T48 reguliert mit Fog/Concertina die Zellformveränderungen während der Mesoderminvagination in Drosophila melanogaster
122
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(26)
(27)
(28)
(30)
(31)
(33)
(34)
(35)
(36)
(37)
(38)
(39)
(40)
Abbildung
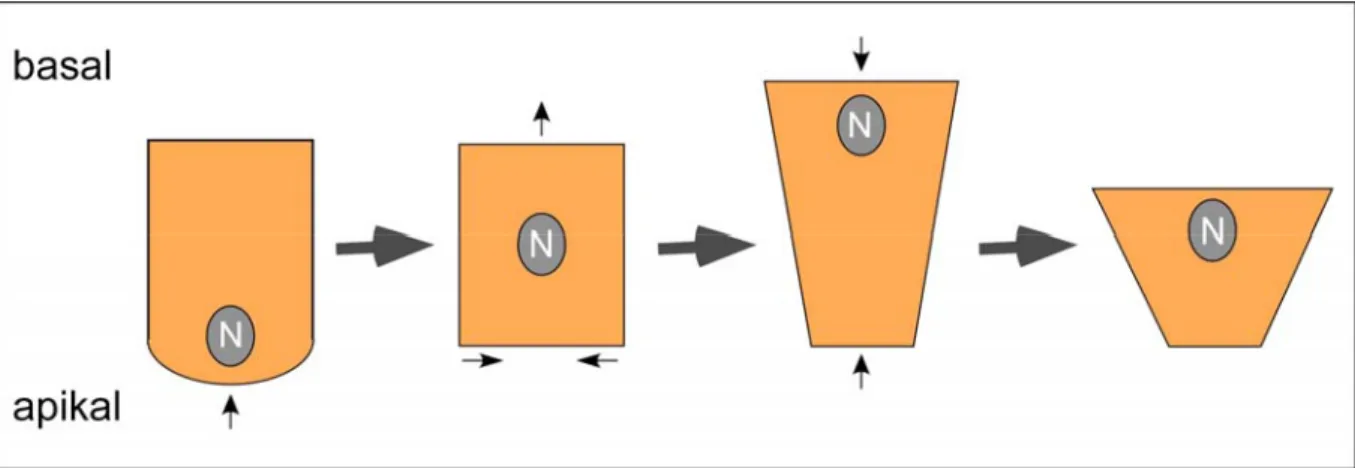
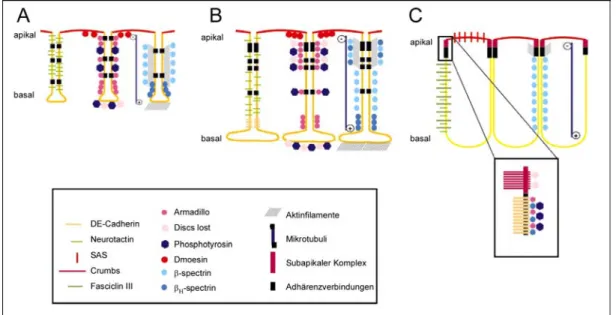
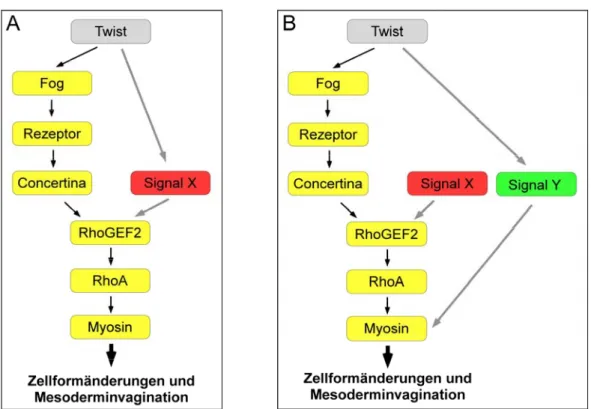
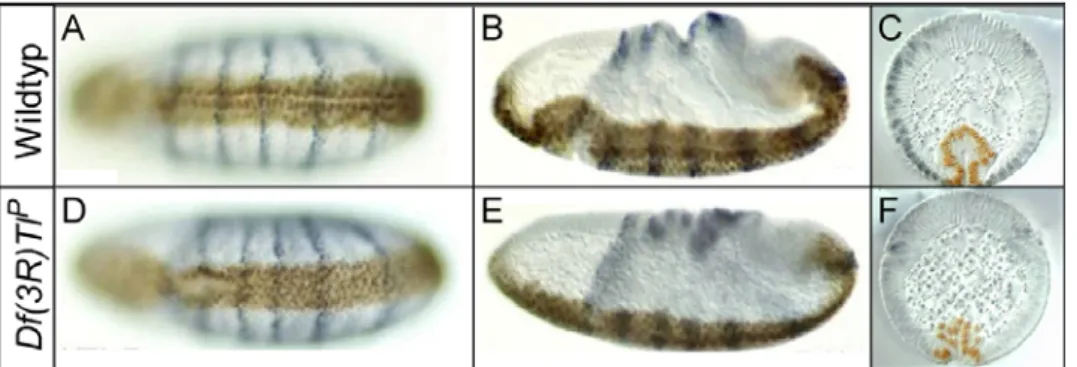
+7
ÄHNLICHE DOKUMENTE
Mutational analysis of a histone deacetylase in Drosophila melanogaster: missense mutations suppress gene silencing associated with position effect variegation.. Types of
Jedoch konnte spä- ter gezeigt werden, dass sowohl die Serratia-Nuklease, als auch die Anabaena- NucA in der Lage sind die synthetische Verbindung
After the population reproduces the complete Hi-C data, we include the vector E (lamina- DamID), again in stages with decreasing contact probability thresholds (λ).... Snapshot of
Das Controlling des Amtes für Betriebswirtschaft und Aufsicht der Justiz-, Gemeinde- und Kir- chendirektion (JGK) zeigt auf, dass die Budgetkredite auf den Kontengruppen 301 (Löhne des
Die etablierte Funktion von Rca1 als Inhibitor des APC/C wurde vor allem durch Überexpression von Rca1-Derivaten während der Embryogenese ermittelt. Für die konservierte F-box
Während eine Aktivierung der Transkription durch den Kofaktor GCN5 mit einer starken Acetylierung des Chromatins in Bereichen GCN5-abhängiger Promotoren einherging, konnten
Bypassing was first identified in the translation of the gene 60 of bacteriophage T4 (Huang et al., 1988; Weiss et al., 1990), where the ribosome translates the mRNA until it
melanogaster, the uniqueness of the X chromosome is reflected by its sex-biased gene content and its special mechanisms of gene expression regulation, such as dosage
