Untersuchungen zum Einfluss der GPI-Verankerung auf das Faltungsverhalten des Prion Proteins
163
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
(39)
(40)
(41)
(42)
(43)
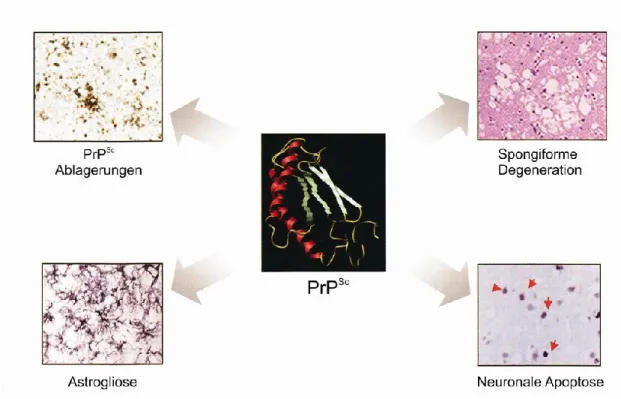
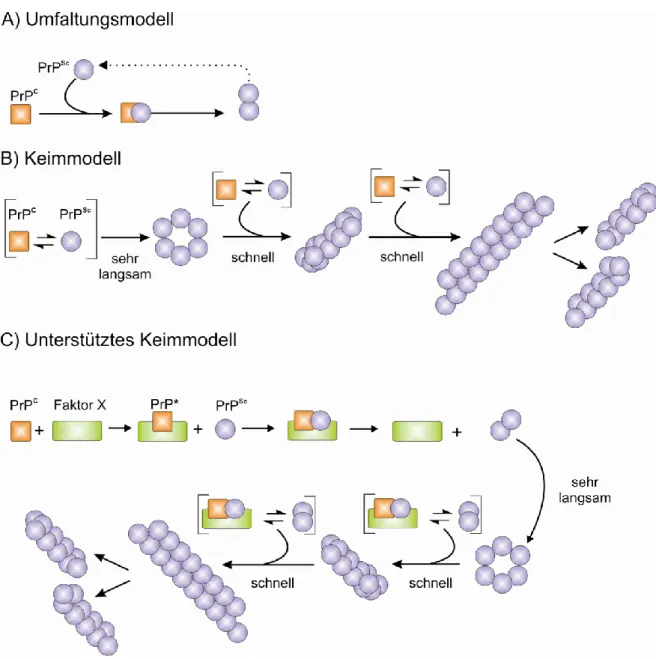
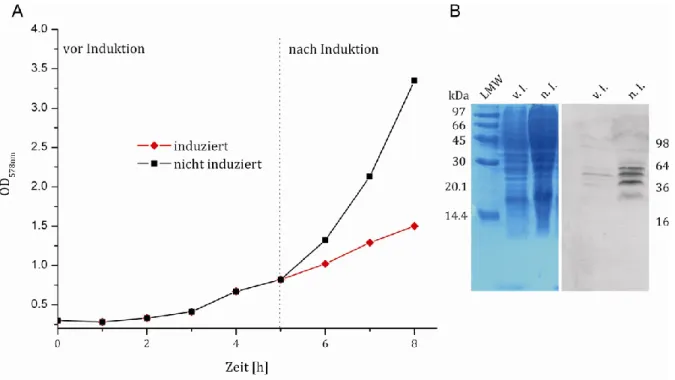
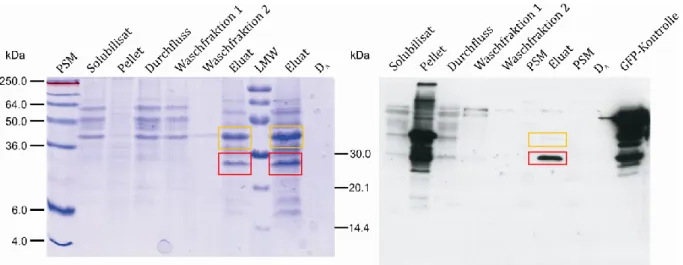
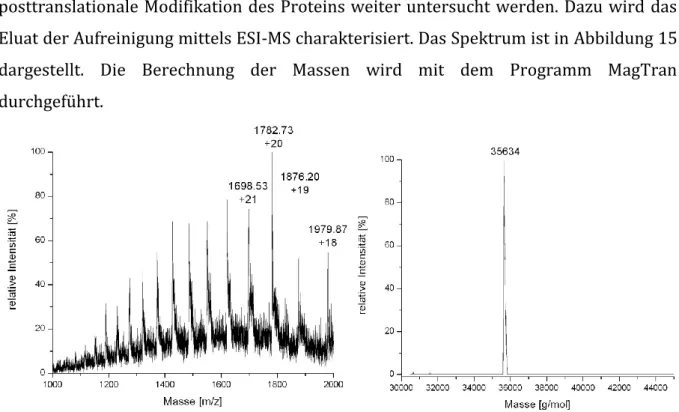
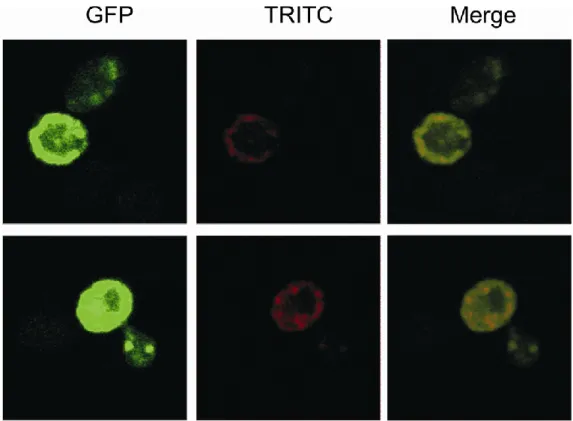
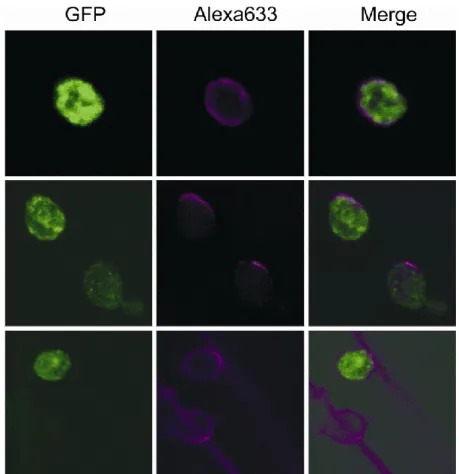
Abbildung


+7

ÄHNLICHE DOKUMENTE
Es wurde gezeigt, dass das erste Drittel des Proteins die Prion- Determinierende-Domäne ist, welche für die Aggregation, nicht aber für die Funktion als Terminationsfaktor
Für den Einsatz frühzeitiger, diagnostischer Testverfahren für Scrapie bei lebenden Schafen, die auf dem Nachweis von PrP Sc in lymphatischen Geweben basieren, ist zunächst genauer
In dieser Arbeit konnte gezeigt werden, dass PrP C eine Hochregulation der LDH-Expression unter Hypoxie in den Zelllinien HEK-293 und Prnp 0/0 , sowie in kortikalen Neuronen (Maus)
In addi- tion to this previously described behavior (Rabl et al. 2017), we also compared ThySynPrP00 with PrP-KO mice showing that the phenotype of transgenic aSyn mice was
Um den Einfluss der PrPSc-Typen auf die Expression der PrPC-Isoformen im Liquor von sCJK-Patienten zu untersuchen, wurden Proben mit Prion Typen 1 und 2, ungeachtet des PRNP-Genotyps
Als weitere potenzielle Wirkstoffklasse wurden kurze Peptide mittels NMR-Spektroskopie auf Interaktionen mit PrP C untersucht. Mit einer Phagendisplay-Bibliothek waren in der
Diese wurden für die Regulation der Expression des PrP C gebraucht und sollten, da sie von einem anderen Organismus stammten, nicht für eine mögliche Funktion des PrP C
scheint eine solche Übertragung zwi- schen den zwei Tiergruppen eher un- wahrscheinlich, jedoch wurden Prio- nen-Erkrankungen in „niederen Orga- nismen“ lange von der Forschung
