Untersuchungen zur Konformation und Dynamik der Reversen Transkriptase von HIV-1 durch ESR- und Einzelmolekülfluoreszenzspektroskopie
147
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
Abbildung
![Abb. 1: Schematische Darstellung des humanen Immunschwächevirus HIV-1 [12] .](https://thumb-eu.123doks.com/thumbv2/1library_info/3654172.1503453/11.892.108.744.296.628/abb-schematische-darstellung-humanen-immunschwächevirus-hiv.webp)
![Abbildung 1.5-1: Kristallstruktur der HIV-1 RT im Komplex mit einer 18/19mer DNA/DNA, die in magenta/hellblau gezeigt ist [26]](https://thumb-eu.123doks.com/thumbv2/1library_info/3654172.1503453/15.892.104.791.102.628/abbildung-kristallstruktur-hiv-komplex-dna-magenta-hellblau-gezeigt.webp)
![Abbildung 1.5-2: Kristallstruktur der Finger- und Daumen-Domäne der p66-Untereinheit von ligandenfreier HIV-1 RT (grün, [27] ) und RT im Komplex mit dsDNA (lila [28] )](https://thumb-eu.123doks.com/thumbv2/1library_info/3654172.1503453/16.892.117.581.258.765/abbildung-kristallstruktur-finger-daumen-domäne-untereinheit-ligandenfreier-komplex.webp)
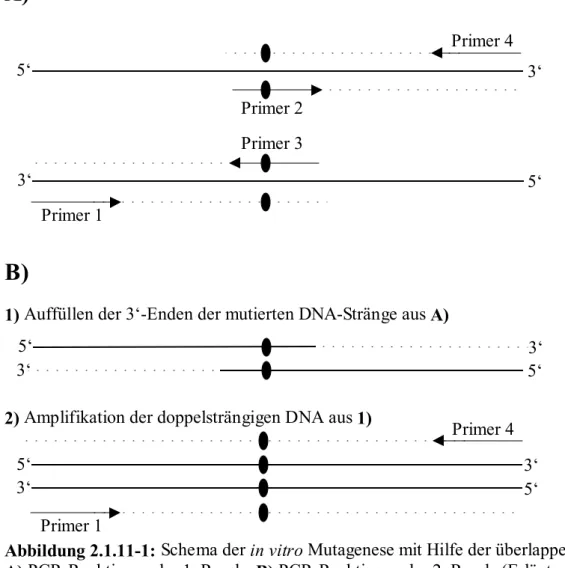
+7
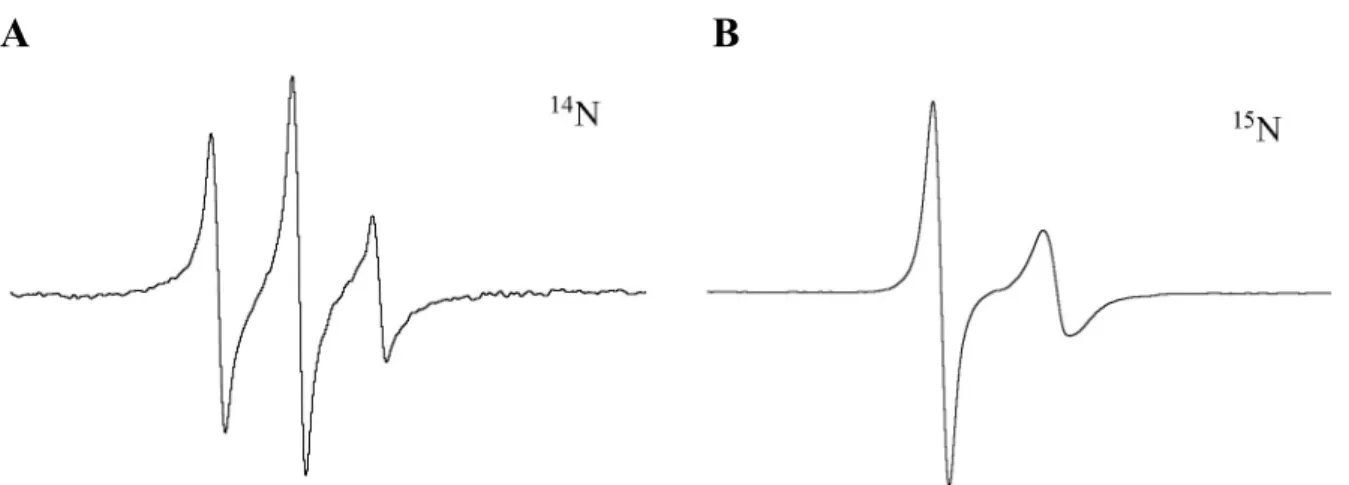
![Abbildung 2.5.1.2-3: Abhängigkeit der Linienform eines ESR-Spektrums von der Beweglichkeit/Rotationskorrelationszeit einer kovalent an ein Protein gebundenen Spinsonde [52] .](https://thumb-eu.123doks.com/thumbv2/1library_info/3654172.1503453/45.892.109.410.426.837/abbildung-abhängigkeit-linienform-spektrums-beweglichkeit-rotationskorrelationszeit-gebundenen-spinsonde.webp)
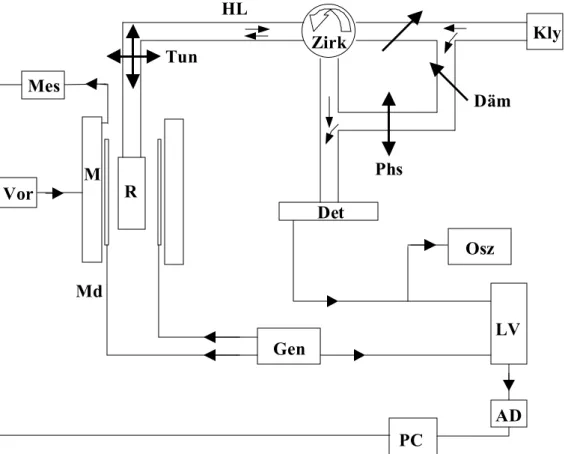
ÄHNLICHE DOKUMENTE
Darunter versteht man die Zeit, die ein Körper oder ein Punkt auf einer Kreisscheibe für eine volle Umdrehung benötigt.. (gemessen in 1 s oder auch
Stelle eine allgemeine Formel auf, mit der du die Flugweite eines schiefen Wurfs mit der Geschwindigkeit v, dem Winkel α und der Höhe h des Abwurfpunktes berechnen kannst.
In der Mathematik wird der Übergang von einer Funktion f(x) zu derjenigen Funktion, welche die Steigung an jedem x-Wert angibt, als „Ableitung der Funktion f“ genannt,
Wenn man 2,5 m über die letzte Sicherung hinausgeklettert ist, fällt man 2,5 m unter die Siche- rung (also 5 m), bis man vom Seil aufgefangen wird. Der Bremsweg setzt sich aus 10
Wenn ihr den Abstand von einem Punkt zum nächsten messt, wirkt sich eine Ungenauigkeit von einem halben Millimeter schon deutlich aus.. Bestimmt besser den Abstand von 5
Mit welcher Geschwindigkeit muss das Auto durch die Kurve rasen, damit es nicht an die Leitplanken gedrückt wird. • Fahrgeschäft
4–8: καί μοι δοκεῖ τὸ τὴν νόησιν ἔχον εἶναι ὁ ἀὴρ καλούμενος ὑπὸ τῶν ἀνθρώπων, καὶ ὑπὸ τούτου πάντας καὶ κυβερνᾶσθαι καὶ πάντων κρα τεῖν· αὐτὸ γάρ μοι τοῦτο
funktionswidrigen Einsatzes der Aktionärsanfechtungsbefugnis. Kapitel Bisherige legislatorische Maßnahmen gegen den funktions-.. widrigen Einsatz der
