Research Collection
Doctoral Thesis
Regulation of DNA Helicases in Homologous Recombination
Author(s):
Grigaitis, Rokas Publication Date:
2021
Permanent Link:
https://doi.org/10.3929/ethz-b-000474764
Rights / License:
In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use.
ETH Library
DISS. ETH NO. 27364
REGULATION OF DNA HELICASES IN HOMOLOGOUS RECOMBINATION
A thesis submitted to attain the degree of DOCTOR OF SCIENCES of ETH ZURICH
(Dr. sc. ETH Zurich)
presented by ROKAS GRIGAITIS
Master of Biochemistry, Vilnius University born on 22.03.1991
citizen of Lithuania
accepted on the recommendation of Prof. Dr. Volodymyr Korkhov
Prof. Dr. Joao Matos Prof. Dr. Petr Cejka
PD Dr. Reinhard Christoph Dechant
2021
2
3
TABLE OF CONTENTS
DECLRATION OF CONTENT 5
SUMMARY 6
ZUSAMMENFASSUNG 8
1. INTRODUCTION 10
1.1 DNA double strand break repair by homologous recombination 10
1.2 Mechanism of homologous recombination 11
1.2.1 DNA end resection 12
1.2.2 Search for homologous template and strand invasion 14
1.2.3 Formation of recombination intermediates 16
1.2.4 Processing of recombination intermediates 18
1.3 Regulation of recombination intermediate processing 21
1.3.1 Regulation of DNA helicases 21
1.3.2 Regulation of DNA nucleases 22
1.4 DNA helicase Sgs1 and its importance for homologous recombination 23
1.5 Goals of the thesis 24
2. EXPERIMENTAL PROCEDURES, RESULTS AND DISCUSSION 26 2.1 Characterization of DNA helicases and nucleases from meiotic extracts of S. cerevisiae 26 2.1.1. Synchronization and harvesting of large meiotic cultures of S. cerevisiae 26
2.1.1.1. Analysis of the results 28
2.1.2. Purification and characterization of the STR complex from prophase I 28 2.1.2.1. Immuno-affinity purification of FLAG-tagged STR components from prophase I lysates 28 2.1.2.2. Elution of Rmi1-FLAG and detection of co-purifying proteins using SYPRO Ruby stain 30
2.1.2.2.1. Data analysis 31
2.1.2.3. On-bead digest followed by MS-based analysis of immuno-affinity purified Top3-FLAG 31
2.1.2.3.1. Analysis of the results 31
2.1.3. Functional analysis of Sgs1 helicase and Mus81-Mms4 nuclease from meiotic extracts 32 2.1.3.1. Analysis of DNA strand-displacement activity in Sgs1-FLAG immunoprecipitates from meiotic
prophase I 32
2.1.3.1.1. Immunoprecipitation of Sgs1-FLAG from prophase I lysates of S. cerevisiae 32
2.1.3.1.2. Preparation of a synthetic gap-DNA substrate 34
2.1.3.1.3. DNA strand-displacement activity assay on agarose beads 35
2.1.3.1.4. Analysis of the results 36
2.1.3.2. Monitoring the activity of semi-purified Mus81-Mms4-FLAG from meiotic metaphase I 36 2.1.3.2.1. Immunoprecipitation of FLAG-tagged Mus81-Mms4 from meiotic yeast lysates 36 2.1.3.2.2. Preparation of a synthetic nicked Holliday junction 37
2.1.3.2.3. Nuclease activity assay on agarose beads 37
2.1.3.2.4. Analysis of the results 37
2.1.4 Adaptation to other DNA processing enzymes and DNA substrates 39 2.2 Regulatory control of RecQ helicase Sgs1/BLM during meiosis and mitosis 40
2.2.1. RESULTS 40
2.2.1.1. Sgs1 is sequentially phosphorylated by Cdc28/CDK and Cdc5/PLK kinases during meiosis 40 2.2.1.2 Cdc28/CDK-mediated phosphorylation enhances the DNA unwinding activity of Sgs1
immunoprecipitates 43
2.2.1.3. Cdc5-mediated hyper-phosphorylation may reduce the DNA unwinding activity of Sgs1 at the
prophase-to-metaphase I transition 44
2.2.1.4 Meiotic phosphorylation of Sgs1 does not regulate the association with Top3-Rmi1 46 2.2.1.5 Phosphorylation enhances the DNA unwinding activity of recombinant Sgs1 47 2.2.1.6 Phosphorylation reduces the affinity of Sgs1 for dsDNA 47 2.2.1.7 Phosphorylation increases the DNA unwinding velocity and processivity of Sgs1 50 2.2.1.8 Phospho-stimulation of Sgs1 activity is required for meiotic DNA joint molecule processing and
chromosome segregation 51
2.2.1.9 Phospho-activation of Sgs1 is required for efficient DNA repair during mitotic proliferation 54
2.2.2 DISCUSSION 56
2.2.2.1 Wiring Sgs1 function to meiotic and mitotic cell cycle progression through phosphorylation 57 2.2.2.2 Concerted regulation of helicase and nuclease activities during recombination in meiotic and
mitotic cells 57
2.2.2.3 Mechanistic basis for orderly formation of noncrossovers and crossovers through sequential
waves of helicase and nuclease activation 58
4
2.2.2.4 Open questions on the regulation of Sgs1/STR 59
2.2.3 SUPPLEMENTARY FIGURES 61
2.2.4 METHODS 66
2.2.4.1 Experimental Model and Subject Details 66
2.2.4.2 Meiotic time courses and vegetative cultures 66
2.2.4.3 FACS Analysis of DNA content 67
2.2.4.4 Protein purification from mitotic and meiotic cultures 67
2.2.4.5 Expression and purification of recombinant proteins 68
2.2.4.6 In vitro phosphorylation and dephosphorylation 68
2.2.4.7 DNA substrate preparation 69
2.2.4.8 DNA unwinding with Sgs1 immunoprecipitates 69
2.2.4.9 DNA unwinding assays with purified Sgs1 70
2.2.4.10 DNA binding assay 70
2.2.4.11 Magnetic tweezers assay 70
2.2.4.12 Transmission electron microscopy 71
2.2.4.13 Analysis of spore viability 71
2.2.4.14 Fluorescence microscopy 71
2.2.4.15 Protein analyses by western blotting 72
2.2.4.16 Analysis of recombination at the HIS4-LEU2 locus 72
2.2.4.17 Quantification and Statistical Analysis 73
3. OUTLOOK 74
4. APPENDIX 76
4.1 Tables 76
ACKNOWLEDGEMENTS 79
REFERENCES 80
5
DECLARATION OF CONTENT
I, Rokas Grigaitis, have written this PhD thesis by myself. The experimental findings described in this thesis represent work performed by myself with contributions from current and past members of the research groups of J. Matos, P. Cejka, R. Seidel and M. Peter. The contributions of others are acknowledged at the beginning of every chapter. Parts of this thesis have been published in:
Grigaitis R, Ranjha L, Wild P, Kasaciunaite K, Ceppi I, Kissling V, Henggeler A, Susperregui A, Peter M, Seidel R, Cejka P, Matos J (2020). Phosphorylation of the RecQ Helicase Sgs1/BLM Controls Its DNA Unwinding Activity during Meiosis and Mitosis. Dev Cell 53(6), 706-723.
Grigaitis R, Susperregui A, Wild P, Matos J (2018). Characterization of DNA helicases and nucleases from meiotic extracts of S. cerevisiae. Methods Cell Biol 144: 371-388.
6
SUMMARY
During their lifetime, cells encounter various forms of DNA lesions, among which – DNA double strand breaks (DSBs). DSBs can arise from exposure to ionizing radiation, genotoxic compounds or replicative and oxidative stress. They can also be introduced deliberately by the cell, as happens in sexually reproducing eukaryotes during meiosis. DSBs are arguably the most dangerous of all DNA lesions: if unrepaired, chromosome discontinuity may lead to cellular death or cancer. DSBs can be repaired by two distinct pathways: non-homologous end joining (NHEJ) and homologous recombination (HR). While NHEJ is a relatively mutagenic process, HR is generally of high fidelity.
HR is a complex biochemical process, which involves multiple steps. The most important ones are 5’ end resection, search for homology, strand invasion, DNA synthesis and processing of recombination intermediates. Interestingly, processing of recombination intermediates can lead to two different outcomes. First, they can be converted into noncrossovers – recombinants where no reciprocal exchange of DNA has occurred. Conversely, a crossover is formed when two recombining chromosomes exchange their arms. In addition, the two outcomes of HR can have different effects on cells. In meiosis, inter-homolog crossing over is required to ensure efficient pairing and segregation of homologous chromosomes, creating novel haplotypes. On the other hand, during mitotic proliferation crossing over between homologous chromosomes might expose recessive deleterious mutations. Thus, noncrossover generation is highly favored.
The ability to form different products of HR is based on the existence of diverse recombination intermediate processing enzymes. In S. cerevisiae, the DNA helicases Sgs1, Srs2 and Mph1 bind to and unwind recombination intermediates generating noncrossovers. The structure- selective endonucleases (SSEs) Mus81-Mms4, Slx1-Slx4 and Yen1 hydrolyze recombination intermediates in a way that produces a mixture of crossovers and noncrossovers. The MutLγ- Exo1 nuclease complex, on the other hand, exclusively generates crossovers. All of these enzymes are present in both mitotic and meiotic cells, thus their existence alone does not explain how cells rewire HR to satisfy specific cellular requirements. It is likely that this is achieved through spatiotemporal regulation of the aforementioned enzymes. Indeed, previous research, has established that the functions of MutLγ-Exo1 complex as well as SSEs are regulated either through post-translational modifications or cell cycle stage-specific interactions with other proteins. However, not much is known about the control of anti- crossover DNA helicases.
Backed by several indirect cues, I postulated that the DNA helicases Sgs1, Srs2 and Mph1 might be regulated in a context-dependent manner, which could be important for the metabolism of recombination intermediates and correct HR outcome. To understand the
7 molecular mechanisms of such regulation, I chose to focus on the RecQ-family DNA helicase Sgs1. Briefly, experimental evidence presented in this thesis reveals that Sgs1 is sequentially phosphorylated by two cell cycle kinases – CDK and polo kinase Cdc5. Phosphorylation by CDK, during S-phase, increases the velocity and processivity of Sgs1, enhancing its DNA unwinding activity in a cell cycle stage-specific manner. In mitotically proliferating cells, S- phase phosphorylation of Sgs1 is required for efficient DNA repair. Whereas during meiosis, the CDK-mediated upregulation of Sgs1 activity is required to prevent the accumulation of multichromatid DNA joint molecules, aberrant crossing over and chromosome nondisjunction.
Overall, the work presented in this thesis demonstrates that Sgs1 is tightly regulated in cells, raising the possibility that other RecQ-family DNA helicases, as well as other anti-crossover DNA helicases, may also be regulated. In addition, this work deepens our understanding on how cells rewire the metabolism of recombination intermediates to achieve the correct outcome of HR.
8
ZUSAMMENFASSUNG
Während ihrer Lebensdauer werden Zellen ständig mit verschiedenen Formen von DNS- Läsionen konfrontiert. Unter anderem mit DNS-Doppelstrangbrüchen (DSB). DSB können durch die Einwirkung von ionisierender Strahlung, genotoxischer Wirkstoffe oder oxidativem Stress hervorgerufen werden. Jedoch können sie auch absichtlich von der Zelle verursacht werden. Dies geschieht zum Beispiel während der Meiose von sich sexuell reproduzierenden Eukaryoten. DSB gehören wohl zu den gefährlichsten aller DNS-Läsionen. Wenn sie nicht repariert werden, können die entstehenden Brüche in den Chromosomen zu Krebs oder auch zum Zelltod führen. DSB können von zwei unterschiedlichen zellulären Mechanismen repariert werden: Nicht-homologe Endverbindung (NHJE) und Homologe Rekombination (HR).
Während eine Reparatur durch NHEJ vergleichsweise oft zu Mutationen führt, zeichnet sich HR durch hohe Genauigkeit bei der Reparatur aus.
HR ist ein komplexer biochemischer Prozess, welcher sich aus mehreren Einzelschritten zusammensetzt. Dabei sind die wichtigsten Schritte die 5’ End-resektion, die Suche nach homologen Chromosomen, DNS Doppelstrang-invasion, DNS Synthese und die Weiterverarbeitung von Rekombinationsintermediaten. Interessanterweise können die Zwischenprodukte des Rekombinationsprozesses zu zwei unterschiedlichen Endprodukten verarbeitet werden – sogenannte Noncrossover und Crossover. Bei der Bildung von Noncrossover-Produkten findet kein reziproker Austausch von DNS statt. Im Gegenteil dazu, werden bei der Crossoverbildung die Arme der rekombinierenden Chromosomen und somit DNS ausgetauscht. Es ist wichtig zu vermerken, dass die unterschiedlichen Endprodukte der Rekombination abweichende Auswirkungen auf die Zelle haben können. Während der Meiose ist die Bildung von Crossover zwischen homologen Chromosomenpaaren erfordert. Dies stellt sicher, dass sich die homologen Chromosomen finden und später korrekt auf die neuen Zellen aufgeteilt werden können. Ausserdem führt dies zuverlässig zur Bildung neuer Haplotypen.
Im Gegenteil dazu, können Crossover während der Mitose dazu führen, dass rezessive, meist schädliche Mutationen zum Vorschein kommen. Um dies zu verhindern, wird während der Mitose die Bildung von Noncossovern bevorzugt.
Die Fähigkeit unterschiedliche Rekombinationsprodukte zu Bilden, beruht auf einer Vielzahl von unterschiedlichen Enzymen welche die Rekombinationsintermediate verarbeiten können.
In S. cerevisiae binden die DNS-Helikasen Sgs1, Srs2 und Mph1 an die Rekombinationsintermediate und entwinden diese, was zur Entstehung von Noncrossovern führt. Struktur-selektive Endonukleasen (SSEs) wie Mus81-Mms4, Slx1-Slx4 und Yen1 produzieren sowohl Crossover als auch Noncrossover, indem sie die Rekombinationsprodukte in einer spezifischen Konformation hydrolysieren. Im Gegensatz dazu produziert der MutLγ- Exo1 Nukleasenkomplex nur Crossover-Endprodukte. Alle diese Enzyme sind sowohl in
9 mitotischen als auch in meiotischen Zellen zu finden. Deshalb erklärt die Expression der Enzyme alleine nicht, wie die Zelle die HR so steuert, dass sie den jeweiligen zellzyklusspezifischen Anforderungen Gerecht wird. Es wird vermutet, dass diese Steuerung durch die räumlich-zeitliche Regulation der verantwortlichen Enzyme erreicht wird. Tatsächlich wurde in vorangehenden Studien gezeigt, dass sowohl der MutLγ-Exo1 Komplex als auch SSEs durch post-translationale Modifikationen und zellzyklusspezifische Interaktionen mit anderen Proteinen reguliert werden. Über die Kontrolle der Noncrossover produzierenden DNS-Helikasen hingegen, ist weniger bekannt.
Auf Grund verschiedener indirekter Hinweise habe ich die Hypothese aufgestellt, dass auch die DNS-Helikasen Sgs1, Srs2 und Mph1 kontextabhängig reguliert sein könnten. Dies wiederum wäre möglicherweise wichtig für die korrekte Verarbeitung von Rekombinationsintermediaten und die Bildung der richtigen Rekombinationsendprodukte. Um die molekularen Grundlagen einer solchen Regulation zu verstehen, habe ich mich auf die RecQ-familien DNS-Helikase Sgs1 fokussiert. Zusammenfassend haben die im Rahmen dieser These präsentierten Experimente gezeigt, dass Sgs1 von zwei Zellzykluskinasen – CDK und Polokinase Cdc5 – sequenziell phosphoryliert wird. Dabei erhöht die erste Phosphorylierung durch CDK während der S-phase die Geschwindigkeit und Prozessivität von Sgs1. Dies steigert die Fähigkeit von Sgs1, DNS zu entwinden. In mitotischen Zellen wird die Phosphorylierung von Sgs1 für die effiziente Reparatur von DNS benötigt. In meiotischen Zellen verhindert die durch CDK verursachte Verstärkung der Sgs1 Aktivität die Anhäufung von multichromosomalen DNS Molekülen, aberranten Crossovern und Chromosom Non- disjunktionen. Die hier präsentierte Arbeit zeigt, dass Sgs1 streng reguliert ist. Dies legt nahe, dass andere RecQ-familien DNS-Helikasen sowie weitere anti-crossover DNS-Helikasen auch reguliert sein könnten. Zusätzlich vertieft diese Arbeit das Verständnis wie Zellen die Verarbeitung von Rekombinationsintermediaten den zellulären Bedürfnissen anpasst und für ein korrektes Ergebnis bei der Homologen Rekombination sorgt.
10
1. INTRODUCTION
1.1 DNA double strand break repair by homologous recombination
Throughout the lifetime of a cell, its DNA can undergo various types of modifications. While some of them are important for the regulation of DNA metabolism, such as transcription (Heberle and Bardet, 2019), others might disrupt or alter cellular processes in a detrimental way (Chatterjee and Walker, 2017). The latter are called DNA lesions. The majority of DNA lesions affect the sequence, structure or continuity of DNA molecules and can cause mutations or chromosomal aberrations, contributing to cellular senescence, death or cancer (Chatterjee and Walker, 2017).
DNA double strand breaks (DSBs) disrupt the continuity of DNA molecules. They occur due to exposure to certain external chemicals as well as ionizing radiation, reactive oxygen species and replication stress (Jeggo and Lobrich, 2007). In some cellular processes, such as meiotic chromosome segregation in gametes and diversification of antibodies or T cell receptors in lymphocytes, DSBs are introduced deliberately (Keeney et al., 1997; Roth, 2014) . Regardless of how they were formed, unrepaired DSBs can cause chromosomal aberrations (Chatterjee and Walker, 2017). The two major pathways that deal with the repair of DSBs are non- homologous end joining (NHEJ) and homologous recombination (HR). NHEJ involves processing and ligation of the broken ends of DNA and therefore is potentially mutagenic (Burma et al., 2006; Lieber, 2010). On the other hand, HR requires a template with a homologous DNA sequence, from which the genetic information is copied. Therefore, DSB repair by HR is generally of high fidelity (Moynahan and Jasin, 2010).
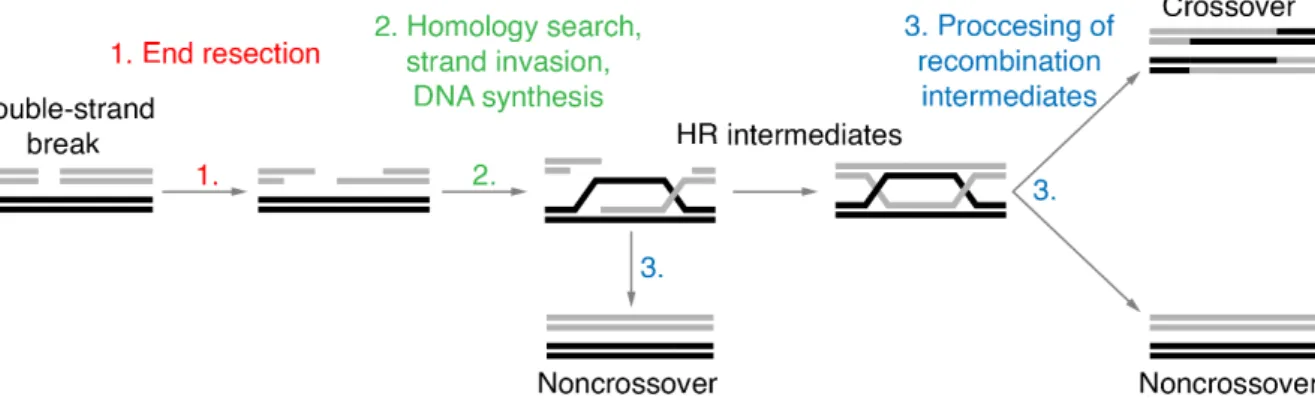
HR is initiated through 5’ to 3’ resection of DNA ends at damage sites (Figure 1) (Cejka, 2015).
Single stranded 3’ overhangs are then captured by recombinases, which facilitate the invasion of a template DNA by the broken chromosome (Bishop et al., 1992; Shinohara et al., 1992).
Eventually, this leads to the formation of recombination intermediates – structures in which the recombining chromosomes are held together by complementary base pairing, otherwise known as DNA joint molecules (Schwacha and Kleckner, 1994). The abnormal persistence of HR intermediates can be detrimental (Hassold and Hunt, 2001; Moynahan and Jasin, 2010) thus they are processed into either crossover or noncrossover recombinants by a subset of specialized enzymes (Boddy et al., 2001; De Muyt et al., 2012; Matos et al., 2011; Ranjha et al., 2014; Rogacheva et al., 2014; Zakharyevich et al., 2012)
.
Interestingly, the outcome of recombination depends on the cellular context and varies between somatic cells and gametes (Allers and Lichten, 2001; Moynahan and Jasin, 2010).In mitotically proliferating cells, DSBs arise inadvertently. Throughout S- or G2/M-phase of the cell cycle, they can be repaired by HR. However, HR can produce crossovers – recombinants with reciprocal exchange of chromosomal arms (Figure 1). Crossing over between non-
11 identical homologous chromosomes can result in the loss of heterozygosity and be deleterious to the cell (Stark and Jasin, 2003). To minimize such a risk, mitotic cells predominantly copy genetic information from sister chromatids, which are identical (Bzymek et al., 2010; Stark and Jasin, 2003). Furthermore, mitotic cells primarily rely on enzymatic machinery that converts recombination intermediates into noncrossovers – gene conversion events without reciprocal exchange. Crossover-generating enzymes act mainly as a backup (Bzymek et al., 2010; Matos et al., 2011; Matos et al., 2013; Symington et al., 2014).
In contrast to mitosis, meiotic inter-homolog crossing over is required for the pairing and separation of homologous chromosomes during meiosis I. It is ensured in several distinct steps. First, DSBs are deliberately introduced throughout the genome by the transesterase Spo11 (Keeney et al., 1997). Depending on the organism, the amount of programmed DSBs differs, e.g. in S. cerevisiae the number is 140-170 DSBs per meiosis (Buhler et al., 2007).
Most of these programmed DSBs are then repaired by inter-homolog recombination (Bzymek et al., 2010; Schwacha and Kleckner, 1997). Processing of the resulting late recombination intermediates by specific enzymes generates crossovers (Allers and Lichten, 2001; Hunter and Kleckner, 2001; Schwacha and Kleckner, 1995).
Figure 1. DNA double strand break repair by homologous recombination (HR). Schematic representation of the key stages of recombinational DNA repair. The ends of a broken DNA molecule are resected to form 3' overhangs, which then with the help of recombinases find and invade a homologous template. Synthesis of the missing genetic information leads to the formation of HR intermediates, which are eventually processed into either crossovers or noncrossovers.
1.2 Mechanism of homologous recombination
Mechanism and pathways of DNA DSB repair by HR are highly conserved, especially among eukaryotes (Wright et al., 2018). Since the data presented in the results section is obtained from S. cerevisiae, the following chapters will be mostly focused on describing the mechanism of HR in this model organism. However, it should be noted that similar mechanisms also apply to higher eukaryotes.
12 1.2.1 DNA end resection
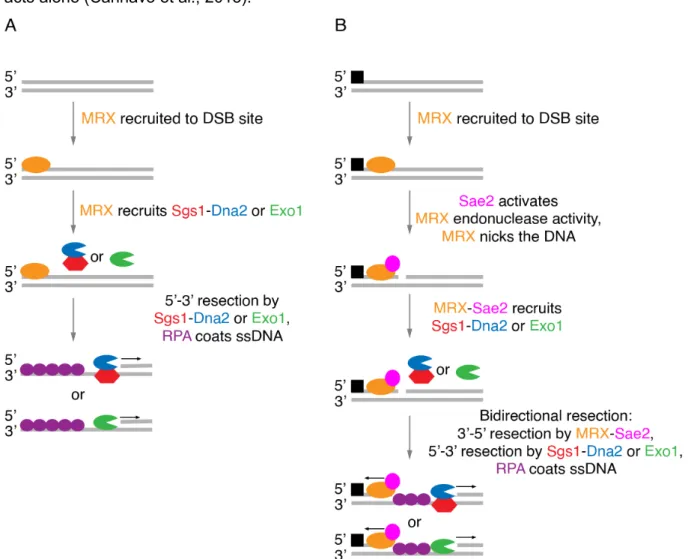
End resection is the initial stage of recombinational DNA repair. It produces 3' DNA overhangs at the break site that serve as a substrate for recombinases. Resection is comprised of two major steps, termed as short- and long-range resection (Cejka, 2015). Short-range resection depends on the MRX-Sae2 complex, comprised of a nuclease Mre11, an ATPase Rad50, a DNA binding protein Xrs2, and the endonuclease Sae2 (Cannavo and Cejka, 2014; Cannavo et al., 2019; Trujillo et al., 2003; Trujillo and Sung, 2001). Long-range resection relies on either the helicase-nuclease complex Sgs1-Dna2 or the 5'-3' exonuclease Exo1 (Cannavo et al., 2013; Niu et al., 2010; Zhu et al., 2008).
The mechanism of the 5' end resection depends on the state of the DSB, as it can either be 'clean' or form secondary structures, have chemical modifications or be tightly bound by proteins (Cejka, 2015; Keeney et al., 1997; Keeney and Kleckner, 1995). In either of the cases, the MRX complex, which shows increased affinity for DNA ends, is recruited to the DSB site (Figure 2) (Lisby et al., 2004; Trujillo et al., 2003). If the DSB is 'clean' MRX serves as an initiation point, although dispensable, for long-range resection by either Sgs1-Dna2 or Exo1 (Cejka, 2015; Llorente and Symington, 2004). On the other hand, if the DSB is 'blocked', e.g.
covalently bound by Spo11 after meiotic DSB formation (Keeney et al., 1997; Keeney and Kleckner, 1995), short-range resection depends on the nuclease activity of the MRX-Sae2 complex. After MRX binds DNA at the break site, Sae2 binds the complex and enhances the endonucleolytic activity of Mre11, which introduces a nick on the 5' end DNA strand in proximity of the DSB (Cannavo and Cejka, 2014; Neale et al., 2005; Zakharyevich et al., 2010). Mre11 also possesses 3'-5' exonuclease activity, which then takes over. The MRX complex proceeds backwards and degrades the DNA towards the 5' end of the DSB. At the same time, the nick serves as the entry point to the long-range resection enzymes (Garcia et al., 2011;
Zakharyevich et al., 2010).
In contrast to short-range resection, there are two non-overlapping long-range resection pathways, defined by the Sgs1-Dna2 complex and Exo1 (Figure 2) (Zhu et al., 2008). The Sgs1-Dna2 complex is comprised of a 3'-5' DNA helicase Sgs1 and Dna2, which possesses ssDNA exonuclease and 5'-3' DNA helicase activities (Bae and Seo, 2000; Cejka and Kowalczykowski, 2010). The Sgs1-Dna2 complex seem to contribute to long-range resection only in proliferating cells, while Exo1 is also active in meiosis (Zakharyevich et al., 2010). Sgs1 unwinds the DNA in the 3'-5' direction, creating ssDNA, which serves as substrate for Dna2 exonuclease activity (Cejka et al., 2010; Zhu et al., 2008). Since Dna2 possesses both 3'-5' and 5'-3' ssDNA exonuclease activities, the direction of the degradation is determined by RPA.
RPA binds the ssDNA generated by Sgs1 unwinding and prevents the degradation of the 3'- terminated ssDNA, while stimulating 5'-3' exonuclease activity of Dna2 (Cejka et al., 2010; Niu
13 et al., 2010). Exo1, on the other hand, can degrade 5'-terminated strand within dsDNA, thus it acts alone (Cannavo et al., 2013).
Figure 2. Schematic representation of DNA' end resection. (A) If the DSB is "clean" the MRX complex binds the DNA at the break site and recruits Sgs1-Dna2 or Exo1, which degrade the DNA in the 5'-3' polarity. RPA covers the exposed ssDNA. MRX nuclease activity is not required for this resection pathway. (B) If the DSB is "blocked"
by a covalent adduct or tightly bound protein (represented by a black square), Sae2 activates the endonuclease activity of the MRX complex. The MRX produces a nick at the break site and recruits Sgs1-Dna2 or Exo1. The MRX complex then degrades the DNA in the 3'-5' direction, while Sgs1-Dna2 or Exo1 degrade the DNA in the opposite direction. RPA coats the exposed ssDNA. Protein names and representations in the illustration are color coded, arrows indicate the direction of movement.
5' end resection governs the choice between DSB repair by HR and NHEJ. It inhibits NHEJ and commits the repair to HR. Thus, cells need to tightly regulate end resection. Resection is strongly controlled by the activity of cyclin dependent kinase (CDK), which activates resection in S- and G2/M-phase (Ira et al., 2004). The S. cerevisiae CDK, Cdc28, phosphorylates Sae2 during S-phase, which then activates Mre11 endonuclease activity (Cannavo et al., 2018;
Huertas et al., 2008). Heterodimeric Top3-Rmi1 and MRX complexes stimulate the long-range resection of the Sgs1-Dna2 complex (Cejka et al., 2010; Kasaciunaite et al., 2019), while Exo1 is stimulated by the MRX complex and Sae2 (Cannavo et al., 2013; Nicolette et al., 2010).
14 1.2.2 Search for homologous template and strand invasion
5' end resection produces relatively long 3' overhangs coated with RPA (Cejka et al., 2010;
Chen et al., 2013). The next step in the DSB repair by HR is search for a homologous template and strand invasion (Figure 1). These processes are facilitated by a certain class of ATPases, termed recombinases. In S. cerevisiae, there are two main recombinases – Rad51, which is expressed in both mitotic and meiotic cells, and the meiosis-specific recombinase Dmc1 (Bishop et al., 1992; Shinohara et al., 1992).
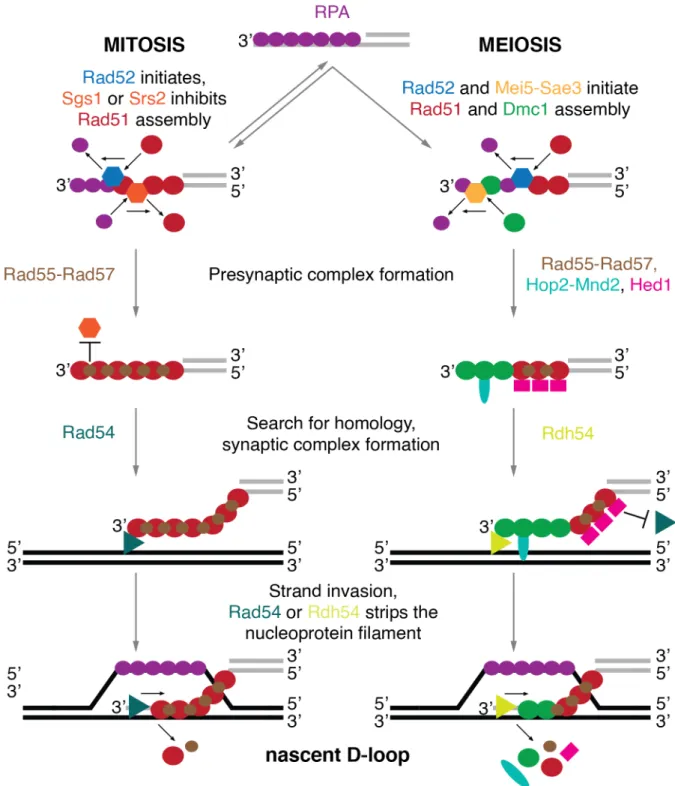
In order to catalyze the strand invasion reaction, recombinases displace RPA and, together with protein cofactors, assemble into a presynaptic complex – a helical nucleoprotein filament that coats long stretches of the 3' ssDNA (Figure 3). Rad51 filament formation is facilitated by Rad52 (Gasior et al., 1998; New et al., 1998), while the Mei5-Sae3 complex aids the loading of Dmc1 (Ferrari et al., 2009; Tsubouchi and Roeder, 2004). Interestingly, the mitotic presynaptic complex is comprised of Rad51, while in meiosis the presynaptic complex contains both Rad51 and Dmc1 (Bishop, 1994; Crickard and Greene, 2018; Yu et al., 2001). The DNA helicase Srs2 and Sgs1 can displace Rad51, but not Dmc1, from DNA, thus destabilizing Rad51 presynaptic complex and preventing hyper-recombination (Crickard, 2019; Krejci et al., 2003; Veaute et al., 2003). On the other hand, the Rad55-Rad57 heterodimer forms a roadblock and prevents Srs2 from Rad51 filament disassembly (Liu et al., 2011).
After the nucleoprotein filament of Rad51 or Dmc1/Rad51 has been assembled, it scans the genomic DNA for a template sequence (Figure 3). In a poorly understood mechanism, the genomic dsDNA is destabilized. This allows the pre-synaptic complex to check the complementarity of the bases. Upon the identification of homology in the template DNA, the synaptic complex forms. It is a structure where base pairing between the invading strand and its complement occurs, but there is no net intertwining of the two. After the formation of the synaptic complex, Rad51 or Dmc1 is stripped from the invading DNA by a DNA translocase Rad54 or Rdh54, respectively (Chan et al., 2019; Wright and Heyer, 2014). The invading DNA strand then intertwines with the template and displacement of the non-template DNA strand occurs, the intermediate known as a nascent displacement loop (D-loop) forms (Wright et al., 2018). Since the 3' end of the invading DNA strand is fully intertwined with the template DNA, DNA synthesis can begin (McVey et al., 2016).
Rad51, although present in mitotic and meiotic presynaptic complexes, only facilitates the homology search and strand-exchange reaction in proliferating cells (Crickard and Greene, 2018). Rad54 has been shown to associate with the presynaptic complex and aids in the process (Heyer et al., 2006). In meiosis, however, the Rad51-Rad54 interaction is disrupted by Hed1, thus resulting in downregulation of Rad51 (Busygina et al., 2012). Therefore, in meiosis, homology search and strand-exchange rely on Dmc1, promoted by Hop2-Mnd2 complex and a Rad54 paralog Rdh54 (Chi et al., 2009; Henry et al., 2006).
15
Figure 3. Schematic representation of search for homology and strand exchange. 5' end resection produces a 3' overhang coated with RPA. RPA is stripped from DNA and presynaptic complex formation is initiated. Rad52 facilitates Rad51 filament formation and Mei5-Sae3 aids Dmc1 loading. DNA helicases Sgs1 and Srs2 can strip Rad51 from ssDNA. Accessory factors bind the Rad51 (Rad55-Rad57) or Dmc1/Rad51 (Rad55-Rad57, Hop2- Mnd2, Hed1) filament and form the presynaptic complex. The presynaptic complex, aided by Rad54 in mitosis and Rdh54 together with Hop2-Mnd2 in meiosis, facilitates search for homology. Once a homologous sequence is found presynaptic complex and template DNA forms the synaptic complex. Rad51 in mitosis or Dmc1 in meiosis facilitate strand invasion. Once strand exchange is accomplished Rad54 strips Rad51 and Rdh54 removes Dmc1 from the invading DNA, resulting in the formation of a nascent displacement loop (D-loop) structure. Protein names and representations in the illustration are color coded, arrows indicate the direction of movement.
16 Another interesting feature of the search for homology and strand exchange processes is the fact that in mitotic cells the synaptic complex, and later D-loops, mostly form between sister chromatids (Bzymek et al., 2010). However, in meiosis, the preferred donor for strand exchange is the homologous chromosome (Hunter and Kleckner, 2001; Schwacha and Kleckner, 1997). While it is not clear if the presynaptic complex has any role in the observed differences, introduction of mismatches in the initial pairing region of the synaptic complex is tolerated only by Dmc1 and not Rad51 (Lee et al., 2015; Qi et al., 2015). Perhaps mitotic presynaptic complex comprised of Rad51 requires perfect sequence homology and only forms a synaptic complexes with and identical sister chromatid template.
1.2.3 Formation of recombination intermediates
After the formation of a nascent D-loop, the 3' end of the invading strand can prime DNA synthesis, further displacing the original dsDNA and extending the D-loop structure (Figure 4).
This is performed by DNA polymerase δ (Pol δ) together with the PCNA-RFC complex (McVey et al., 2016). Both nascent and extended D-loops can be disassembled. Once it is disrupted, the invading strand can go through another round of homology search and strand invasion, as described in section 1.2.2 (Figure 3) (Smith et al., 2007). Otherwise, if DNA synthesis has been long enough, the disrupted strand can be repaired through synthesis-dependent strand annealing (SDSA), described in the section 1.2.4 (Figure 5) (Wright et al., 2018).
On the other hand, during the extension process the displaced ssDNA, coated with RPA, can capture the second resected DSB end (Figure 4). Annealing of RPA coated ssDNA is facilitated by Rad52 (Lao et al., 2008; Sugiyama et al., 2006). The captured end can be extended through DNA synthesis by a DNA polymerase, likely either by Pol δ or Pol ε together with the PCNA- RFC complex (McVey et al., 2016). Eventually, this results in the formation of a recombination intermediate containing a double Holliday junction (HJ) (Bzymek et al., 2010; Hunter and Kleckner, 2001; Schwacha and Kleckner, 1995). The second DSB end can also invade the template trough recombinase-mediated strand exchange and form its own D-loop simultaneously with the synthesis of the first end (Figure 4). If this happens, the two ends can still be displaced and repaired through SDSA, however, there is a much greater chance that they will mature into a double HJ intermediate (Wright et al., 2018). In some cases, recombination results in the formation of atypical recombination intermediates, such as multichromatid DNA joint molecules (Jessop et al., 2006; Oh et al., 2007; Piazza et al., 2017).
17
18
Figure 4.Schematic representation of the formation and maturation of recombination intermediates. Strand exchange produces a nascent displacement loop (D-loop) structure, which is stripped by either Rad54 or Rdh54. 3' end of the invading end primes DNA synthesis by DNA polymerase δ (Pol δ) and the PCNA-RFC complex. It displaces the original dsDNA and extends the D-loop. At the same time the second end, coated with RPA, can be captured by Rad52. Rad52 then facilitates annealing of the second 3' end and RPA coated displaced DNA strand.
Pol δ or Pol ε and the PCNA-RFC complex extends the second end. The presynaptic complex can also form on the second 3' end and then it initiates strand exchanges, producing another D-loop. In both cases, extension of both ends eventually leads to the formation of a recombination intermediate containing a double Holliday junction (HJ).
It is not clear which proteins are involved in this process. Protein names and representations in the illustration are color coded, arrows indicate the direction of movement.
The variety of recombination intermediates reflect specialized cellular requirements. In mitotic cells, only a subset of intermediates mature to form double HJs as the majority are disengaged at the D-loop stage (Symington et al., 2014). In meiosis, however, the ZMM proteins (Zip1, Zip2, Zip3, Zip4, MutSγ and Mer3) promote the maturation of DNA joint molecules into double HJs (Figure 5) (Borner et al., 2004; Lynn et al., 2007).
1.2.4 Processing of recombination intermediates
The abnormal persistence of HR intermediates can be detrimental, as they might trigger chromosomal rearrangements or obstruct chromosome segregation and lead to aneuploidy (Hassold and Hunt, 2001; Moynahan and Jasin, 2010; Wang et al., 2017). Therefore, cells are able to channel recombination intermediates into specialized HR products – noncrossovers or crossovers (Figure 5).
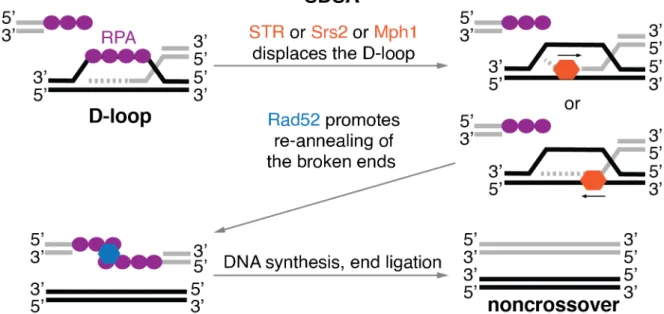
Anti-crossover DNA helicases can disassemble early recombination intermediates, such as nascent and extended D-loops. There are 3 such enzymes in S. cerevisisae: Sgs1, which acts as part of Sgs1-Top3-Rmi1 (STR) complex, Srs2 and Mph1 (Bennett et al., 2000; Cejka and Kowalczykowski, 2010; Fricke et al., 2001; Gangloff et al., 1994; Ira et al., 2003; Liu et al., 2017; Mullen et al., 2005; Prakash et al., 2009). In order to disrupt the D-loop, the helicases bind to the 3' end of the invading strand or to the template strand at the branch point (Figure 5). They then translocate in the 3'-5' direction on ssDNA using ATP hydrolysis for energy and unwind the D-loop. It is likely that each of the DNA helicases unwind slightly different types of D-loops, marked by interacting proteins or the topological status (Piazza et al., 2019).
If there is sufficient extension of the displaced strand, it can be repaired through SDSA (Figure 5). First, Rad52 facilitates annealing of the displaced strand to the second resected end of the DSB (Figure 5) (Lao et al., 2008; Sugiyama et al., 2006). Then, either Pol δ or Pol ε with the PCNA-RFC complex synthesizes the missing genetic information on both strands. Finally, the remaining nicks are ligated and a noncrossover is produced (McVey et al., 2016; Wright et al., 2018).
19
Figure 5. Schematic representation of the synthesis-dependent strand annealing (SDSA) pathway. DNA helicases Sgs1 (part of STR), Srs2 or Mph1 bind and then disassemble the displacement loop (D-loop) structure by unwinding the DNA in 3'-5' direction. Rad52 then binds the two RPA coated ssDNA ends and re-anneal them.
Synthesis of the missing genetic information and ligation produces a noncrossover recombinant. Protein names and representations in the illustration are color coded, arrows indicate the direction of movement.
Recombination intermediates that escape early processing by anti-crossover DNA helicases can mature into late recombination intermediates, which contain double HJs (Bzymek et al., 2010; Hunter and Kleckner, 2001; Schwacha and Kleckner, 1995). These can be disassembled in several different ways. First, recombination intermediates can be converted into noncrossovers in a process termed dissolution (Figure 6) (Bizard and Hickson, 2014).
Although the exact mechanistic details of double HJ dissolution is not known, it is clear that the DNA unwinding and topoisomerase activities of the STR complex are required for this process. One likely possibility is that Sgs1 translocates on ssDNA migrating the HJs and fusing them into a hemicatenated intermediate. This intermediate is then decatenated by Top3, resulting in the formation of a noncrossover. Rmi1 stimulates the dissolution process (Bizard and Hickson, 2014; Cejka et al., 2012; Mullen et al., 2001).
Double HJ containing recombination intermediates can also be processed by DNA nucleases.
The structure-selective endonucleases (SSEs) Mus81-Mms4, Slx1-Slx4 and Yen1 bind and hydrolyze the crossing or non-crossing strand in HJs and, as such, generate a mixture of crossovers and noncrossovers in a process known as resolution (Figure 6) (de los Santos et al., 2003; De Muyt et al., 2012; Matos et al., 2011). During meiosis, the MutLγ-Exo1 nuclease complex can also process double HJs, producing exclusively crossover recombinants (Figure 6) (Wild et al., 2019; Zakharyevich et al., 2012). The mechanism of such cleavage is not fully understood. However, it is has been proposed that hydrolysis of DNA involves polymerization of MutLγ filament along the DNA (Manhart et al., 2017; Ranjha et al., 2014; Rogacheva et al., 2014). The ZMM proteins, especially the MutSγ complex, which promotes the maturation of
20 early recombination intermediates during meiosis, also play a role in this process. They are thought to recruit the MutLγ-Exo1 complex and stimulate its DNA cleavage activity (Borner et al., 2004; Cannavo et al., 2020; Jessop et al., 2006; Kulkarni et al., 2020; Lynn et al., 2007).
PCNA-RFC complex has also been shown to stimulate activity of the MutLγ-Exo1 complex in vitro and modulate its function in vivo (Cannavo et al., 2020; Kulkarni et al., 2020).
Figure 6. Schematic representation of double Holliday junction processing pathways. Late recombination intermediates involving double Holliday junctions (HJs) can be processed by three distinct pathways. The dissolution pathway is defined by the activity of the STR complex, comprised of a DNA helicase Sgs1, a topoisomerase Top3 and an auxiliary protein Rmi1. Sgs1 promotes convergent migration of the HJs. Top3 then decatenates the newly formed hemicatenane and a noncrossover recombinant is generated. Double HJs can also be processed in the resolution pathway, which involves structure-selective endonucleases (SSEs) Mus81-Mms4, Slx1-Slx4 and Yen1. These enzymes recognize the HJs and hydrolyze them producing either crossovers or noncrossovers. The last pathway involves the MutLγ-Exo1 nuclease, which recognize double HJs bound by the ZMM proteins. The MutLγ-Exo1 then cleaves the HJs in an unknown mechanism to exclusively produce crossovers.
This pathway is meiosis specific. Protein names and representations in the illustration are color coded, arrows indicate the direction of movement or site of DNA cleavage.
21 1.3 Regulation of recombination intermediate processing
As stated previously, in mitosis noncrossovers are favored over crossovers. To avoid crossing over, mitotic cells mainly use anti-crossover DNA helicases and utilize SDSA (Figure 5) and dissolution pathways (Figure 6) (Bzymek et al., 2010; Dayani et al., 2011; Symington et al., 2014). However, SSEs act as a safeguard in case the anti-crossover pathways are compromised and recombination intermediates persist until anaphase, where they would directly interfere with chromosome segregation (Matos and West, 2014). On the other hand, a certain amount of crossovers is required to segregate homologous chromosomes successfully during meiosis. To ensure crossing over, meiotic cells utilize the MutLγ-Exo1 complex, as well as SSEs (Figure 6) (Matos et al., 2011; Zakharyevich et al., 2012). Anti-crossover pathways are also important during meiosis – by generating noncrossovers they ensure that the number of crossovers is not too high and they are evenly distributed along the chromosomes (Youds and Boulton, 2011; Kohl and Sekelsky, 2013; McMahill et al., 2007; Zickler and Kleckner, 2016). To achieve such careful orchestration of noncrossover and crossover formation, cells need to regulate the recombination intermediate processing machinery. Although it is not fully clear how this is achieved in mitosis and meiosis, known means of control in S. cerevisiae will be described in the following two chapters.
1.3.1 DNA helicases
DNA helicases Sgs1 (part of the STR complex), Srs2 and Mph1 are involved in noncrossover generation. In mitosis, all three disassemble nascent or extending D-loops, which can then be repaired into noncrossovers through SDSA (Figure 5). Furthermore, the STR complex generates noncrossovers by 'dissolving' double HJs (Figure 6). These two pathways handle the majority of recombination intermediates during mitotic proliferation (Dayani et al., 2011; Ira et al., 2003; Prakash et al., 2009; Symington et al., 2014).
During meiosis, on the other hand, the STR complex produces the majority of the noncrossovers while the roles of Srs2 and Mph1 in recombination intermediate processing are not clear. The STR complex is known to function during S-phase and prophase I, prior crossing over, which takes place as cells exit prophase I to enter the first meiotic division (De Muyt et al., 2012; Zakharyevich et al., 2012). It is not clear how such temporal organization is achieved.
Although, it is known that the ZMM proteins bind to fraction of recombination intermediates and in an unknown way protect them from disassembly by the STR complex (Borner et al., 2004;
Lynn et al., 2007).
It is not known whether the anti-crossover DNA helicases are regulated. Nevertheless, some data suggest that they might be. Sgs1 and Srs2 are post-translationally modified in proliferating cells: Sgs1 is sumoylated (Bermudez-Lopez et al., 2016; Bonner et al., 2016) and phosphorylated (Hegnauer et al., 2012), while Srs2 is phosphorylated in response to DNA
22 damage (Liberi et al., 2000). Work from our laboratory has also shown that they are post- translationaly modified during early stages of meiosis (Wild et al., 2019).
1.3.2 DNA nucleases
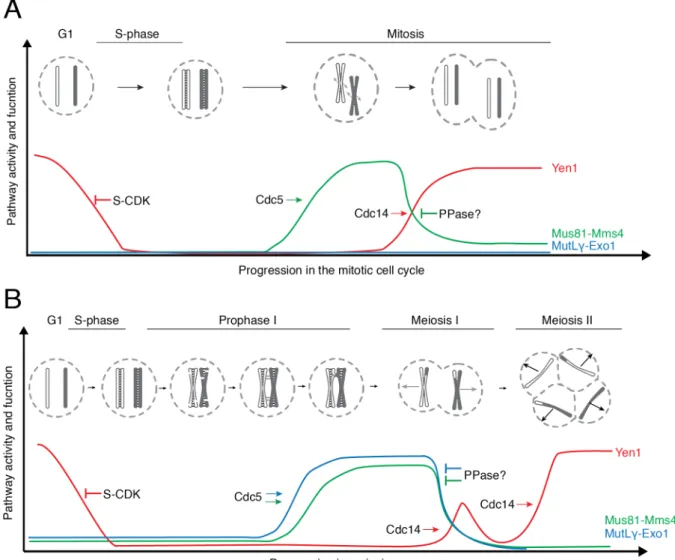
During mitotic proliferation, the SSEs Mus81-Mms4, Slx1-Slx4 and Yen1 act as backup for the noncrossover producing pathways. Mus81-Mms4 and Yen1 are tightly regulated by stage- specific phosphorylation (Figure 7) (Matos and West, 2014). They are both kept inactive throughout S-phase, as they can cleave replication forks. During mitotic proliferation, at the G2/M transition, the M-phase cyclin-dependent kinase (M-CDK), polo kinase Cdc5 and Dbf4- Cdc7 kinase phosphorylate Mms4 – the regulatory subunit of the Mus81-Mms4 nuclease. This hyper-activates the Mus81 nuclease within the complex, which can then hydrolyze branched DNA molecules, including HJs, more efficiently (Gritenaite et al., 2014; Matos et al., 2011;
Matos et al., 2013; Szakal and Branzei, 2013). S-phase CDK (S-CDK), on the other hand, phosphorylates Yen1. This decreases the DNA binding affinity of Yen1 and excludes it from the nucleus, neutralizing it (Blanco et al., 2014; Matos et al., 2011). As cells enter anaphase, Mms4 can no longer be phosphorylated, thus Mus81-Mms4 complex is inactivated (Matos et al., 2011; Matos et al., 2013). At the same time, Cdc14 phosphatase dephosphorylates Yen1, making it active and allowing it back into the nucleus (Blanco et al., 2014; Matos et al., 2011;
Matos et al., 2013).
In meiosis, on the other hand, the MutLγ-Exo1 complex and SSEs generate crossovers that are crucial for the segregation of homologous chromosomes (Arter et al., 2018; Matos et al., 2011; Zakharyevich et al., 2012). The function of MutLγ-Exo1 complex is thought to be regulated by interactions with different proteins (Figure 7). First, the complex can only cleave recombination intermediates that have been captured by the ZMM proteins, suggesting that the latter are involved in the recruitment or activation of the complex (Borner et al., 2004;
Cannavo et al., 2020; Kulkarni et al., 2020; Lynn et al., 2007; Zakharyevich et al., 2012).
Furthermore, a chromatin remodeler Chd1 binds to the MutLγ-Exo1 at the end of prophase I, and promotes its ability to generate crossovers by an unknown mechanism (Wild et al., 2019).
Finally, the function of MutLγ-Exo1 is also regulated in a temporal manner by the polo kinase Cdc5, but the mechanism remains unknown (Allers and Lichten, 2001; Clyne et al., 2003;
Sanchez et al., 2020; Sourirajan and Lichten, 2008). The SSEs Mus81-MMS4 and Yen1 are also regulated in meiosis. Mus81-Mms4 is activated upon M-CDK and Cdc5 phosphorylation at the end of prophase I, while Yen1 is activated upon dephosphorylation at the onset of meiosis II (Arter et al., 2018; Matos et al., 2011).
23
Figure 7. Regulation of pro-crossover DNA nucleases. (A) Regulation of pro-crossover DNA nucleases during mitotic proliferation. Mus81-Mms4 is inactive until G2/M transition, when M-phase cyclin dependent kinase (M-CDK) and polo kinase Cdc5 phosphorylate Mms4 and hyperactive activate the complex. It is then deactivated at the transition to anaphase, by an unknown protein phosphatase (PPase). On the other hand, S-phase CDK (S-CDK) phosphorylation of Yen1 excludes it from the nucleus and makes it inactive. Yen1 is activated at the onset of anaphase by dephosphorylation by Cdc14. (B) Regulation of pro-crossover nucleases during meiosis. MutLγ-Exo1 becomes functional at the end of prophase I and this process is dependent on Cdc5. Mus81-Mms4 and Yen1 are activated at the end of prophase I and the beginning of meiosis II respectively. Activation mechanisms are similar to those in mitosis.
1.4 DNA helicase Sgs1 and its importance for homologous recombination RecQ-family DNA helicases are conserved from bacteria to humans. They play important roles in the maintenance of genomic integrity, acting as key factors in HR. Sgs1 is a RecQ-family DNA helicase present in S. cerevisiae (Bernstein et al., 2010). As discussed above, it possesses an ATP dependent 3'-5' DNA unwinding activity. In vitro, Sgs1 binds to and unwinds a variety of DNA substrates: dsDNA, RNA-DNA duplexes, forked DNA structures, G quadruplexes, D-loops and HJs, etc (Bennett et al., 1998; Cejka and Kowalczykowski, 2010).
It possesses a typical RecQ-family domain structure, comprised of a RecQ helicase core,
24 which contains ATPase and RecQ-C terminal domains, and a HRDC domain at the C-terminus.
Its N-terminus contains a disordered tail, which is important for protein-protein interactions (Bernstein and Keck, 2003; Bernstein et al., 2003). Together with Top3 and Rmi1, Sgs1 forms the STR complex (Cejka et al., 2012; Gangloff et al., 1994; Mullen et al., 2005). It is unknown whether Sgs1 has functions that are independent of the STR complex.
Sgs1 is involved in many cellular processes. Cells lacking Sgs1 display increased extrachromosomal rDNA circle accumulation, increased mitotic recombination, DNA damage sensitivity, gross chromosomal rearrangements, defective checkpoint signaling, decreased replicative lifespan, decreased meiotic tetrad formation and lower spore viability (Bernstein et al., 2010). Its most prominent roles lie in the repair of DNA DBSs by HR. As already mentioned throughout the previous chapters, Sgs1 plays a role in almost every stage of HR. It is involved in long-range resection (Figure 2), regulation of search for homology and strand invasion (Figure 3) and most notably – processing of recombination intermediates (Figures 5 and 6).
Although Sgs1 contributes to HR in many ways, the consensus view was that Sgs1 is not regulated and is constitutively active in both meiosis and mitosis. However, Sgs1 has been found to be post-translationally modified in response to replication stress and in meiosis (Bermudez-Lopez et al., 2016; Bonner et al., 2016; Hegnauer et al., 2012; Wild et al., 2019).
Lastly, although directly contributing to the generation of noncrossovers, Sgs1 has a pro- crossover role in meiotic HR, although it is not yet understood (Zakharyevich et al., 2012).
1.5 Goals of the thesis
The principal goal of my thesis was to obtain mechanistic insight into how mitotic and meiotic cells rewire HR to their specialized needs. In order to ensure that the correct outcome (noncrossover vs crossover) is formed in the right context (mitosis vs meiosis), cells directly regulate the HR machinery at various stages, most notably – recombination intermediate processing. Although, it is known that pro-crossover enzymes are under stringent spatiotemporal control, anti-crossover DNA helicases have not been implied in any sort of direct regulation. Given the evidence that these enzymes are involved in complex interaction networks and can be post-translationally modified, I postulate that there may be molecular mechanism that directly regulate the biochemical properties of the anti-crossover DNA helicases.
I chose to investigate the regulation of Sgs1 due to its prominent roles in DNA joint molecule metabolism. Compared to Srs2 and Mph1, Sgs1 is also better characterized biochemically.
Finally, Sgs1 undergoes specific PTMs in both mitotic and meiotic cells. By utilizing a variety of molecular biology methods and approaches, during the time of my PhD, I have addressed the following questions:
1. Is Sgs1 activity regulated in mitotic and meiotic cells?
25 2. If so, what is the mechanism of such regulation?
3. How does regulation of Sgs1 affects the metabolism of recombination intermediates and contribute to the overall outcome of HR?
26
2. EXPERIMENTAL PROCEDURES, RESULTS AND DISCUSSION
2.1 Characterization of DNA helicases and nucleases from meiotic extracts of S.
cerevisiae
One major challenge in describing the regulation of an enzyme is lack of methods that allow direct assessment of its enzymatic activity in different stages of the cell cycle. For that purpose, I have adopted a modified a method that had been previously used to characterize the cellular activity of the SSEs Mus81-Mms4 and Yen1 (Arter et al., 2018; Blanco et al., 2014; Matos et al., 2011; Matos and West, 2017). It enables semi-purification of Sgs1 from meiotic cultures of S. cerevisiae, followed by in vitro DNA unwinding reactions using synthetic DNA substrates.
The method, experimental data and protocol has been published as
Grigaitis R, Susperregui A, Wild P, Matos J (2018). Characterization of DNA helicases and nucleases from meiotic extracts of S. cerevisiae. Methods Cell Biol 144: 371-388.
A. Susperregui and P. Wild from the laboratory of J. Matos contributed to this paper by performing the experiments in Figure 8 and 9.
2.1.1 Synchronization and harvesting of large meiotic cultures of S. cerevisiae
Budding yeast constitutes an ideal model organism to study meiosis. Diploid yeast cells can be stimulated to undergo meiosis by nitrogen starvation leading to the formation of an ascus with four haploid cells. As a result, the properties of the meiotic products can be studied directly. A second advantage of budding yeast is that its genome can be easily modified, and mutations combined, to study gene/protein function (e.g. gene deletions, point mutations and gene tagging). Furthermore, such mutants can also be used to halt meiotic progression at well- defined cell-cycle stages. Finally, upscaling of culture size is easy to achieve and inexpensive, enabling biochemical approaches to study protein function. Below, we provide a detailed protocol that can be used to generate any desired volume of sporulating SK1 strains.
Depending on the inclusion of specific mutations in the strain background (e.g. ndt80∆ or PCLB2- CDC20), cells can be released to initiate sporulation and accumulate at specific stages of meiosis (pachytene or metaphase I, respectively) (Clyne et al., 2003; Lee and Amon, 2003;
Petronczki et al., 2006; Xu et al., 1995) (Figure 8A).
Notes:
• Liquid cultures are prepared using a yeast fermenter (10 l) in which pressurized (1.0 bar), filtered and humidified air serves to mix and aerate the culture (Oelschlaegel et al., 2005).
To monitor meiotic progression, samples are collected for immunofluorescence analysis of spindle morphology and chromosome synapsis, as well as DNA content, at 2 h intervals (see Figure 8A).
27 Solutions and reagents:
• YPD plates: 1% Yeast Extract, 2% Peptone, 2% Glucose, 2% Agar
• YPGlycerol plates: 1% Yeast Extract, 2% Peptone, 2% Glycerol, 2% Agar
• YEPA medium: 1% Yeast Extract, 2% Peptone, 2% Potassium acetate
• Sporulation medium (SPM) (30°C): 2% Potassium acetate
• 2 mM PMSF in water (ice-cold)
• Yeast SK1 strains YML3105, YML2924, YML2917 and YML324 (see Table 1 for details)
Figure 8. Generation of synchronous meiotic cultures. (A) Schematic representation of the steps required for the generation of the large-scale synchronous meiotic cultures. (B) FACS analysis of DNA content showing the kinetics of DNA replication upon transfer of ndt80∆ cells into sporulation medium (SPM). To follow chromosome synapsis and accumulation in pachytene, Zip1 is stained on chromosome spreads. The fraction (%) of cells with fully synapsed chromosomes, 8 h after transfer to SPM, is shown. (C) FACS profile showing the kinetics of DNA replication during meiosis for PCLB2-CDC20 cells. The accumulation of cells in metaphase I is evaluated by in situ immunofluorescence analysis of spindle morphology by tubulin staining. The proportion of cells showing a bipolar metaphase I spindle after 8 h in SPM is shown. (D) Protein extracts from cells in (B) and (C) were analyzed by western blotting using anti-Cdc5 antibodies (sc-6732).
Procedure:
1. As illustrated in Figure 8A, plate the strain of choice on YPGlycerol and incubate at 30°C for 48 h.
2. Pick a colony, streak it as a small patch onto a YPD plate and leave it incubating for 24 h.
28 3. Expand the small patch to a thin layer on a full YPD plate and leave it incubating for 24 h.
4. Expand the plate on 8 new full YPD plates and leave incubating for 24 h.
5. Resuspend the cells in 6 l of YEPA medium at an OD600 = 0.3 and incubate with vigorous aeration in the 10 l fermenter for 15 h at 25°C.
6. Monitor the G1 arrest by inspecting cell morphology in a light microscope. More than 90%
of cells are expected to be unbudded.
7. Centrifuge cells at 5895 rcf, 8 min at RT and wash once with 500 ml of pre-warmed SPM.
8. To induce sporulation, resuspend cells in 3 l of pre-warmed SPM to a final OD600 of 3.5.
9. Cells are harvested at any desired time after induction of meiosis. For pachytene (ndt80∆) or metaphase I (PCLB2-CDC20), cells should be collected approximately 8 h after induction of meiosis (Figure 8B). Harvest cells by centrifugation at 5895 rcf for 8 min at RT; wash once with 2 mM PMSF and snap-freeze the pellet in liquid nitrogen (storage at -80°C).
2.1.1.1 Analysis of the results:
FACS analysis of DNA content reveals that prior to induction of meiosis (t = 0 h) cultures have, as expected, a predominantly G1 DNA content (Figure 8B-C). 8 h after induction of sporulation
>80% of ndt80∆ mutants display fully synapsed chromosomes (Figure 8B) and >60% of PCLB2- CDC20 mutants have a metaphase I spindle (Figure 8C). In contrast to ndt80∆ mutants, PCLB2- CDC20 strains accumulate polo kinase Cdc5 (Figure 8D).
2.1.2 Purification and characterization of the STR complex from prophase I
In order to purify individual homologous recombination enzymes from meiotic cultures, we have engineered yeast strains in which the respective genes are modified at the endogenous locus to encode for a tag that allows tandem-affinity purification: 6xHis-6xFLAG (see Table 1).
While we exclusively use FLAG-affinity in the subsequent sections, the 6xHis tag provides added flexibility for additional purification steps or alternative purification strategies.
The protocols described in this section have been optimized for the purification of chromatin- associated proteins from meiotic and mitotic cultures. FLAG-affinity purified proteins following this protocol can be analyzed by SYPRO Ruby staining after elution with FLAG peptide (section 2.1.2.2). Alternatively, on-bead digestion of the immobilized proteins can be performed to study sample composition by shotgun-MS (section 2.1.2.3). Depending on the cellular abundance of the target protein, appropriate scaling of the input material may be needed.
2.1.2.1 Immuno-affinity purification of FLAG-tagged STR components from prophase I lysates
Notes:
• PMSF is dissolved in DMSO freshly before use.
29
• 2 mM PMSF, tablets of cOmplete™ EDTA-free Protease Inhibitor Cocktail (Roche) and 1 mM DTT are freshly added to the Lysis buffer before use.
• FLAG M2 Magnetic Beads are washed 3 times with Wash buffer and resuspended in Wash buffer 1:1 (V/V) prior to use.
• 2 versions of the Lysis buffer are used in this protocol. For SYPRO Ruby staining, cell lysis is performed in the presence of 500 mM KCl. For shotgun-MS, the lysis buffer contains 150 mM KCl.
Solutions and reagents:
• Lysis buffer (MS): 25 mM HEPES (pH 8.0 at 25°C), 150 mM KCl, 15% Glycerol, 0.1% NP- 40, 2 mM MgCl2, 0.1 mM EDTA, 0.5 mM EGTA, 1 mM NaF, 20 mM β-glycerophosphate, 2 mM PMSF, 1 tablet/50 ml of cOmplete™ EDTA-free Protease Inhibitor Cocktail (Roche), 1 mM DTT.
• Lysis buffer (SYPRO Ruby): identical to Lysis Buffer (MS) but containing 500 mM KCl.
• Wash buffer: 25 mM HEPES pH 8.0, 150 mM KCl
• Anti-FLAG M2 Magnetic Beads (Sigma-Aldrich). 1:1 (V/V) slurry in Wash buffer
• Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad)
• Freezer Mill (SPEX SamplePrep 6870 Freezer/Mill®)
• 100 kU Nuclease (Pierce Universal Nuclease)
• Magnet (biotool.com, B23804, 50 ml*8) Procedure:
1. Resuspend the frozen cell pellet in 40 ml of Lysis buffer.
2. Decant the solution dropwise into liquid nitrogen and collect the solidified droplets.
Immediately store the droplets at -80°C.
3. Mechanically pulverize the droplets using the Freezer Mill (under recommended settings, pre-cool (2 min), run time (3 min), cool time (2 min), cycles (6), rate (15 CPS). At this point, the generated powder can be stored at -80°C.
4. Resuspend the yeast powder in 40 ml Lysis buffer.
5. Add 100 kU Nuclease and incubate 1 h at 4°C with rotation.
6. Centrifuge solution at 3220 rcf for 10 min at 4°C, followed by a second centrifugation at 38800 rcf, 30 min, at 4°C.
7. Determine protein concentration of the cleared lysate and dilute it to obtain 65 ml at a concentration of 10 mg/ml.
8. Add 200 µl of pre-equilibrated slurry of Magnetic anti-FLAG beads and incubate for 90 min on an orbital rotor at 4°C.
30 9. Apply magnetic field to pull beads to the side of the tube. Carefully remove the supernatant
by aspiration.
10. Wash beads 3 times with 30 ml of Lysis buffer and 3 times with 30 ml of Wash buffer.
Apply magnetic field to remove the supernatant.
11. After the final wash, centrifuge for 1 min at 100 rcf, 4°C, using a benchtop centrifuge.
Carefully remove the supernatant, using a hypodermic needle connected to a vacuum pump, without completely drying the beads.
12. Resuspend the beads in 200 µl of Wash buffer and transfer them to a 1.5 ml Eppendorf tube. For SYPRO Ruby staining, follow immediately the steps described in section 2.2.
For characterization of the sample composition by shotgun-MS analysis, beads are snap- frozen in liquid nitrogen and stored at -80°C in preparation for section 2.1.2.3.
2.1.2.2 Elution of Rmi1-FLAG and detection of co-purifying proteins using SYPRO Ruby stain
Notes:
• SYPRO Ruby Protein Gel Stain is filtered with a Steritop® 0.22 µm (GP Millipore) before use.
• MilliQ water is used for the preparation of all solutions.
Solutions and reagents:
• 3xFLAG Peptide (5 mg/ml) (F4799, Sigma-Aldrich)
• Fixative solution: 50% methanol, 7% acetic acid
• Steritop® 0.22 µm (GP Millipore)
• SYPRO Ruby Protein Gel Stain (Thermo Fisher)
• NuPAGETM 4-12% Bis-Tris Protein Gel, 1.0 mm, 10-well (Thermo Fisher)
• Illustra ProbeQuant G-50 Micro Column (GE Healthcare)
• Protein sample buffer: 4x NuPAGE™ LDS Sample Buffer (Invitrogen), 200 mM DTT Procedure:
1. Pipet the bead suspension from step 12 (section 2.1) into an Illustra ProbeQuant Micro Column.
2. Spin the column at 100 rcf for 1 min in a table-top centrifuge in order to completely remove the Wash buffer from the beads.
3. Seal column tightly with Parafilm, load 60 µl of 2 mg/ml 3xFLAG peptide on the column and incubate for 25 min at RT rotating (orbital rotor). Place the column in a new 1.5 ml Eppendorf tube and collect the eluate by spinning down (100 rcf, 2 min) in a table-top centrifuge.
4. Repeat step 3 and pool the two elutions.
31 5. Add 4x Sample Buffer and load on a NuPAGETM 4-12% Bis-Tris Protein Gel. Run the
electrophoresis for 1 h at 200 V.
6. After electrophoresis, place the gel in a clean container with 100 ml of Fixative solution.
Incubate for 30 min on a rocking shaker. Repeat once more with fresh Fixative solution.
7. Add 60 ml of pre-filtered SYPRO Ruby gel stain and shake overnight.
8. After staining, wash the gel with 100 ml of wash solution (10% methanol, 7% acetic acid) and incubate for 30 min. Rinse the gel in MilliQ water at least twice, 5 min each time.
2.1.2.2.1 Data analysis:
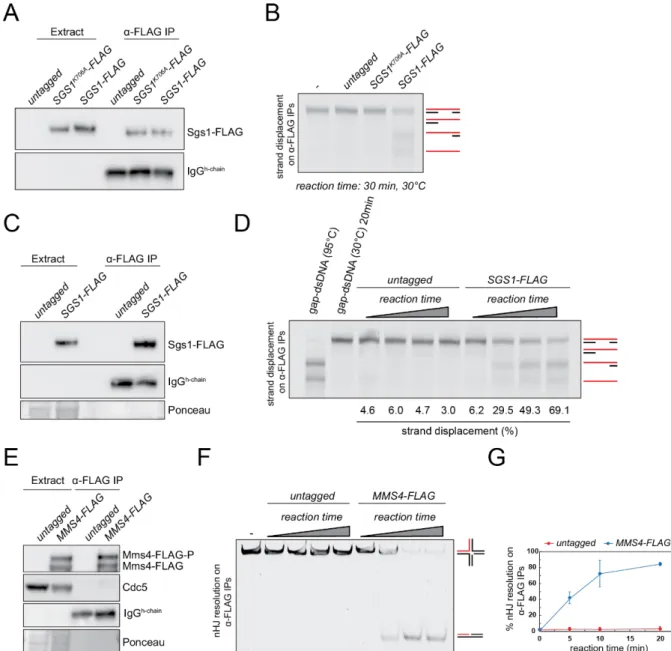
Rmi1-FLAG was affinity-purified using the protocol described above. SYPRO Ruby staining of the eluted material revealed the prominent enrichment of Rmi1-FLAG and Top3 in the purified material. The presence of Sgs1 is not detected above background levels. Proteins that bind nonspecifically to the FLAG beads and are eluted with the FLAG peptide, are detected in a control purification using an untagged strain (Figure 9B).
2.1.2.3 On-bead digest followed by MS-based analysis of immuno-affinity purified Top3- FLAG
Procedure:
1. To the beads from step 12 in section 2.1 add: 90 µl of buffer (10 mM Tris/2 mM CaCl2, pH 8.2) and 10 µl of trypsin (100 ng/µl in 10 mM HCl).
2. Microwave for 30 min at 60oC.
3. Collect the supernatant and wash the beads with 150 µl buffer (0.1% TFA, 50% ACN).
4. Combine the supernatants, dry and dissolve in 20 µl of 0.1% formic acid diluted 10x and transfer to autosampler vials for LC/MS/MS. Inject 1 µl.
2.1.2.3.1 Analysis of the results:
As depicted in Figure 9B, shotgun-MS analysis revealed that other members of the STR complex, Sgs1 and Rmi1, can be effectively co-purified with Top3-FLAG from meiotic cultures.
32
Figure 9. SYPRO Ruby and shotgun-MS analysis of immuno-affinity purified STR components from meiotic prophase I. (A) SYPRO Ruby staining of FLAG-affinity purifications from cell lysates of strains with the indicated genotypes (left panel). 10% of the purified material was loaded. *Sgs1 cannot be detected by SYPRO Ruby.
Western blot analysis of the purified material (1% loaded on the gel), with an anti-FLAG antibody, is used to confirm the enrichment for Rmi1-FLAG (right panel). (B) Immuno-affinity purified (IP) Top3-FLAG from a 3 l meiotic culture was analyzed by Western blotting (1% loaded of the IP was loaded on the gel) using an anti-FLAG antibody. 5% of the purified material was analyzed by shotgun-MS for the indicated STR components. Displayed are the total number of spectral counts detected for each of the indicated proteins.
2.1.3 Functional analysis of Sgs1 helicase and Mus81-Mms4 nuclease from meiotic extracts
In this section, we describe how to assess the enzymatic activities of Sgs1 and Mus81-Mms4, when semi-purified from prophase I and metaphase I extracts, respectively. The general approach relies on immobilizing Sgs1-FLAG or Mus81-Mms4-FLAG on FLAG-affinity beads and assaying for enzymatic activity directly using synthetic DNA substrates (Matos et al., 2011;
Matos and West, 2017).
2.1.3.1 Analysis of DNA strand-displacement activity in Sgs1-FLAG immunoprecipitates from meiotic prophase I
The purification conditions and amounts described below were optimized for further biochemical assessment of strand displacement activity of Sgs1-FLAG immunoprecipitates on a fluorescently labeled gap-DNA substrate.
2.1.3.1.1 Immunoprecipitation of Sgs1-FLAG from prophase I lysates of S. cerevisiae Notes:
• All procedures should be performed on ice or at 4°C.
33
• ANTI-FLAG® M2 Affinity Gel agarose beads are washed and incubated in the Blocking buffer for at least 45 min to decrease nonspecific binding of proteins. After blocking, the beads are adjusted to 1:1 (V/V) with the Blocking buffer.
Solutions and reagents:
• ANTI-FLAG® M2 Affinity Gel agarose beads (Sigma-Aldrich)
• Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad)
• Blocking buffer: 40 mM Tris-HCl (pH 7.5 at 25°C), 150 mM NaCl, 15% (V/V) Glycerol, 0.1%
(V/V) NP-40, 1 mg/ml BSA
• ⌀ 0.5 mm glass beads (Carl Roth)
• Lysis buffer: 40 mM Tris-HCl (pH 7.5 at 25°C), 150 mM NaCl, 15% Glycerol, 0.1% NP-40, 0.1 mM EDTA, 0.5 mM EGTA, 1 mM NaF, 20 mM M2glycerophosphate, 2 mM PMSF, 1 tablet/50 ml solution of cOmplete™ EDTA-free Protease Inhibitor Cocktail (Roche), 1 mM DTT
• Protein sample buffer: 2x NuPAGE™ LDS Sample Buffer (Invitrogen), 200 mM DTT
• Reaction buffer: 40 mM Tris-AcO (pH 7.5 at 25°C), 4 mM Mg(AcO)2, 4 mM ATP, 2 mM DTT
• Wash buffer: 40 mM Tris-HCl (pH 7.5 at 25°C), 150 mM NaCl, 15% Glycerol, 0.1% NP-40 Procedure:
1. Thaw 0.5-0.8 g of yeast cells (obtained as described in section 2) and add 400 μL of Lysis buffer. Mix well to ensure that the cells are completely resuspended.
2. Add 500 μL of glass beads to the solution and vortex shortly.
3. Lyse the cells with a homogenizer (FastPrep-24TM 5G; MP Biomedicals) under the recommended settings for S. cerevisiae. Confirm the efficiency of cell lysis using a light microscope.
4. Punch a hole into the bottom of the 1.5 ml tube using a needle and place it into a 2 ml tube. To separate the extract from the glass beads, centrifuge the assembly in a precooled tabletop centrifuge for 1 min at 500 rcf.
5. Centrifuge the whole cell lysate at >15,000 rcf for 30 min in a precooled tabletop centrifuge and carefully transfer the cleared lysate to a new 1.5 ml tube.
6. Measure the total protein concentration of the lysate (e.g. by the Bradford assay).
7. Normalize the protein input concentration of proteins to 10 mg/ml in a total volume of 600 μl (this amount is sufficient to immunoprecipitate Sgs1-FLAG for one strand displacement reaction and western blot analysis).
8. Transfer 20 μl of the normalized extract into a microcentrifuge tube containing 20 μl of the protein Sample buffer. Boil the sample for 5 min, snap-freeze in liquid nitrogen and store