Synthese und Anwendung katalytisch aktiver polymerer Nanopartikel
199
0
0
Volltext
(4)(5)
(6)(7)
(8)(9)
(10)(11)
(12)
(13)
(14)(15)
(19)
(23)
(26)
(27)
(28)
(30)
(31)
(33)
(34)
(35)
(36)
(37)
(38)
Abbildung
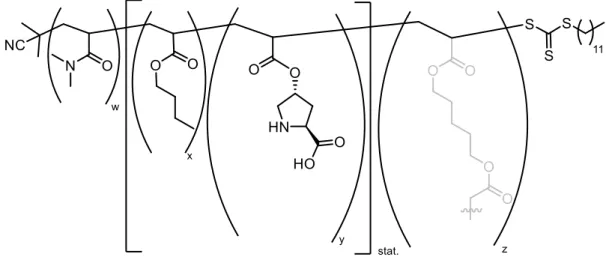
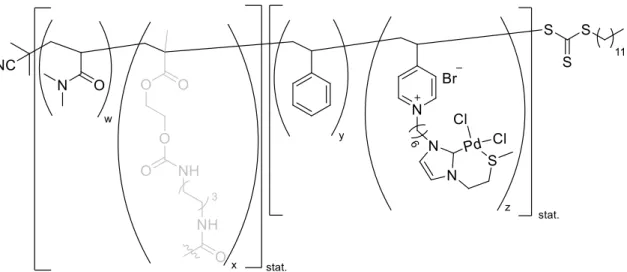
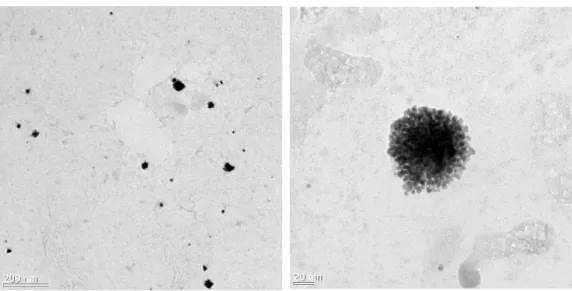
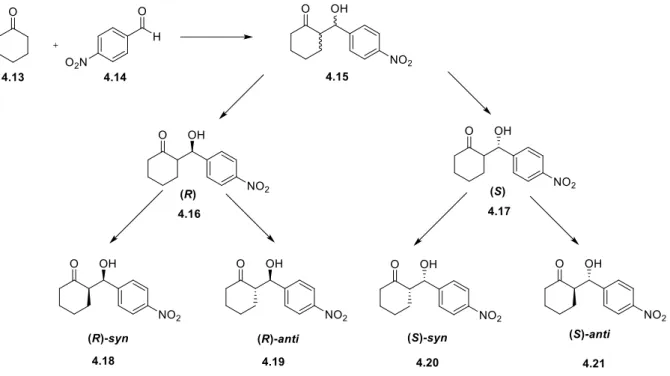
+7
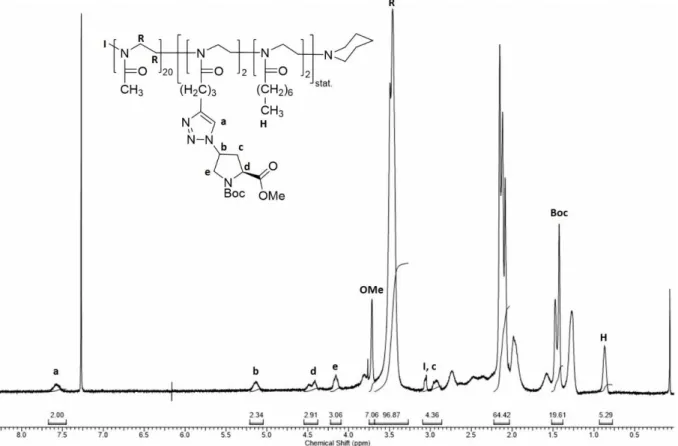
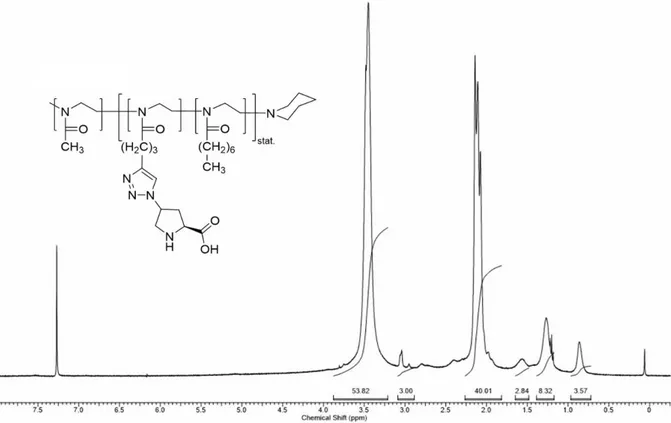
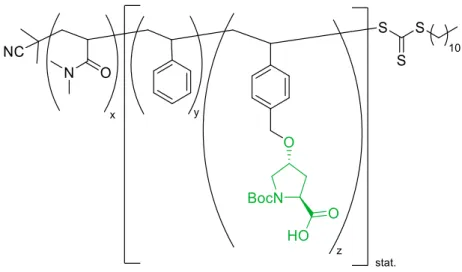
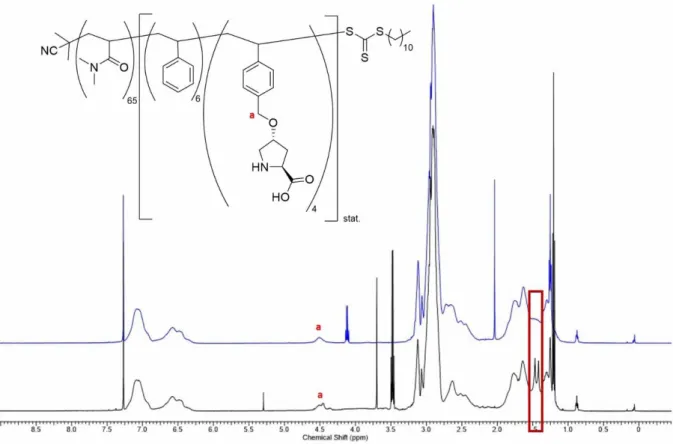
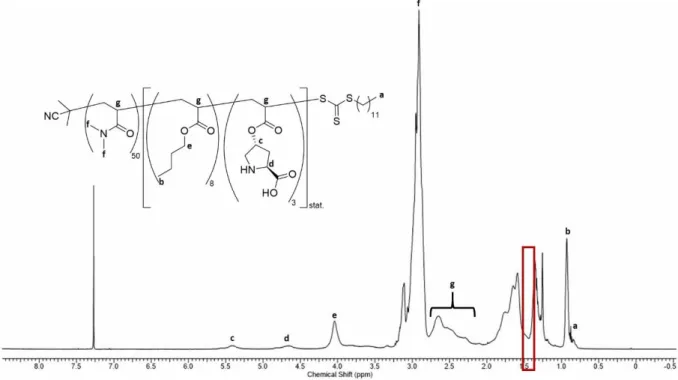
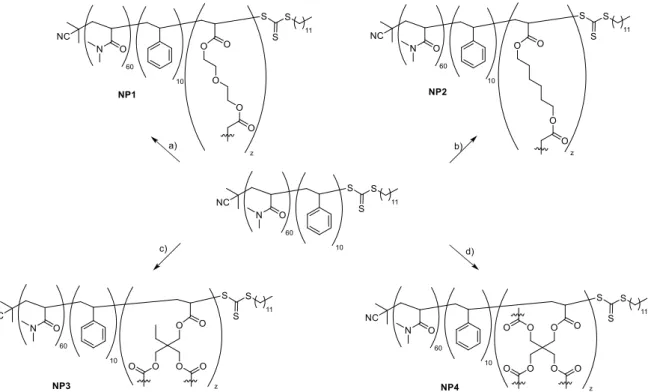
ÄHNLICHE DOKUMENTE
Die Lösung des Säurechlorids 49 wird bei -11 °C zugetropft und 90 Minuten gerührt, um eine leicht trübe, gelbe, hochviskose Suspension zu erhalten.. Die Suspension des
Brett, Messer, kleine Schälchen mittelgroßer Topf, Pfannenwender, Waage, EL. Messbecher, Waage, Rührlöffel, Kurzzeitmesser, TL, Abschmecklöffel,
bloqueio de transporte [1-9] Suporte para bits [1-10] Gancho para cinto. [1-11] Áreas de pega isoladas
Dabei zeigte sich, dass auch eine Reihe von Nitrobenzol-Derivaten selektiv durch die Einwirkung von sichtbarem Licht und PbBiO 2 Br reduziert
Propagieren Röntgenstrahlen durch eine Substanz, führen unterschiedliche Prozesse zu Beugungsphänomenen. Kohärente oder Thomson-Streuung führt zur Ausbreitung
Tabelle 18 zeigt, dass in HisAF der Austausch D127V zu einer vergleichbaren Aktivität führt wie der analoge Austausch in HisF und diese Aktivität durch den zusätzlichen
Obwohl sowohl die Boc- als auch die Z-Schutzgruppe für die Synthese und anschließende Polymerisation von Makromonomeren geeignet sein sollten, erschien die Abspaltung
Aufbauend darauf beschreibt Hibler 1977 die MÄ oglichkeit, das plastische Mo- dell durch ein viskoses Verhalten im Inneren der Flie¼°Ä ache zu ergÄ anzen (s. Dabei geht er von
