in Kohlekraftwerken. Dort kann es als geringer Teil des Kamingases (bis zu 420-3.000 ppm) in die Umwelt gelangen [1, 2]. SO2 verschmutzt die Luft, verursacht durch Einatmen Lungenerkrankungen und durch Bildung von saurem Regen schwerwiegende Umwelt- und Gebäudeschäden [2]. Zur Vorbeugung muss das SO2 aus dem Kamingas herausgefiltert werden, was herkömmlicher- weise mit einer Wäsche durch Wasser und Kalkstein geschieht. SO2 wird gelöst und greift sauer den Kalkstein an, welcher nach und nach zersetzt wird. Durch diese Methode wird zwar ein enormer Teil (85-95%) [2] des schädlichen SO2 herausgefiltert, jedoch sind im Kamingas weiterhin um die 50-450 ppm SO2 enthalten, welches in die Atmosphäre gelangt [1, 2]. Alleine durch Energiegewinnung, beispielsweise durch die Verbrennung von Kohle, werden so jährlich etwa 80 Mt (2015) SO2 freigesetzt [2]. Zusätzlich stört das verbleibende SO2 im weiteren Verlauf der Abgasreinigung, wenn Stickoxide (NOx) katalytisch behandelt oder Kohlenstoffdioxid (CO2) chemisch gespeichert werden soll [2]. Bei der Separation und Speicherung von Gasen haben sich in der Vergangenheit die sogenannten metallorganischen Gerüstverbindungen (metal-organic frameworks, MOFs) als erfolgreich erwiesen, daher könnten sie in Zukunft eine erfolgreichere Alternative für die Entfernung von SO2, aus dem Kamingas, darstellen (Abb. 1.1) [1, 2].
Abbildung 1.1: Entfernung von SO2 aus Kamingas, durch einen MOF (Abb. aus [2]).
Ein MOF ist ein Gerüstmolekül aus anorganischen Zentren, welche von organischen Linkern miteinander verbunden werden. Dank der dabei entstehenden Hohlräume bildet sich eine enorm große Oberfläche aus, mit welcher das MOF mit anderen Molekülen wechselwirken und diese adsorbieren kann. In vielen Fällen lässt sich dieser Prozess durch Erhitzen unter Vakuum umkehren, sodass das MOF wiederholt zur Adsorption eingesetzt werden kann [2, 3]. SO2 und Wasserdampf im Kamingas stellen für MOFs ein Problem dar, da es, je nach Aufbau, durch Korrosion nachhaltig degradieren kann [1]. DMOF-1 basiert auf Zink, Triethylendiamin (1,4-diazabicyclo[2.2.2]octane, DABCO) und Terephthalsäure (1,4-benzenedicarboxylic acid, BDC). Während die Metallzentren an den Achsen a und b über BDC verbunden werden, verbindet DABCO diese zweidimensionalen Lagen über die c-Achse (Abb. 1.2) [1, 4]. Durch Variation der Linker und des Metalls lassen sich die Eigenschaften von DMOF-1 beeinflussen.
Abbildung 1.2: Aufbau (Abb. aus [5], verändert) und Ausschnitt eines DMOFs.
2 Stand der Forschung
Hungerford et al. konnten zeigen [1], dass verschiedene DMOF-Spezies unterschiedli- che Stabilitäten gegenüber Wasserdampf und SO2 aufweisen. Dabei wurden die Metall- zentren Zink (Zn), Cobalt (Co), Nickel (Ni), Kupfer (Cu), sowie verschiedene Linker (Abb. 2.1) untersucht. So halten z.B. die zusätzlichen Methylgruppen von TMBDC (TM in Abb. 2.1) durch sterische Hinderung Wassermoleküle von den Metallzentren des DMOFs fern und verhindern so die Degradation durch Hydrolyse. Ebenso wur- den die Linker Dimethylterephthalsäure (2,5-dimethyl terephthalic acid, DMBDC) 1,4- Naphthalendicarbonsäure (1,4-naphthalenedicarboxylic acid, NDC) und 9,10-Anthracendi- carbonsäure (9,10-anthracenedicarboxylic acid, ADC) (Abb. 2.1) an einem Zn-DMOF auf ihre Stabilität durch wasserabweisende Eigenschaften in feuchter SO2-Atmosphäre untersucht.
Abbildung 2.1: Strukturen der verschiedenen benutzten Linker (Abb. aus [1]).
Die Studie zeigte auch, dass die Stabilitäten der DMOFs durch die Metalle (M) nach der Irving-Williams-Reihe zunehmen (Co < Ni < Cu > Zn) und so Kupfer und Nickel die stabilsten Spezies darstellen, während die MOFs mit Co und Zn als Zentralatom ungefähr gleich stabil in feuchter SO2-Atmosphäre waren. Für die verschiedenen Linker stellten ADC und TMBDC die stabilsten Spezies dar.
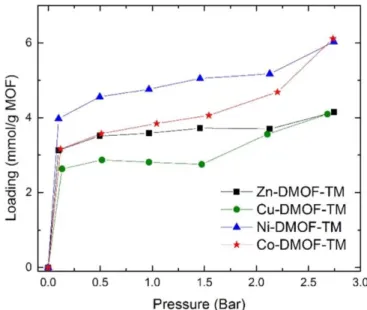
Für die untersuchten M-DMOF-Spezies wurde die Adsorption von SO2 unter trockenen Bedingungen gemessen. Abb. 2.2 zeigt das SO2-Adsorptionsverhalten der DMOFs bei
konstanter Temperatur (T=25 °C) und Drücken von null bis drei bar auf der x-Achse. Die Menge des adsorbierten SO2 lässt sich in mmol/g MOF auf der y-Achse ablesen.
Abbildung 2.2: SO2-Adsorptionsverhalten verschiedener M-DMOFs bei konstanter Tem- peratur (Abb. aus [1]).
Bei Drücken bis zu einem Bar adsorbiert das stabile Cu-DMOF-TM am wenigsten SO2, das zweitstabilste Ni-DMOF-TM adsorbiert am erfolgreichsten SO2. Ab einem Druck von ca. 0,25 bar ist der Anstieg der SO2-Adsorption nicht mehr so groß wie bei kleineren Drücken. Da das Zn-DMOF-TM durch SO2 degradiert, liegt der Sättigungswert der Ad- sorption unter dem von Ni-DMOF-TM. Bei den verschiedenen Linkervariationen stellt sich das DMOF mit TMBDC als Linker am erfolgreichsten heraus, da es bereits bei kleinen Drücken eine erhöhte Adsorption aufweist [1]. Aus diesen Gründen wurde für diese Arbeit das Ni-DMOF und der Linker TMBDC gewählt. Die MOFs mit Zn als Metallzentrum und BDC als Linker dienen dabei als Vergleichswerte.
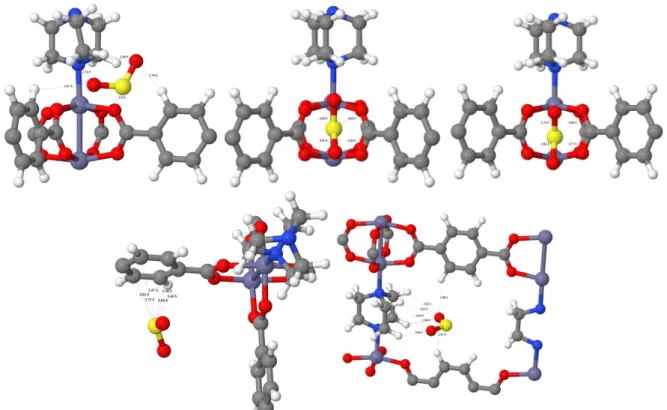
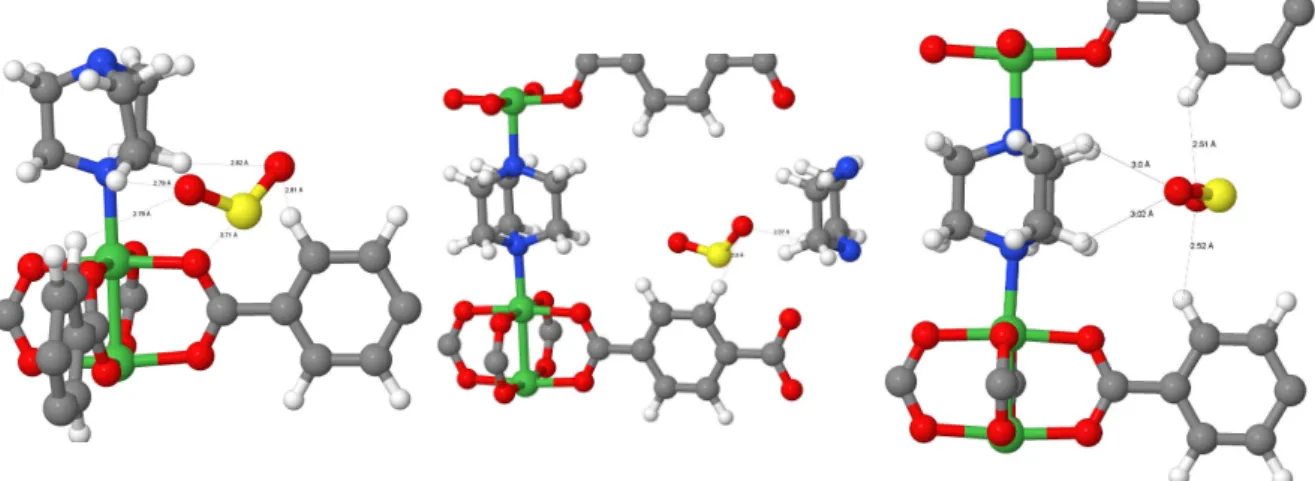
Tan et al. [3] berechneten mit DFT-Methoden die Bindungsenergien der Adsorption von SO2 an Zn-DMOF und ermittelten Werte von 5 kJ/mol für die Assoziation an DABCO und Phenyl-H (Abb. 2.3 d), 22 kJ/mol für die π-Bindung zum Phenylring (Abb. 2.3 b), sowie 61 und 66 kJ/mol für Bindung von SO2 an die Carboxy-Gruppe bzw. zu den H-Atomen von DABCO (Abb. 2.3 c und a). Für die Bindungsenergien bei Ni-DMOF wären aufgrund der isomorphen Struktur ähnliche Werte zu erwarten.
Abbildung 2.3: SO2-Bindungsstellen bei Ni/Zn-DMOF, mittels DFT berechnet (Abb. aus [3]).
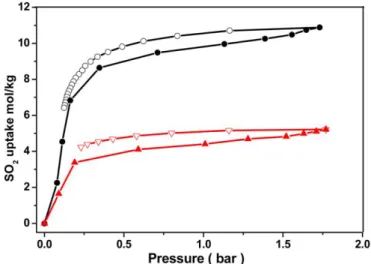
Für Zn- und Ni-DMOF wurde die Adsorption unter trockenen Bedingungen gemessen.
Abb. 2.5 zeigt das SO2-Adsorptions- und Desorptionsverhalten von Ni- und Zn-DMOF bei konstanter Temperatur (T=298 K) und Drücken von null bis zwei bar. Die schwarze Farbe steht hierbei für das Ni-DMOF, die rote Farbe für das Zn-DMOF. Die farbig gefüllten Symbole stehen für Adsorption und die leeren für die Desorption von SO2 [3].
Abbildung 2.4: SO2-Adsorptionsverhalten zweier DMOFs bei konstanter Temperatur. Rot:
Zn-DMOF, schwarz: Ni-DMOF. Offene Symbole: Desorption, geschlossene Symbole: Adsorption (Abb. aus [3]).
Es wird deutlich, dass SO2 stärker an Ni-DMOF als an Zn-DMOF adsorbiert. Aufgrund dessen dass das Zn-DMOF durch SO2 stärker degradiert. Der Anstieg der SO2-Adsorption ist ab einem Druck von ca. 0,25 bar nicht mehr so groß wie bei kleineren Drücken [3].
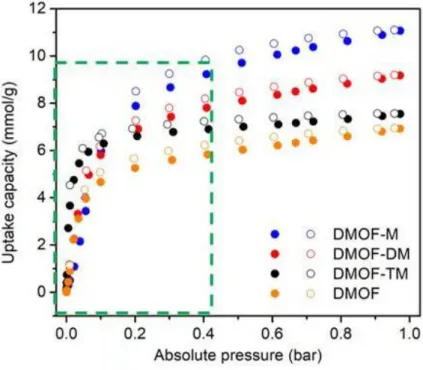
In einer aktuellen Studie [6] synthetisierte Dr. Shang Hua Xing im AK Janiak Ni- DMOFs mit un-, mono-, di- und tetra-methylierten BDC Linkern. Abb. 2.5 zeigt das SO2-Adsorptions- und Desorptionsverhalten von Ni-DMOF-X Spezies unter trockenen
Bedingungen bei konstanter Temperatur (T=293 K). Die farbig gefüllten Symbole stehen für die Adsorption und die leeren für die Desorption von SO2.
Abbildung 2.5: SO2-Adsorptionsverhalten verschiedener Ni-DMOF-X-Varianten bei kon- stanter Temperatur. Offene Symbole: Desorption, geschlossene Symbole:
Adsorption (Abb. aus [6]).
Bei geringen Drücken ist Ni-DMOF-TM in der SO2 Adsorption von am erfolgreichsten. Ab Drücken von mehr als 0,1 bar jedoch liegen die Sättigungswerte der DMOF-Varianten mit einfach und zweifach methylierten Linkern über dem Sättigungswert von Ni-DMOF-TM.
Die geringere SO2 Adsorption des Ni-DMOF ist mit der Degradation der DMOF-Struktur durch SO2zu erklären. Der starke Anstieg der SO2-Adsorption flacht ab einem Druck von ca.
0,2 bar deutlich ab [6]. Im Druckbereich 0-0,4 bar steigt dabei die SO2-Aufnahmekapazität am stärksten an. Daher wurde dieser Bereich für die Adsorptionssimulation dieser Arbeit genauso gewählt.
Des Weiteren fand Dr. Xing mittels DFT-Berechnungen an Clustermodellen verschiedene Bindungspostionen und damit verbundene Bindungsenergien von SO2 an Ni-DMOF und Ni-DMOF-TM. Dabei fällt auf, dass die Bindungsenergien des SO2 in Ni-DMOF-TM (33,6- 51,8 kJ/mol) deutlich größer sind als bei Ni-DMOF (28,4-32,0 kJ/mol) [6]. Abbildungen der Bindungspositionen und die dazugehörigen Bindungsenergien befinden sich im Anhang (Abb. 9.1 und 9.2).
Tan et al. [7], Glomb et al. [8], Carter et al. [9] und Zhang et al. [10, 11] beschäftigten sich in ihren Arbeiten ebenfalls mit Fragestellungen, welche Kombination von Metallzentren und Linkern für eine möglichst wirkungsvolle SO2-Adsorption bzw. Kamingasreinigung mit MOFs geeignet sein könnte.
3 Zielsetzung
Im Rahmen dieser Arbeit wird das SO2-Adsorbtionsverhalten in modifizierten DMOF-1- Strukturen simuliert, um neue Möglichkeiten für die Kamingasreinigung zu finden. DMOF- Strukturen gelten als besonders vielversprechend, da sie Gase trennen und speichern können. Zur Simulation der SO2-Adsorption werden DMOF-Strukturen mit Zink und Nickel als Metallzentren und BDC und Tetramethylterephthalsäure (tetramethyl-1,4- benzenedicarboxylic acid, TMBDC) als Linker betrachtet. Die Kombination von Ni und TMBDC erwies sich als besonders stabil gegenüber der Degradation durch SO2 und der Hydrolyse aus den Kamingasen.
Die Strukturen der DMOFs werden dabei mit Hilfe von Computerprogrammen generiert und mittels Dichtefunktionaltheorie(density functional theory, DFT)-Verfahren mit einer Basis aus ebenen Wellen optimiert. Des Weiteren werden Bindungspositionen von SO2 innerhalb der DMOF-Strukturen analysiert und ihre Bindungsenergien bestimmt. Ihre SO2- Adsorption wird mittels Monte-Carlo-Rechnungen simuliert und Adsorptionsisothermen erstellt.
4 Theorie
4.1 Metallorganische Gerüstverbindungen
Ein MOF ist ein poröses, dreidimensionales, periodisch kristallines Gerüst aus Metall- zentren, den sekundären Baueinheiten (secondary building units, SBU), sowie organischen Linkern, welche die SBUs miteinander verbinden. Die Zwischenräume der Linker bilden ein definiertes Geflecht aus Kanälen und Kammern, den Poren. Damit kann das MOF trotz geringer Masse eine extrem große Oberfläche (mehrere tausend m2/g) erhalten.
Durch diese Oberfläche tritt das MOF in Wechselwirkung mit anderen Verbindungen und kann sie beispielsweise adsorbieren. Je nach Linker und SBU besitzen MOFs verschiedene physikalische und chemische Eigenschaften. So kann z.B. die Reaktivität in den Poren oder die Porengröße selbst durch die Wahl der Linker modifiziert und gesteuert werden. Zudem lassen sich auch die SBUs der MOFs variieren, weshalb die Möglichkeiten, verschiedene MOFs zu erstellen, nahezu unendlich groß sind (Abb. 4.1) [12, 13, 14].
Abbildung 4.1: Beispiele von MOF-Strukturen (Abb. aus [6]).
MOFs sind kristalline Festkörper, jedoch besitzen einige von ihnen eine gewisse Flexibilität, wodurch sie unter Druck oder durch Gastmoleküle wie z.B. Lösungsmittel, verformbar sind [4, 5, 15]. MOFs finden Anwendung bei der Gastrennung, Gasspeicherung, Katalyse und in vielen anderen Bereichen [12, 13, 14].
Binden ein oder mehrere Moleküle an ein MOF, so lässt sich deren Bindungsenergie EB berechnen [7]:
EB = (E[M OF +n·X]−E[M OF]−n·E[X])/n (4.1) Hierbei steht n für die Beladung der betrachteten MOF-Zelle, also wie viele Moleküle X in dieser binden, z.B. SO2 [7].
4.2 Dichtefunktionaltheorie
Die Anzahl der Elektronen N ist gleich dem Integral der Dichte ρ über den Raum~r [16]:
N =
Z
ρ(~r)d~r (4.2)
Nach dem Theorem von Hohenberg und Kohn lässt sich die Energie E im Grundzustand als Energiefunktional der Dichte E[ρ] bestimmen. Daraus folgt die Dichtefunktionaltheorie (density functional theory, DFT). Da jedoch das genaue Energiefunktional, welches die Dichte und die Energie verknüpft, nicht bekannt ist, werden Näherungslösungen genutzt [17, 18]. Der Ansatz von Kohn-Sham teilt das Energiefunktional in einen Teil für die kinetische Energie TS[ρ] auf, bei welcher die Elektronenwechselwirkungen vernachlässigt werden. Damit werden ca. 99% der realen Energie erfasst. Zusätzlich wird es aufgeteilt in die Funktionale der Kern-Elektronen-Wechselwirkung Ene[ρ], den Coulombanteil der ElektronenwechselwirkungJ[ρ] und eine Korrektur für die realen Wechselwirkungen, welche mit dem Term des Austauschkorrelationsfunktional Exc[ρ] beschrieben wird [17].
EDF T[ρ] =TS[ρ] +Ene[ρ] +J[ρ] +Exc[ρ] (4.3) Dabei steht der Ausdruck der ersten Klammer für die Korrektur der vorhergegangenen Näherung der kinetischen Energie und die Korrekturen der Korrelations- und Austausch- wechselwirkungen werden in der zweiten Klammer zusammengefasst. [16, 17]:
Exc[ρ] = (T[ρ]−TS[ρ]) + (Eee[ρ]−J[ρ]) (4.4) Das Funktional Exc[ρ] ist nun das einzige Funktional, welches sich nicht genau bestimmen lässt. Daher wird das Problem, das "wahre" Funktional zu finden, von E[ρ] auf Exc[ρ]
verschoben. Somit lässt sich ein verhältnismäßig großer Anteil der Energie bestimmen und sie muss nur um einen kleinen Teil korrigiert werden.
4.2.1 Funktionale
Für die Wahl des Austauschkorrelationsfunktionals gibt es eine Reihe von Möglichkeiten.
Bezieht sich das Funktional nur auf die Dichte an einem Punkt ρ(~r), so wird von der lokalen Dichtenäherung (local density approximation, LDA) gesprochen. Werden zusätzlich die Spinzustände α und β mit einbezogen, wird die genauere lokale Spin-Dichtenäherung (local spin density Approximation, LSDA) erhalten. Durch das Einbeziehen des Gradienten der Dichte ∇ρ = ∂ρ/(∂~r) wird ein gradientenkorrigiertes Dichtefunktional (generalized gradient approximation, GGA) erhalten. Bei diesem werden die Änderungen der Dichte besser beschrieben, die Orbitale des betrachteten Systems werden "flexibler". Somit lassen sich GGAs, nutzen um inhomogene Ladungsverteilungen besser beschreiben zu können [16, 17, 18]. Das Funktional, welches in dieser Arbeit verwendet wurde, ist ein GGA nach Perdew, Burke und Ernzerhof (PBE).
4.2.2 Dispersionskorrektur
Dispersionswechselwirkungen, z.B. London-Wechselwirkungen, werden durch induzierte Dipole verursacht. Sie können einen erheblichen Einfluss auf die Energie eines System haben.
Insbesondere, wenn diese schwachen und kurzreichweitigen Wechselwirkungen kumulativ auftreten, können berechnete Energien stark von experimentellen Werten abweichen. Daher sollte nach Möglichkeit bei der Rechnung mittels DFT eine Dispersionskorrektur eingefügt werden:
EDF T−D3 =EDF T −EDisp (4.5)
Die Dispersionsenergien werden anhand von empirischen Parametern und meist mit Dämpfungsfunktionen, beschrieben [17, 19]. Die Dispersionskorrektur, welche in dieser Arbeit genutzt wurde, ist die D3 Korrektur nach Grimme et al [19].
4.3 Ebene Wellen
4.3.1 Basisfunktion der ebenen Wellen
Bei lokalisierten Basen hängen die Orbitale des Systems von der Linearkombination der Atomorbitale ab. Für ebene Wellen wird hingegen ein periodisches System betrachtet. Aus einer Einheitszelle mit periodischen Randbedingungen bildet sich ein bis ins unendliche fortlaufendes System. Dieses lässt sich durch die äußersten Valenzelektronen beschreiben.
Diese liegen in Metallen als fast freie Elektronen vor. Die Lösung der Schrödinger Gleichung
für ein freies Elektron in einer Dimension kann als komplexe Exponentialfunktion oder trigonometrische Funktionen dargestellt werden [17]:
φ(x) =Aeikx+Be−ikx (4.6)
φ(x) =Acos (kx) +Bsin (kx) (4.7)
Die Energie hängt quadratisch vom Faktor k ab [17]:
E = 1
2k2 (4.8)
Für unendlich große Systeme "verschmelzen" die Molekülorbitale zu Bändern, da die definierten Energieunterschiede der Orbitale verschwinden. Die Elektronen eines Bandes können mithilfe von Orbitalen eines Basissatzes durch ebene Wellen beschrieben werden.
Diese lassen sich in drei Dimensionen darstellen als [17]:
χk(r) =eikr (4.9)
Der Wellenvektor k spielt hierbei die gleiche Rolle wie der Exponent ζ in Gauß-ähnlichen Orbitalen. Über den Translationsvektor t lassen sich die Werte des Vektors k bestimmen [17]:
k·t = 2πm (4.10)
Hierbei steht m für eine positive ganze Zahl.
Die Anzahl an ebenen Wellen lässt sich aus der Energie und dem Volumen der Einheitszelle bestimmen, dabei ergibt sich nach Gleichung 3.8 die maximale kinetische Energie [17]:
MP W = 1
2π2V Ecut2/3 (4.11)
Durch Erhöhen des Parameters Ecut, dem Cutoff der kinetischen Energie, wird die Qualität des Basissatzes erhöht. So entspricht ein Basissatz durch ebene Wellen mit einem Cutoff bei 200 meV und einer Zellenlänge von 15 Å einem Basissatz mit 20.000 Funktionen [17].
4.3.2 Pseudopotentiale
Mit den Basisfunktionen der ebenen Wellen lassen sich Valenzelektronen gut beschreiben.
Jedoch sorgen die kernnahen Elektronen mit ihren schnellen Bewegungen für Probleme bei der Berechnung, vor allem bei schweren Elementen mit vielen Elektronen wird sehr
viel Rechenaufwand benötigt. Diese kernnahen Elektronen sind, im Gegensatz zu den Valenzelektronen, nicht an der Bindungsbildung beteiligt. Da diese aber die Valenzorbitale deformieren, sind sie zu wichtig, um vernachlässigt zu werden. Daher werden diese Elek- tronen über effektive Rumpfpotentiale, den Pseudo-Potentialen (PP), genähert dargestellt.
Die PPs geben durch ein Coulomb-Potential am Kern elementspezifisch die Wechselwirkun- gen mit den kernnahen Elektronen wieder, sodass sich die Valenzelektronen mit weniger Rechenaufwand realistisch darstellen lassen. Eine weitere Verbesserung stellen die ultrawei- chen PPs dar, durch welche der Rechenaufwand weiter verringert wird. Dies geschieht über die Näherung des kernnahen Bereiches, indem die dort auftretende starke Dichteänderung durch Hilfsfunktionen beschrieben wird. Somit wird das Potentialmaximum verringert, wodurch der Rechenaufwand sinkt und die Genauigkeit der Potentialbeschreibung, bei größerem Abstand zum Kern, erhalten bleibt [17, 20]. Die ultraweichen PPs, welche in dieser Arbeit verwendet wurden, sind PPs nach der Rappe-Rabe-Kaxiras-Joannopoulos (RRKJ)-Methode [21] (Die genauen PPs für alle verwendeten Elemente befinden sich im
Anhang unter Abschnitt 9.3).
4.3.3 k-Raum
Der k-Raum steht für den reziproken Raum nach der ersten Brillouin-Zone, welcher sich durch den Wellenvektork aufspannt, da an allen Grenzpunkten der Wiegner-Seitz Zelle die Laue/Braggsche Bedingung erfüllt ist. Um z.B. die Elektronendichte zu bestimmen, muss über die Wiegner-Seitz Zelle integriert werden. Dabei wird ein Gitter, das k-Punktegitter, um den Ursprung angelegt und anhand dieser Punkte wird über die Zelle integriert.
Das Gitter lässt sich nach Monkhorst und Pack für ganze Zahlen ni = 1,2...Ni in drei Dimensionen wie folgt aufstellen:
kn1,n2,n3 ≡
3
X
i
2ni−Ni−1
2Ni Gi (4.12)
Gi steht dabei für die Vektoren des reziproken Gitters. Je feiner die Größe des Gitters gewählt wird, umso genauere Ergebnisse werden erzielt. Jedoch müssen dadurch mehr Punkte berechnet werden. Der Punkt (0,0,0) wird dabei Gamma genannt. Zudem gilt, je größer die Einheitszelle ist, um so kleiner ist diese im reziproken Raum [20]. In dieser Arbeit wird der Punkt Gamma und ein 2x2x2 k-Punktegitter [22] verwendet.
4.4 BFGS-Geometrieoptimierung
Die potentielle Energie eines Moleküls ist abhängig von seinen Koordinaten (3N-6, 3N-5 wenn linear). Aus allen Möglichkeiten der Anordnungen von Koordinaten eines Moleküls ergibt sich eine Hyperfläche der potentiellen Energie. Das globale Minimum dieser Hyper- fläche stellt den stabilsten Aufbau des Systems in den gegebenen Koordinaten dar. Die Funktion der Hyperfläche ist dabei so komplex, dass sich nicht alle Punkte auf dieser unendlich großen Fläche bestimmen lassen. Um zum Minimum zu gelangen, muss daher ein besonderes Verfahren angewendet werden, die Geometrieoptimierung. Eine Methode der Geometrieoptimierung bilden die Quasi-Newton-Methoden, wie es auch die Methode nach Broyden-Fletcher-Goldfarb-Shanno (BFGS) ist. Bei Quasi-Newton-Methoden wird zusätzlich zu den Gradienten eine genäherte, inverse Hessematrix benutzt, um schneller zum Minimum zu gelangen. Es gibt unterschiedliche Verfahren, während der Berechnung ein Update der Hesse-Matrix zu erhalten [16]. Die BFGS-Methode basiert auf folgender Gleichung [23]:
H+ =Hc+yyT
yTs −(Hcs)(Hcs)T
sTHcs (4.13)
Dieses Verfahren wurde bei den Geometrieoptimierungen dieser Arbeit eingesetzt.
4.5 Magnetisierung
Als diamagnetisch werden Übergangsmetall-Atome bezeichnet, wenn alle Elektronen mit gepaarten Spins vorliegen, wie bei der d10- Konfiguration des Zn. Bei Ni hingegen liegen durch die d8- Konfiguration ungepaarte Elektronen vor. Diese lassen sich in ihrem Spin unterschiedlich ausrichten. In paramagnetischen Materialien wie Ni liegen die Spins in zufälliger Ordnung vor. Diese lassen sich jedoch durch äußere Einflüsse wie Magnetfelder oder Temperatur ausrichten. Richten sich die Spins in die gleiche Richtung parallel aus, so entsteht ein gerichtetes magnetisches Moment (ferromagnetisch). Wenn sich die Spins gleichmäßig antiparallel in unterschiedliche Richtungen ausrichten, hebt sich die Magnetisierung auf (antiferromagnetisch) (Abb. 4.2) [24]. In dieser Arbeit wurden die ungepaarten Spins von Ni sowohl ferromagnetisch als auch antiferromagnetisch betrachtet.
Abbildung 4.2: a) paramagnetische b) ferromagnetische c) antiferromagnetische Spins.
4.6 Reales Gas
4.6.1 Van der Waals-Gleichung
Innerhalb dieser Arbeit wird nur das Verhalten von Reinstoffgasen betrachtet.
Nach dem Gesetz für ideale Gase gilt:
pV =nRT (4.14)
Dabei steht p für den Druck, V für das Volumen, n für die Stoffmenge, R für die Gas- konstante und T für die Temperatur. Ideale Gasteilchen besitzen untereinander keine Wechselwirkung und Eigenausdehnung, daher beschreiben sie reales Gas nur näherungs- weise. Daher werden zusätzliche Korrekturterme eingeführt, welche das reale Verhalten der Gase besser beschreiben sollen [25]. Diesen Ansatz verfolgt die Van der Waals-Gleichung [24]:
p= nRT V −nb −a
n V
2
(4.15) Das Volumen V bei großen Drücken entspricht nicht dem tatsächlichen Volumen, in dem sich ein Gasteilchen bewegen kann. Die Gasteilchen können sich einander nicht beliebig nähern, wodurch das Volumen um den Betrag des Eigenvolumens pro Gasteilchen subtrahiert wird (V-nb). Um die anziehenden Wechselwirkungen mit einzubeziehen, wird angenommen, dass das Volumen für kleine Drücke dem Volumen des idealen Gases entspricht, da dabei das Volumen V sehr viel größer als das Eigenvolumen nbist. Da durch die Zunahme der attraktiven Wechselwirkungen zwischen den Gasteilchen der Druck nach außen abnimmt, wird angenommen, dass sich dieser um das Quadrat der Konzentration n/V verringert. Zusätzlich wird die Proportionalitätskonstante a eingefügt, welche die Stärke der anziehenden Wechselwirkungen beschreibt. Die Van-der-Waals-Konstanten a und b stehen hierbei für empirische Parameter, welche für jedes Gas verschieden sind [24, 26]. Eingefügt in die Zustandsgleichung des idealen Gases ergibt sich die Van der Waals-Zustandsgleichung [24, 26]:
p+an2 V2
!
(V −nb) =nRT (4.16)
4.6.2 Peng-Robinson-Gleichung
Die 1976 veröffentlichte Peng-Robinson-Zustandsgleichung stellt eine Erweiterung der Van der Waals-Zustandsgleichung dar [27]:
p= RT
V¯ −b − a(T)
V¯V¯ +b+bV¯ −b (4.17) Dabei steht ¯V in der Zustandsgleichung für das molare Volumen (V/n), die Parameter a(T) für die Energie und b für das Eigenvolumen. Diese berechnen sich nach [27]:
a(T) = 0,45724R2Tk2α(T)
pk (4.18)
b = 0,07780RTk
pk (4.19)
α(T) =
1 + (0,37464 + 1,5422ω−0,26992ω2)
1−qT /Tk
2
(4.20) α(T) ist ein Teil von a(T), wobei ω für den azentrischen Faktor steht [27].
4.6.3 Chemisches Potential und Fugazität
Das chemische Potentialµ ist die partielle molare Gibbs-Energie, welche für einen Reinstoff die Änderung der Gibbs-Energie ∆Gbei Änderung der Stoffmenge ∆n darstellt [25, 28]:
µ= dG dn = G
n =Gm (4.21)
Für das Modell des idealen Gases lautet das chemische Potential [28]:
µid=µ◦+RT ·ln p p◦
!
(4.22) Dabei steht µ◦ für das chemische Standard-Potential des idealen Gases bei Standarddruck p◦ (p◦ = 1bar) [25, 28]. Das chemische Potential von realen Gasen errechnet sich analog dazu, jedoch wird hier eine Korrektur für den Druck eingeführt, welche das reale Verhalten beschreiben soll, die Fugazität fg. Die Fugazität fg errechnet sich nach der folgenden Gleichung [28]:
fg =ϕ·p (4.23)
Der Fugazitätskoeffizient ϕist eine druckabhängige Größe und lässt sich aus Zustands- gleichungen wie der Peng-Robinson-Gleichung bestimmen [28, 29]:
ln(ϕ) = pV¯ RT −1
!
−ln p( ¯V −b) RT
!
+ a(T) 2,828bRTln
V¯ + 2,414b V¯ −0,414b
!
(4.24)
4.7 Lennard-Jones-Potential
Anhand des Lennard-Jones-Potentials lässt sich die Wechselwirkung (und Viskosität) von Gasen darstellen [30].
VLJ(r) =−α r6 + β
r12 (4.25)
Dabei hat der erste Term die Form der magnetischen Dipol-Dipol-Wechselwirkung und der zweite stellt eine empirische Anpassung der kurzreichweitigen Pauli-Abstoßung dar [30]. Somit steht ein Term für die anziehende Wechselwirkung der Teilchen, wenn sich diese entfernt von einander befinden und einer für deren Abstoßung, wenn sie sich zu nahe kommen [24].
Eine andere Schreibweise für das Lennard-Jones-Potential ist:
V(r) = 4 σ12 r12 −σ6
r6
!
( >0) (4.26)
Dabei steht σ für den Abstand der Atome, bei dem gilt: V(r = σ) = 0. ist die Tiefe des Potentialtopfes [24, 30, 31]. Die Beschreibung 1/r12 für den abstoßenden Term würde durch die Exponentialfunktion e−r/σ besser beschrieben werden. Der Rechenaufwand ist damit jedoch viel größer, was zu einem Problem bei der Erfassung der Wechselwirkungen von Systemen mit vielen Teilchen, wie Gasen oder Flüssigkeiten, führt [24].
4.8 Monte-Carlo-Simulationen
Monte-Carlo-Verfahren basieren auf der Erzeugung von Zufallsereignissen. Diese Methodik lässt sich auch für die Bestimmung der Adsorption von Gasmolekülen in Gitterstrukturen anwenden. Dabei wird z.B. ein MOF betrachtet, in welchem sich Gasmoleküle anordnen.
Dies geschieht anhand von Zufallszahlen, welche bestimmen, ob und wie sich ein Molekül bewegt. Mit der Monte-Carlo-Methode werden neue Konfigurationen mit neuen geo- metrischen Anordnungen der Moleküle generiert. Diese werden mit der damit verbundenen Änderung der potentiellen Energie z.B. molekülmechanisch (unter anderem durch das
Lennard-Jones-Potential) berechnet und mithilfe eines Filters überprüft und gegebenenfalls verworfen [18, 24]. Ist die potentielle Energie der neuen Konfiguration kleiner als die der Ursprungskonfiguration, so wird diese akzeptiert. Ist sie jedoch größer, so wird ein Kriterium benötigt, welches über die Konfiguration entscheidet. Dafür wird die Boltzmann-Verteilung genutzt. Diese besagt, dass in zwei Zuständen mit einer Energiedifferenz ∆E bei der TemperaturT das Verhältnis der Besetzungszahlen im Gleichgewicht gegeben ist. Dies stellt die folgende Formel dar, wobei k für die Boltzmann-Konstante steht [24]:
e−∆E/kT (4.27)
Der Exponentialfaktor wird für jede Konfiguration in der Monte-Carlo-Methode ausgerech- net und mit einer Zufallszahl zwischen 0 und 1 verglichen. Liegt der Wert dabei unter der Zufallszahl, so wird er verworfen und eine neue Konfiguration erzeugt. Ist er andernfalls größer, wird die Konfiguration akzeptiert [24]. Dies wird mehrere tausend Mal wiederholt, sodass sich eine Boltzmannverteilung einstellt [18].
Die Adsorbtion von Gasmolekülen wird häufig im großkanonischen Ensemble (µ, V, T= const.) betrachtet [24, 32]. Hier geht in die Wahrscheinlichkeit P für das Auftreten eines bestimmten Mikrozustandes in das chemische Potential µ mit ein [32]:
P =e
Ω +µN−E
kT (4.28)
Dabei ist N die Teilchenzahl und Ω das großkanonische Potential. Dieses kann über die großkanonische Zustandssumme Z ausgedrückt werden[32]:
Ω =−kT ln(Z) (4.29) Die Wahrscheinlichkeit, mit der eine Operation, z.B. Translation eines Gasmoleküles, in einer Simulation gelingt, sollte dabei so hoch wie möglich liegen. Zusätzlich können die Parameter für die entsprechenden Monte-Carlo-Schritte equilibriert werden, um so die Genauigkeit der Methode zu erhöhen.
4.9 Adsorption und Adsorptionsisotherme
Die Adsorption beschreibt das reversible Anlagern eines Teilchens an eine Grenzfläche wie z.B. eine Phasengrenze [26]. Bei der Physisorption, einer Form der Adsorption, wechsel-
wirken das Adsorbat und das Adsorbens schwach über Van-der-Waals-Kräfte. Im Gegensatz dazu bindet das Adsorbat bei der Chemisorption chemisch an das Adsorbens, womit es stärker gebunden ist als bei der Physisorption. Die Chemisorption spielt eine wichtige Rolle in der heterogenen Katalyse [33, 34]. Die Physisorption kann als Vorstufe der Chemisorption agieren, indem diese energetisch ein lokales Minimum auf dem Weg zum globalen Minimum der Chemisorption darstellt [33]. Da es sich bei den Kräften, welche für die Adsorption sorgen, nicht um weitreichende Kräfte handelt, bilden sich meist nur monomolekulare Lagen an den Oberflächen aus. Je höher die spezifische Oberfläche (A/g) eines Stoffes ist, um so mehr kann von diesem adsorbiert werden, da es sich um eine Oberflächenreaktion handelt. Da diese Reaktion reversibel abläuft, gibt das Adsorbens das Adsorbat wieder ab, dies wird Desorption genannt [26]. Die Energie, welche aufgebracht werden muss, um ein durch Physisorption gebundenes Teilchen zu desorbieren, ist kleiner als bei der Chemisorption [33]. Erfolgen Adsorption und Desorption in gleicher Geschwindigkeit, stellt sich das Adsorptionsgleichgewicht ein, womit die Anzahl der adsorbierten Teilchen konstant bleibt. Die Größe der maximalen Oberflächenkonzentration im Gleichgewicht hängt hierbei zusätzlich von der Temperatur und der Adsorbatkonzentration bzw. dem Partialdruck ab. Wird die gesamte Oberfläche bedeckt, sodass keine weiteren Teilchen adsorbieren können, ist der Sättigungswert erreicht und das Absorbens liegt "gesättigt" vor.
Über einen hohen Partialdruck lässt sich der Sättigungswert näherungsweise berechnen.
Unter isothermen Bedingungen lässt sich aus der Beziehung der Oberflächenkonzentration a (mol/m2) zu dem Partialdruckp bzw. der Konzentrationcdes Adsorbats eine Langmuir- Adsorptionsisotherme aufstellen [26]:
a =k1· p
p+k2 (4.30)
a=k1· c c+k2
(4.31) Dabei stehen k1 und k2 für Konstanten, welche von Temperatur, Adsorbat und Adsorbens abhängen. Für große Partialdrücke wird a = k1 und damit der Sättigungswert erreicht [26].
5 Durchführung
5.1 Erstellung der DMOF-Strukturen
Die Struktur der Zn-DMOF-Einheitszelle (Abb. 5.1) wurde freundlicherweise von Simon Hédé als Eingabedatei für Quantum ESPRESSO [21] zur Verfügung gestellt. Um das methylierte Zn-DMOF-TM zu erhalten, wurden mit dem ZMAT-Editor des Programms gmolden [35] die Wasserstoffe des Linkers durch CH3-Fragmente ersetzt. Die Ni-DMOF- Varianten wurden generiert, indem die Zn-Atome in den Strukturen des Zn-DMOFs und Zn-DMOF-TM mit Ni-Atomen ersetzt wurden.
Abbildung 5.1: Einheitszellen der untersuchten DMOF-Varianten (Grün=Zn/Ni).
5.2 Strukturoptimierung
Die vier Einheitszellen der verwendeten DMOF-Varianten wurden unter periodischen Randbedingungen mit dem Programm Quantum ESPRESSO [21] optimiert. Als Basis wurden ebene Wellen mit ultraweichen RRKJ-PPs verwendet [21] (Die genauen PPs für alle verwendeten Elemente befinden sich im Anhang unter Abschnitt 9.3). Die Berechnungen der Energien erfolgte anhand des PBE-Funktionals mit und ohne Dispersionskorrektur
(D3) [19]. Die DMOF-Strukturen wurden sowohl mit einem k-Punktegitter (2x2x2) [22]
als auch mit nur einem k-Punkt (dem Gamma-Punkt) beschrieben. Die Geometrieoptimie- rungen mit dem BFGS-Algorithmus erfolgte in zwei Varianten: ohne Restriktionen und mit experimentellen Zellparametern als Konstanten. Die Konvergenz der Energie wurde mit verschiedenen Cutoff-Werten (zwischen 300 und 900 Ry für die kinetische Energie der Ladungsdichte, bzw. 30-90 Ry für die Wellenfunktionen) überprüft. Zur Berechnung der Energien der Zn-DMOFs wurde die restricted DFT-Methode genutzt. Die Ni-DMOFs wur- den mit der unrestricted DFT-Methode im ferromagnetischen und antiferromagnetischen Zustand bei unterschiedlichen Startmagnetisierungen untersucht. In die Einheitszellen der optimierten DMOF-Strukturen wurde jeweils ein SO2-Molekül eingebracht (siehe z.B.
Abb. 6.6), um verschiedene Bindungsfälle zu erzeugen. Um die Bindungsenergie nach Gl.
4.1 zu berechnen, wurde das SO2-Molekül in einer leeren Einheitszelle optimiert. Für das Ni-DMOF-TM wurde aus der optimierten Einheitszelle mit dem ProgrammGDIS [36] eine Superzelle (2x2x2) mit einer Kantenlänge von ca. 20 Å erstellt. Die Geometrieoptimierungen wurden für diese Superzelle mit und ohne SO2 wiederholt.
5.3 Bindungsstellen
Die Bindungslängen wurden mit dem Programm Jmol [37] für alle Bindungspositionen der Zn-DMOFs und der Ni-DMOFs gemessen. Bindungslängen zwischen dem Sauerstoff des SO2-Moleküls und den Wasserstoffen der MOFs mit einer Länge von über 3,2 Å wurden dabei als nicht-bindend gewertet. Bindungslängen zwischen dem Schwefel des SO2-Moleküls und dem Sauerstoff der MOFs oder den Kohlenstoffatomen der Phenylringe der MOFs mit einer Länge von über 4 Å wurden dabei ebenfalls als nicht-bindend gewertet.
5.4 Berechnung der Ladungen
Mit dem Programm cp2k [38] wurden die ESP-gefitteten Ladungen der Atome der Ein- heitszellen mit der REPEAT-Methode berechnet [39]. Die Berechnungen erfolgten in der DZVP-MOLOPT-GTH Basis mit entsprechenden GTH-PBE Pseudopotentialen. Für die Nickelatome erfolgte die Ladungsberechnung nur für den antiferromagnetischen Zustand.
Die Sortierung der Atomgruppen mit dem Programm Mercury4 [40] erschließt sich aus der Abb. 5.2. Bei den DMOF-TM-Varianten wurden die Kohlenstoffe der Methylgruppen der Atomgruppe C5 und die Wasserstoffe der Methylgruppen der Atomgruppe H3 zugeordnet.
Abbildung 5.2: Zuordnung der Atomgruppen in Zn/Ni-DMOFs. (Abb. aus [41], verändert).
5.5 Monte-Carlo-Simulation
Für die Monte-Carlo-Simulation der SO2-Adsorption wurden mit dem ProgrammGDIS[36]
Superzellen (4x4x4) mit einer Kantenlänge von ca. 40 Å erstellt. Dies erfolgte anhand der optimierten Einheitszellen ohne Restriktionen und anhand von optimierten Einheitszellen nach experimentellen Zellparametern der DMOF-TM-Varianten (Die CCDC-IDs [42] der Kristallstrukturen befinden sich im Anhang, Abschnitt 9.4). Die berechneten Ladungen wurden den Atomen in den Superzellen zugeordnet. Die intermolekularen Wechselwirkungen wurden anhand von Lennard-Jones Parametern aus dem UFF-Kraftfeld [43] für die Atome N, O, C und H beschrieben. Die Parameter für die Ni- und Zn-Atome stammen aus dem UFF4MOF-Kraftfeld [44]. Die Parameter für SO2 wurden der Arbeit von Ketko et al.
[45] entnommen. (Die Parameter befinden sich im Anhang, Abschnitt 9.5) Das chemische Potential von SO2 wurde nach der Peng-Robinson-Zustandsgleichung (Gl. 4.17) für Drücke im Bereich von 0-40 kPa [6] berechnet. Mit dem Programm Cassandra [46] wurden bei einer Temperatur von 293 K [6] für verschiedene Druckpunkte Monte-Carlo-Rechnungen im großkanonischen Ensemble durchgeführt. Dabei wurden für alle sechs Superzellen 20.000.000 Equillibrierungsschritte und 20.000.000 Produktionschritte durchgeführt. Die Anzahl der adsorbierten SO2-Moleküle pro Druckpunkt wurde über die Mittelung der Produktionsschritte erhalten. Mit diesen Informationen lassen sich Adsorptionsisothermen konstruieren (Abb. 6.12 bis 6.14).
6 Ergebnisse
6.1 Voruntersuchungen
Um ein geeignetes Protokoll für die Optimierung der modifizierten DMOF-Strukturen und der SO2-Adsorption zu finden, wurde die Zn-DMOF Struktur zunächst mit einem Standard- Cutoff von 400 Ry geometrieoptimiert. Dabei wurde der Einfluss der Dis-persionskorrektur anhand von fünf exemplarischen SO2-Bindungspositionen und ihren Adsorptionsenergien untersucht (siehe Abb. 6.5 für eine grobe Übersicht). Die Analyse der final bestimmten Bindungsstellen findet sich in Kapitel 6.3. Des Weiteren wurde die Konvergenz der Ge- samtenergie hinsichtlich des Cutoffs für die kinetische Energie anhand der modifizierten DMOF-Strukturen bestimmt. Weitergehend beinhaltet Kapitel 6.1.3 eine Untersuchung der magnetischen Eigenschaften der nickelhaltigen DMOF-Varianten.
6.1.1 Dispersionskorrektur
Die folgenden Ergebnisse sollen den Effekt der Dispersionskorrektur anhand des Zn-DMOFs verdeutlichen. Bei der Betrachtung der daraus resultierenden Bindungsenergien mit und ohne Korrektur kommt es zu wesentlichen Unterschieden welche in den Tabellen 6.1 und 6.2 zusammengefasst sind.
Tabelle 6.1: Bindungsenergien des opti- mierten Zn-DMOF ohne D3 in kJ/mol.
Gamma 2x2x2 Pos. 1 -9,32 -9,34 Pos. 2 -10,09 -10,00 Pos. 3 -5,68 -5,66 Pos. 4 -7,47 -7,44 Pos. 5 -7,53 -7,50
Tabelle 6.2: Bindungsenergien des opti- mierten Zn-DMOF mit D3 in kJ/mol.
Gamma 2x2x2 Pos. 1 -37,62 -29,10 Pos. 2 -32,88 -33,02 Pos. 3 -31,69 -31,74 Pos. 4 -23,60 -23,49 Pos. 5 -29,89 -29,40
Die Anwendung der Korrektur bewirkt eine deutliche Vergrößerung der Bindungsenergien (-5,66 bis -10,9 kJ/mol ohne Korrektur und zwischen -23,49 und -37,62 kJ/mol mit Korrektur). In einigen Fällen versechsfacht sich der Wert für die Bindungsenergien sogar.
Eine Vernachlässigung der Dispersionskorrektur führt somit zu großen Fehlern, weshalb sie in den nachfolgenden Rechnungen stets berücksichtigt wurde.
6.1.2 Konvergenz bei verschiedenen Cutoffs
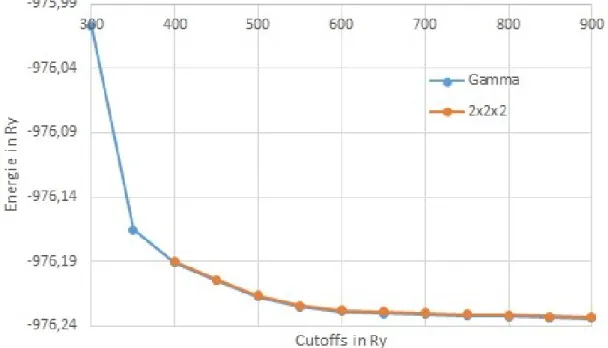
Um möglichst genaue Energieberechnung zu erhalten, wurden die Strukturen der DMOF- Varianten bei verschiedenen Cutoffs berechnet. Im Folgenden wird dies anhand des Zn- DMOF-TM vorgestellt. Für Berechnungen um den Gamma-Punkt wurden bei dieser DMOF-Variante zusätzlich die Cutoffs 300 und 350 Ry verwendet, um die Wirkung des Cutoffs auf die Energie, welche in Abb. 6.1 zu sehen ist, zu verdeutlichen. Die y-Achse zeigt dabei die Gesamtenergie in Ry, die x-Achse zeigt die Cutoffs in Ry.
Abbildung 6.1: Energien des optimierten Zn-DMOF-TM bei unterschiedlichen Cutoffs.
Die Graphiken aller weiteren DMOF-Varianten befinden sich im Anhang (Abb. 9.3-9.5).
Bei Betrachtung aller Graphiken wird ersichtlich, dass die Gesamtenergie mit der Erhöhung des Cutoffs sinkt. Die Kurven des Gamma-Punkts und des 2x2x2 k-Punktegitters verlaufen sehr ähnlich, zeigen bei den Ni-DMOFs jedoch einen sehr kleinen Versatz in der Energie von
ca. 0,013 Ry. Da die Geometrieoptimierung mit einem hohen Cutoff zu langen Rechenzeiten führt, wurde der niedrigste konvergente Wert für die Berechnungen gesucht. Vorherige Rechnungen wurden bei einem Cutoff von 400 Ry durchgeführt, dieser Wert liegt jedoch deutlich unter der Konvergenzschwelle. Wie in Abb. 6.1 zu erkennen ist, liegt diese Schwelle bei etwa 650 bis 700 Ry.
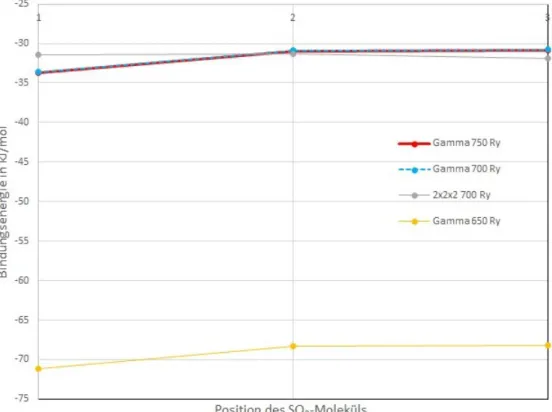
Um den Einfluss des Cutoffs auf die Gittergrößen und SO2-Bindungsenergien der drei Bindungspositionen für das Ni-DMOF (siehe Abb. 6.7 für eine grobe Übersicht) zu bestimmen, wurde die Optimierung mit höheren Cutoffs durchgeführt. Die Ergebnisse dieser Berechnungen werden in Abb. 6.2 zusammengefasst. Die y-Achse zeigt dabei die Bindungsenergien in kJ/mol, die x-Achse zeigt die drei Bindungspositionen des SO2.
Abbildung 6.2: Bindungsenergien des optimierten Ni-DMOF bei Cutoffs von 650 bis 750 Ry.
Die x-Achse zeigt die Bindungspositionen von SO2 am DMOF (Abb. 9.26- 9.31).
Dabei fällt auf, dass die Berechnungen, in denen nur der Gamma-Punkt bei 650 Ry berücksichtigt wurde, zu einer deutlichen Überschätzung der Bindungsenergien führt (gelbe Punkte in Abb. 6.2). Dieser Effekt tritt bei der Wahl eines Cutoffs von 700 bzw 750 Ry nicht mehr auf. Es konnte abschließend nicht geklärt werden, welchen Ursprung diese massive Überschätzung hatte. Da sich keine wesentliche Änderung der Bindungsenergie bei
der Erhöhung auf 750 Ry ergab, wurde für weitere Berechnungen ein Cutoff von 700 Ry verwendet.
Der Wert des Cutoffs hatte auf die Strukturparameter der MOFs ebenfalls keine wesentli- chen Auswirkungen (Tab. 6.3).
Tabelle 6.3: Zellparameter Ni-DMOF bei verschiedenen Cutoffs.
650 Ry 700 Ry 750 Ry
a in Å 11.000482471 10.995836932 10.995836932 b in Å 11.008065580 11.003464761 11.003464761 c in Å 9.269771682 9.265544729 9.265544729
6.1.3 Magnetisierung der Ni-DMOFs
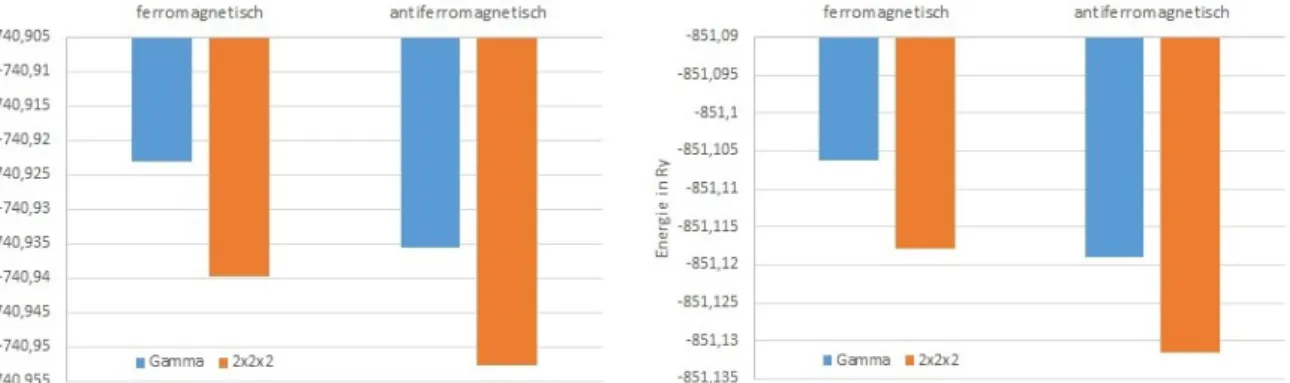
Die nickelhaltigen DMOF-Varianten wurden sowohl ferromagnetisch als auch antiferro- magnetisch betrachtet. Dabei wurde ein Cutoff von 700 Ry verwendet. Bei der ferro- magnetischen Betrachtung für beide Ni-Atome wurde ein Wert von +0,7 µB eingestellt, während hingegen für die antiferromagnetische Betrachtung jeweils bei einem Nickel eine Startmagnetisierung von +0,7 µB und bei dem benachbarten eine von -0,7 µB eingestellt wurde. In Abb. 6.3 werden die Energien unter ferromagnetischer und antiferromagnetischer Betrachtung der Ni-DMOF-Varianten gegenübergestellt.
Abbildung 6.3: Gesamtenergie von Ni-DMOF (links) und Ni-DMOF-TM (rechts) in ferro- magnetischer und antiferromagnetischer Betrachtung mit verschiedenen Gitterparametern.
Dabei lässt sich erkennen, dass die Werte der Energie bei antiferromagnetischer Betrachtung für beide Ni-DMOF-Varianten etwas energetisch tiefer liegen, als bei der ferromagnetischen
Betrachtung. Daher wurden die Ni-Atome in allen weiteren Berechnungen antiferromagne- tisch betrachtet. Des Weiteren fällt auf, dass die Energien bei allen Betrachtungsweisen für das k-Punkte Gitter tiefer liegen als für den Gamma-Punkt.
Die finale Magnetisierung der Ni-Atome liegt bei 1.2398 µB für ein Ni-Atom und -1.2431 µB für das jeweilig benachbarte. In den Arbeiten von Tan et al. finden sich die Werte 1,76 µB [47] und etwa 1 µB [3] als Magnetisierungswerte pro Ni-Atom. Damit liegt der gefundene Wert von ca. 1.2 µB dazwischen.
6.2 Vergleich der Strukturen
Im Folgenden werden die frei optimierten DMOF-TM Geometrien mit den Strukturen verglichen, bei denen die Zellparameter nach experimentellen Daten fixiert wurden. Abb. 6.4 zeigt die optimierten Einheitszellen der DMOF-TM-Varianten entlang der c-Achse. Im Bild links ist die frei optimierte Geometrie gezeigt, während im rechten Bild, bei der Optimierung, die Zellparameter aus dem Experiment eingestellt und fixiert wurden.
Abbildung 6.4: Optimierte Einheitszelle des Zn-DMOF-TM (ohne Restriktionen, links) (nach experimentellen Zellparametern, rechts) (Die des Ni-DMOF-TM sehen sehr ähnlich aus) (Abb. erstellt mit dem ProgrammXCrySDen [48]).
Bei der freien Optimierung ist eine deutliche Deformation der Einheitszelle zu erkennen.
Die Winkel α und β, zwischen den Achsen a und b, betragen hier nicht 90°. Auch die Länge der Zellachsen verändert sich, wie in Tab 6.4 zu erkennen ist.
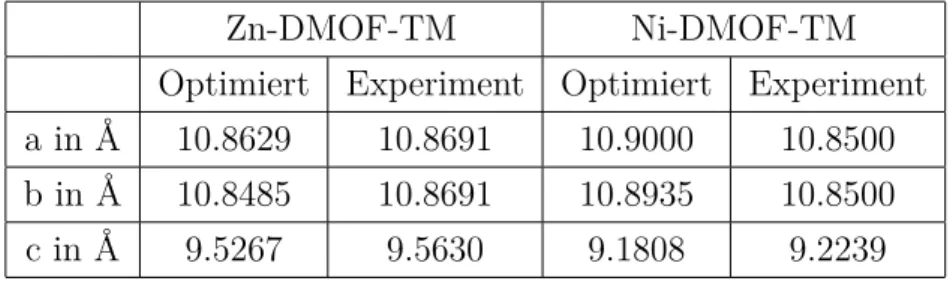
Tabelle 6.4: Zellparameter aller DMOF-TM-Varianten nach Optimierung ohne Restrik- tionen und experimentelle Daten.
Zn-DMOF-TM Ni-DMOF-TM
Optimiert Experiment Optimiert Experiment a in Å 10.8629 10.8691 10.9000 10.8500 b in Å 10.8485 10.8691 10.8935 10.8500
c in Å 9.5267 9.5630 9.1808 9.2239
Im frei optimierten Zn-DMOF-TM werden alle Zellachsen geringfügig kürzer, im Ni- DMOF-TM vergrößern sich die Beträge leicht entlang der a- und b-Achsen, während der Betrag der c-Achse etwas kleiner wird. Zudem fällt auf, dass die Zellachsen der frei optimierten Zellen unterschiedliche Längen aufweisen. Die Deformation bewirkt in jedem Fall eine Verkleinerung der Poren, somit kann die frei optimierte Struktur prinzipiell weniger Adsorbat binden.
6.3 Adsorption
6.3.1 Zn-DMOF-Varianten
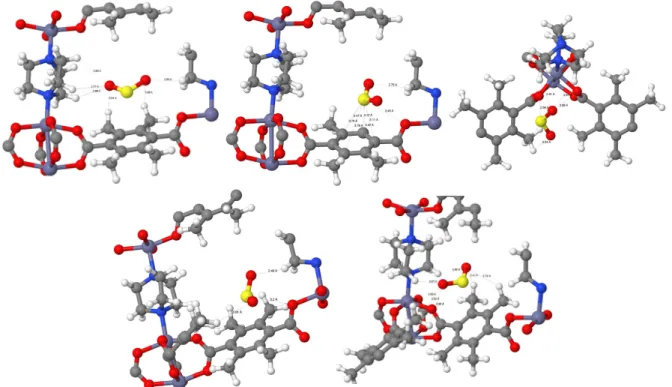
Abb. 6.5 bietet einen Überblick über die fünf betrachteten Bindungspositionen von SO2 im Zn-DMOF. Die Abbildung ist nach den Positionen 1-5 geordnet (vergrößerte Abbildungen aller einzelnen Positionen befinden sich im Anhang, Abb. 9.6-9.15).
Abbildung 6.5: Überblick über die betrachteten Bindungspositionen von SO2im Zn-DMOF (vergrößerte Abbildungen befinden sich im Anhang, Abb. 9.6-9.15).
Während der Optimierung wanderten die SO2-Moleküle von unterschiedlichen Startpunkten in sehr ähnliche Positionen, welche sich gering in Bindungslängen und Bindungsenergien unterscheiden. Das SO2 bindet mit seinen Sauerstoffatomen im Zn-DMOF an die Wasser- stoffatome der Phenylringe und des DABCOs (Position 1 und 5). Zusätzlich bindet der Schwefel des SO2-Moleküls über eine π-Bindung an den Phenylring (Position 4) und an die Sauerstoffatome der Carboxygruppen der BDCs.
Die Bindungsenergien an den verschiedenen Bindungspositionen (Pos. 1-5) werden in Tabelle 6.5 dargestellt.
Tabelle 6.5: Bindungsenergien des opt. Zn-DMOF, mit einem Cutoff von 700 Ry in kJ/mol.
Gamma 2x2x2 Pos. 1 -37,61 -36,42 Pos. 2 -33,01 -33,11 Pos. 3 -31,67 -31,81 Pos. 4 -23,70 -23,68 Pos. 5 -29,87 -29,80
Die Bindungsenergien des Gamma-Punktes und des 2x2x2 k-Punktegitters unterscheiden sich nur wenig voneinander. Position 4 gleicht unbeabsichtigt in Energie (22 kJ/mol) und Struktur der von Tan et al. [3] gefundenen Orientierung des SO2 mit π-Bindung an den Phenylring (vgl. Abb. 2.3 b). Für alle anderen Positionen konnte keine entsprechende Bindungsstelle zu denen von Tan et al. [3] gefunden werden.
Abb. 6.6 bietet einen Überblick über die fünf betrachteten Bindungspositionen von SO2 im Zn-DMOF-TM (vergrößerte Abbildungen aller einzelnen Positionen befinden sich im Anhang, Abb. 9.16-9.25).
Abbildung 6.6: Überblick über die betrachteten Bindungspositionen von SO2im Zn-DMOF- TM (vergrößerte Abbildungen befinden sich im Anhang, Abb. 9.16-9.25).
Das SO2 bindet mit seinen Sauerstoffatomen im Zn-DMOF-TM an die Wasserstoffatome der Methylgruppen (Position 1, 3, 4 und 5) und des DABCOs (Position 1 bis 5). Zusätzlich bindet der Schwefel des SO2-Moleküls über eineπ-Bindung an den Phenylring (Position 2) und an die Sauerstoffatome der Carboxygruppen der BDCs (Position 2, 3 und 4).
Die Bindungsenergien an den verschiedenen Bindungspositionen (Pos. 1-5) werden in Tabelle 6.6 dargestellt.
Tabelle 6.6: Bindungsenergien des opt. Zn-DMOF-TM, mit einem Cutoff von 700 Ry in kJ/mol.
Gamma 2x2x2 Pos. 1 -46,29 -61,65 Pos. 2 -54,06 -54,59 Pos. 3 -59,30 -59,28 Pos. 4 -52,55 -53,48 Pos. 5 -53,61 -52,62
Die Bindungsenergien des Gamma-Punktes und des 2x2x2 k-Punktegitters unterscheiden sich, außer in Position 1, kaum. Dies liegt an einer veränderten Bindungsposition, in der Gamma optimierten Struktur liegt das SO2-Molekül leicht verschoben vor. Die Bindungs- energien des unmethylierten DMOFs sind im Vergleich zum methylierten DMOF jedoch deutlich geringer.
6.3.2 Ni-DMOF-Varianten
Abb. 6.7 bietet einen Überblick über die drei betrachteten Bindungspositionen von SO2 im Ni-DMOF (vergrößerte Abbildungen aller einzelnen Positionen befinden sich im Anhang, Abb. 9.26-9.31).
Abbildung 6.7: Überblick über die betrachteten Bindungspositionen von SO2 im Ni-DMOF (vergrößerte Abbildungen befinden sich im Anhang, Abb. 9.26-9.31).
Das SO2 bindet mit seinen Sauerstoffatomen im Ni-DMOF an die Wasserstoffatome der Phenylringe und des DABCOs (Position 1 bis 3). Zusätzlich bindet der Schwefel des
SO2-Moleküls an die Sauerstoffatome der Carboxygruppen der BDCs (Position 1).
In Tabelle 6.7 werden die Bindungsenergien der verschiedenen Bindungspositionen (Pos.
1-3) dargestellt.
Tabelle 6.7: Bindungsenergien des opt. Ni-DMOF, mit einer antiferromagnetischen Start- magnetisierung von 0,7 und einem Cutoff von 700 Ry in kJ/mol.
Gamma 2x2x2 Pos. 1 -33,62 -31,45 Pos. 2 -30,86 -31,28 Pos. 3 -30,74 -31,85
Trotz der verschiedenen Optimierungsmethoden des Gamma-Punktes und des 2x2x2 k-Punktegitters unterscheiden sich die Bindungsenergien kaum voneinander. Trotz ande- rer Bindungspositionen ähneln sie den Bindungsenergien aus der Arbeit von Dr. Xing (Abb. 2.7) [6] sehr. Es fällt zudem auf, dass sich die Bindungsenergien des Zn-DMOFs und
des Ni-DMOFs ähneln.
Einen Überblick über die drei betrachteten Bindungspositionen von SO2 im Ni-DMOF-TM bietet Abb. 6.8 (vergrößerte Abbildungen aller einzelnen Positionen befinden sich im Anhang, Abb. 9.32-9.37).
Abbildung 6.8: Überblick über die betrachteten Bindungspositionen von SO2im Ni-DMOF- TM (vergrößerte Abbildungen befinden sich im Anhang, Abb. 9.32-9.37).
Das SO2 bindet mit seinen Sauerstoffatomen im Ni-DMOF-TM an die Wasserstoffatome der Methylgruppen und des DABCOs (Position 1 bis 3). Zusätzlich bindet der Schwefel des SO2-
Moleküls über eine π-Bindung (Position 2) an den Phenylring und an die Sauerstoffatome der Carboxygruppen der BDCs (Position 1 bis 3).
Die Bindungsenergien an den verschiedenen Bindungspositionen (Pos. 1-3) werden in Tabelle 6.8 dargestellt.
Tabelle 6.8: Bindungsenergien des opt. Ni-DMOF-TM, mit einer antiferromagnetischen Startmagnetisierung von 0,7 und einem Cutoff von 700 Ry in kJ/mol.
Gamma 2x2x2 Pos. 1 -45,42 -44,45 Pos. 2 -56,99 -58,33 Pos. 3 -54,15 -53,26
Die Bindungsenergien des Gamma-Punktes und des 2x2x2 k-Punktegitters unterscheiden sich kaum voneinander. Trotz anderer Bindungspositionen ähneln sie den Bindungsenergien aus der Arbeit von Dr. Xing (Abb. 2.8) [6]. Die Bindungsstelle und die Energie aus Abbildung 2.8 b) bilden dabei eine Ausnahme, da diese Bindungsstelle mit denen aus dieser Arbeit nicht vergleichbar ist. Auch hier fällt auf, dass sich die Bindungsenergien der zink- und nickelhaltigen MOFs ähneln.
6.3.3 Ni-DMOF-TM Superzelle
Abb. 6.9 zeigt die Größenrelation zwischen der Einheitszelle (links) und der Superzelle (rechts) vom Ni-DMOF-TM.
Abbildung 6.9: Größenrelation zwischen Einheitszelle (links) und Superzelle (rechts) vom Ni-DMOF-TM.
Abb. 6.10 bietet einen Überblick über die drei betrachteten Bindungspositionen von SO2 in einer Superzelle vom Ni-DMOF-TM (vergrößerte Abbildungen aller einzelnen Positionen befinden sich im Anhang, Abb. 9.38-9.40).
Abbildung 6.10: Überblick über die betrachteten Bindungspositionen von SO2 in einer Ni-DMOF-TM Superzelle (vergrößerte Abbildungen befinden sich im Anhang, Abb. 9.38-9.40).
Die Berechnung der Superzelle (2x2x2) aus dem Ni-DMOF-TM ergab eine Veränderung der ursprünglichen Bindungspositionen (Abb. 6.8), was eine Änderung der Bindungsenergien verursachte (Tab. 6.9).
Tabelle 6.9: Bindungsenergien des opt. Ni-DMOF-TM, als Superzelle und Einheitszelle in kJ/mol.
Superzelle Einheitszelle Pos. 1 -56,58 -44,45 Pos. 2 -58,85 -58,33 Pos. 3 -56,73 -53,26
Diese Bindungsenergien in der Superzelle befinden sich jedoch in einer ähnlichen Größen- ordnung wie die der Einheitszelle.
Tabelle 6.8 zeigt die Ähnlichkeit der Energien von acht Einheitszellen, verglichen mit der Energie der Superzelle.
Tabelle 6.10: Energieunterschied zwischen der Superzelle und acht Einheitszellen in Ry.
Superzelle Einheitszellen Abweichung ∆E MOF -6809,05681167 -6809,05183944 0,004972230000021
SO2 -84,65882767 -84,65903062 0,000202950000002 Pos. 1 -6893,75873991 -6893,74473208 0,004972230000021 Pos. 2 -6893,76046788 -6893,75530263 0,005165249999664 Pos. 3 -6893,75885059 -6893,75144221 0,007408380000925
Dabei wird deutlich, dass die Energie der Superzelle von der Energie der Einheitszellen nur gering abweicht.
6.3.4 Adsorptionsisothermen
Abb. 6.11 zeigt die SO2-Beladung der Zn-DMOF-Superzelle nach der Monte-Carlo- Rechnung, bei den Drücken 1, 3, 5, und 40 kPa.
Abbildung 6.11: SO2-Beladung der Zn-DMOF-Superzelle bei den Drücken 1, 3, 5, und 40 kPa.
Mit den Ergebnissen der Monte-Carlo-Rechnung lassen sich Adsorptionsisothermen kon- struieren, wie sie in den folgenden Abbildungen zu sehen sind.
Abb. 6.12 zeigt die Adsorptionsisotherme beider M-DMOF-Varianten. Die y-Achse zeigt die SO2-Adsorption in mmol/g, die x-Achse zeigt den Druck von 0-40 kPa.
Abbildung 6.12: Adsorptionsisotherme beider M-DMOF-Varianten.
Es ist deutlich erkennbar, dass die Zn-DMOF-Varianten mehr SO2 als die Ni-DMOF- Varianten adsorbieren. Die Adsorptionskurven steigen ab einem Druck von ca. 3 kPa steil an. Der starke Anstieg der Adsorption flacht bei einem Druck von ca. 7 kPa ab. Die Sättigungswerte der DMOFs liegen bei ca. 14 und 13 mmol/g. Die Adsorption der DMOFs wird im Vergleich zu den Ergebnissen von Tan et al. (Abb. 2.4) und Dr. Xing (Abb. 2.5) [3, 6] überschätzt.
Abb. 6.13 zeigt die Adsorptionsisotherme beider M-DMOF-TM-Varianten, welche ohne Restriktionen optimiert wurden.
Abbildung 6.13: Adsorptionsisotherme beider M-DMOF-TM-Varianten, optimiert ohne Restriktionen.
Auch hier ist deutlich erkennbar, dass die Zn-DMOF-Varianten mehr SO2 als die Ni-DMOF- Varianten adsorbieren. Schon bei sehr niedrigen Drücken steigen die Adsorptionskurven sofort steil an. Der starke Anstieg der Adsorption flacht jedoch schon bei einem Druck von ca. 2 kPa ab. Die Sättigungswerte der DMOFs liegen bei ca. 4 und 3,8 mmol/g. Damit liegt der Sättigungswert des Ni-DMOF-TM deutlich unter den Ergebnissen von Hungerford et al. (Abb. 2.2) und Dr. Xing (Abb. 2.5) [1, 6].
Abb. 6.14 zeigt die Adsorptionsisotherme beider M-DMOF-TM-Varianten, welche nach experimentellen Zellparametern optimiert wurden.
Abbildung 6.14: Adsorptionsisotherme beider M-DMOF-TM-Varianten, optimiert nach experimentellen Zellparametern.
Es ist ebenfalls deutlich erkennbar, dass die Zn-DMOF-Varianten mehr SO2 als die Ni- DMOF-Varianten adsorbieren. Ebenso steigt hier die Adsorption schon bei sehr niedrigen Drücken steil an, flacht jedoch bei einem Druck von ca. 3 kPa wieder ab. Die Sättigungs- werte der DMOF-TM-Varianten, welche nach experimentellen Daten optimiert wurden, liegen bei ca. 7,5 und 7 mmol/g. Damit wird der Sättigungswert des Ni-DMOF-TM, wie in den Ergebnissen von Hungerford et al. (Abb. 2.2) und Dr. Xing (Abb. 2.5) [1, 6], erreicht.
Die unmethylierten DMOFs adsorbieren gesättigt am meisten SO2, die tetramethylierten DMOFs, optimiert nach experimentellen Zellparametern, adsorbieren gesättigt am zweit- besten SO2 und dabei ungefähr doppelt so viel wie die DMOFs, welche ohne Restriktionen optimiert wurden. Bei niedrigen Drücken (bis ca. 3 kPa) adsorbieren die tetramethylierten DMOFs mehr SO2 als die unmethylierten DMOFs, jedoch ist ihre Sättigung sehr schnell erreicht. Die Zn-DMOFs adsorbieren SO2 stärker als Ni-DMOFs, was im Widerspruch zu den Ergebnissen von Hungerford et al. (Abb. 2.2) und Tan et al. (Abb. 2.4) [1, 3] steht.