Apoptosemodulation nach selektiver Inhibition der Cyclooxygenase-2 (COX-2) in humanen
144
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(27)
(28)
(29)
(30)
(31)
(32)
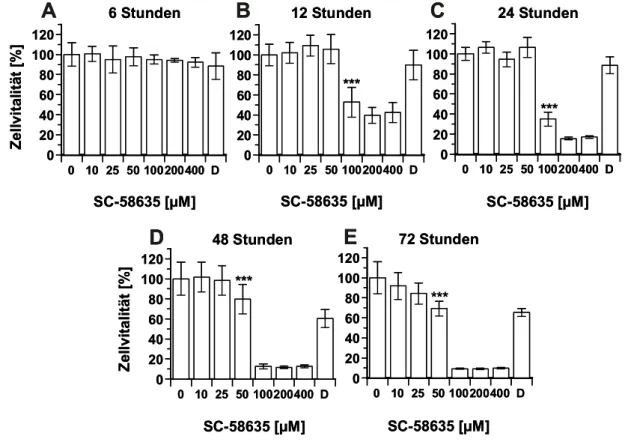
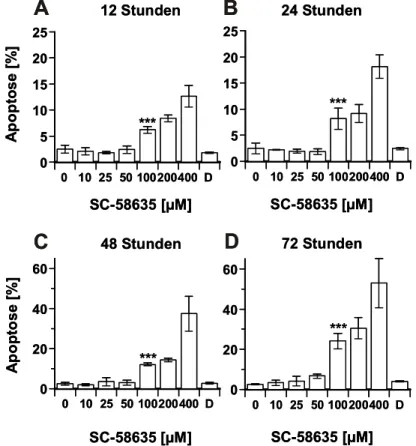
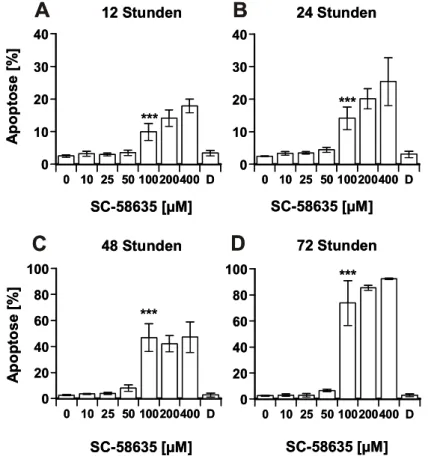
Abbildung
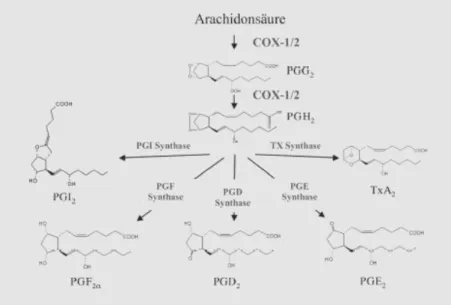
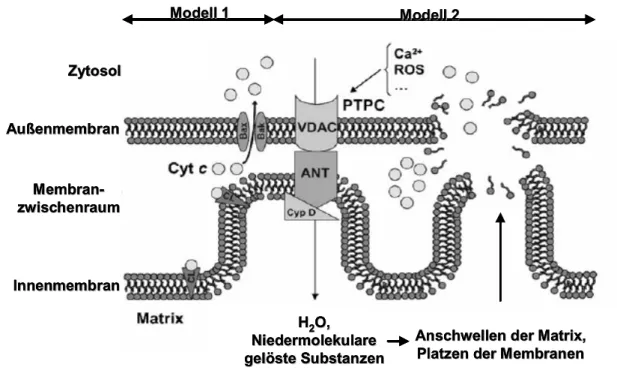
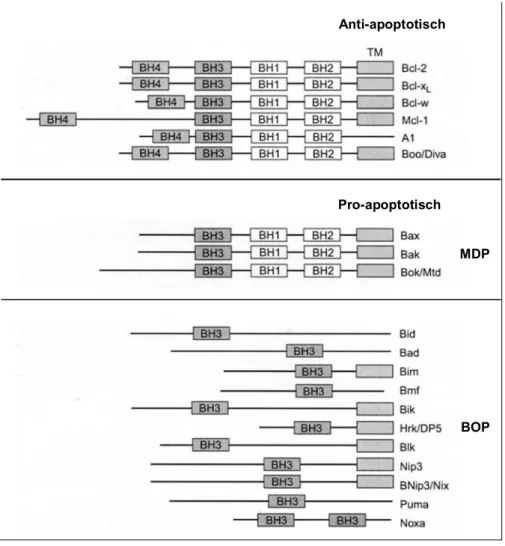
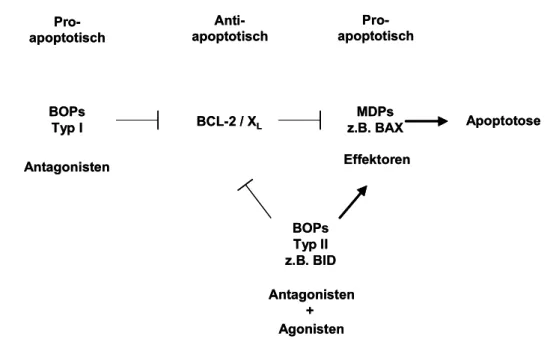
+7



ÄHNLICHE DOKUMENTE
mit "Lonesome Cowboys" Story eines unverschämt hüb- den akuten Fall dar: Der Film schen Heroinsüchtigen und ist eine Parodie auf den We- seiner von einem Mann ge- stern,
„Ich, Julia Buchwald, erkläre, dass ich die vorgelegte Dissertationsschrift mit dem Thema: „Die Co-Expression der Cyclooxygenase-2 (COX-2) und der Rezeptoren der Epidermal Growth
Gestaltet bitte pro Kirchenkreis ein gemeinsames Feld auf einer Pinnwand der Arbeitsfelder AmK und Jugendarbeit?. - Größe maximal 2 X A3 = A2 - werdet
Clemens Bethge, Konsistorium, Referat 2.2 Kirchliches Leben im Anschluss Gespräch der Konferenz mit Herrn Bethge: Die Entwicklung und Weiterentwicklung im Arbeitsbereich Arbeit
Die Strafe für die Übertretung wird entweder den Göttern überlassen, indem sich das verletzte Tabu von selbst rächt, oder die Gesellschaft bestraft jenen Verwegenen, der sie
2.4.3 Reflexion historischer Anamnesekonzepte der Sozialen Arbeit ..... Anamnesekonzepte in der Sozialen Arbeit
Hierzu trägt die Autorin doppelt bei: zum einen durch die systematische Aufarbeitung der Konzepte und des Forschungsstands, zum anderen durch ihre subtile Fallstudie im Rahmen
Neben dem Präsidenten des Bayerischen Landesamtes für Umwelt und einem Vertreter des Sachverständigenrates für Umweltfragen kommen auch Politiker*innen zu Wort?. Der Bürgermeister
