Synthese und Charakterisierung kernvernetzter, polymerer
Nanopartikel zur Anwendung in der Biomedizin
Dissertation
zur Erlangung des Doktorgrades der Naturwissenschaften
(Dr. rer. nat.)
Technische Universität Dortmund Fakultät für Chemie und Chemische Biologie
Arbeitsgruppe Polymere Hybridsysteme
vorgelegt von
Anne-Larissa Kampmann aus Schwerte
Dortmund, 2017
Die vorliegende Arbeit wurde in der Zeit von Oktober 2013 bis Januar 2017 unter der Leitung von Herrn Prof. Dr. Ralf Weberskirch an der Fakultät Chemie und Chemische Biologie der Technischen Universität Dortmund erstellt.
Teile dieser Arbeit wurden bereits veröffentlicht:
A.-L. Kampmann, M. Luksin, I. Pretzer, R. Weberskirch, Macromol. Chem. Phys. 2016, 15, 1704-1711.
A.-L. Kampmann, T. Grabe, C. Jaworski, R. Weberskirch, RSC Adv. 2016, 6, 99752- 99763.
1. Gutachter: Prof. Dr. Ralf Weberskirch 2. Gutachter: Prof. Dr. Jörg C. Tiller Eingereicht am 16.01.2017
Danksagung
Herrn Prof. Dr. Ralf Weberskirch danke ich für das interessante Forschungsthema, den hilfreichen Diskussionen und Anregungen und der jederzeit gewährten Forschungsfreiheit.
Herrn Prof. Dr. Jörg Tiller möchte ich herzlich für die freundliche Übernahme des Koreferates danken.
Zudem möchte ich den Mitgliedern des Arbeitskreises, Hanne Petersen, David Pelzer, Patrick Bolduan und Omar Sallouh für die freundschaftliche Arbeitsatmosphäre, sowie die gute Zusammenarbeit danken. Zusätzlich danke ich ganz herzlich Omar Sallouh für die Bereitstellung des in der Arbeit eingesetzten Peptids.
Ein besonderer Dank gilt Dr. Andrea Ernst, ohne die ich die Promotion nicht überstanden hätte. Durch ihre ausgeglichene, ruhige und besonnene Art hat sie mich aus so manchem Motivationstief und Aggressionshoch befreit.
Natürlich geht auch ein großer Dank an die Miterfinder -Platzes. Irene Pretzer und Henning Sand, ich danke euch für die tolle Zeit, nicht nur im Labor und Büro, sondern auch in Kiel, Halle, Essen und Marl!
Und wir warten immer noch auf Sir Linus und den Wein
Weiter möchte ich den Mitarbeitern der Fakultät Chemie und chemische Biologie, Katja Weber, Silvia Lessing, Andreas Hammer, Andrea Bockelmann, Dr. Ljuba Iovkova, Dr Alexandra Behler und Heidi Auer, für ihre stete Hilfsbereitschaft danken.
Genau wie den fleißigen Helfern, Monika Meuris, Iris Henkel, Dr. Wolf Hiller, Dr. Patrick Degen, Hannes Raschke, Marvin Heil und Anna Dantas, die mir bei der Lösung der vielen analytischen Problemstellungen geholfen haben. Zudem möchte ich den Mitarbeitern der Arbeitskreise von Prof. Tiller und Prof. Rehage für die Nutzung etlicher Geräte und das mir entgegengebrachtes Vertrauen bedanken.
Ein besonderer Dank gilt zudem Britta Glowacki und Herrn Prof. Klaus Jurkschat für die Kooperationsbereitschaft und die tolle Zusammenarbeit.
In diesem Zuge danke ich auch Herrn Prof. Ralf Schirrmacher und seinem Arbeitskreis für die Durchführung der Markierungsexperimente und die tollen PET-Aufnahmen.
Natürlich darf die Frau nicht vergessen werden, die mich bis zur Promotion gebracht hat. Ein großer Dank geht an Dr. Nadine Engelhardt, die eine wunderbare Betreuerin während meiner Bachelor- und Masterzeit war.
Ebenso danke ich allen meinen Praktikanten, Bacheloranten und Masteranten, die ich während meiner Promotion betreuen durfte. Ohne eure Hilfe wäre ich nicht so schnell vorangekommen. Ein ganz großer Dank an Michael Luksin, Irene Pretzer, Sascha Wilhelm, Simon Kotnig, Tobias Grabe, Carolin Jaworski, Laura Nowak, Carina Seitz, Andre Kemna, Kim Schlippkötter und Max Wiesehahn.
Natürlich danke ich auch meinen Korrekturleserinnen Andrea, Irene und Hanne dafür, dass sie sich die Zeit genommen haben und mit vielen hilfreichen Anmerkungen und Tipps zur Verbesserung der vorliegenden Arbeit beigetragen haben.
Meiner Familie danke ich für ihre Unterstützung und Hilfe während des Studiums und der Promotion. Kathi, Benni, Tobi, Bonnie, Lisa, Omi und Opa, ihr macht die verrückteste Familie zu der Besten der Welt.
Ein großer Dank gilt außerdem meinen langjährigen Freunden, für die Abwechslung und außerhalb des Labors. Elena Tschirbs, Siobhan Loftus, Fabian Wißing und Lukas Winkler, danke für eure Freundschaft und die verrückten, lustigen und teils turbulenten Zeiten der vergangenen Jahre. Ohne euch wäre ich nicht die, die ich heute bin!
INHALTSVERZEICHNIS
I
Inhaltsverzeichnis
1. Motivation ...1
2. Einleitung: Nanopartikel ...2
2.1 Methoden zur Nanopartikel-Synthese...4
2.1.1 Dispersionen aus vorgeformten Polymeren ...4
2.1.2 Polymerisationen von Monomeren ...8
2.2 Anforderungen an polymere Nanopartikel in der Biomedizin ... 11
2.3 Verwendetes Polymersystem ... 14
2.3.1 Die kationische Polymerisation von 2-Oxazolinen ... 14
2.3.2 2-Oxazolin-Monomere ... 16
2.3.3 Endgruppenfunktionalisierung ... 18
3. Aufgabenstellung und Zielsetzung ... 20
4. Nanopartikelsynthese mit Hilfe der Mikroemulsion ... 22
4.1 Ergebnisse und Diskussion Mikroemulsion I ... 23
4.1.1 Synthese von Alkin-funktionalisierten amphiphilen Poly(2-oxazolin)en ... 23
4.1.2 Nanopartikelsynthese mittels der Mikroemulsionspolymerisation und amphiphiler, Alkin-funktionalisierten Poly(2-oxazolin)-Makro-monomere 26 4.1.3 Zusammenfassung ... 34
4.2 Ergebnisse und Diskussion Mikroemulsion II ... 35
4.2.1 Synthese von Acrylat-funktionalisierten amphiphilen Poly(2-oxazolin)en ... 35
4.2.2 Nanopartikelsynthese mittels der Mikroemulsionspolymerisation und amphiphiler, Acrylat-funktionalisierten Poly(2-oxazolin)-Makro-monomere ... 40
4.2.3 Nanopartikelfunktionalisierung mit biorelevanten Molekülen ... 42
4.2.4 Zusammenfassung ... 55
5. Synthese funktionalisierter Nanopartikel für die Magnetresonanz- tomographie (MRT) ... 56
5.1 Einleitung: Molekulare Bildgebung... 56
5.2 Einführung in die Magnetresonanztomographie ... 57
5.3 Ergebnisse und Diskussion: Darstellung von Poly(2-oxazolin)-DO3A[Gd]- basierten Kontrastmitteln ... 63
5.3.1 Darstellung des Chelat-Liganden DO3A(tBu)3 ... 63
5.3.2 Darstellung von DO3A[Gd]-basierten Homopolymeren ... 64
5.3.3 Darstellung eines DO3A[Gd]-Blockcopolymers ... 73
INHALTSVERZEICHNIS
II
5.3.4 Darstellung eines DO3A[Gd]-basierten polymeren Nanopartikels... 78
5.3.5 Zusammenfassung ... 86
6. Synthese funktionalisierter Nanopartikel für die Positronen-Emissions- Tomographie (PET) ... 87
6.1 Einführung in die Positronen-Emissions-Tomographie ... 87
6.2 Ergebnisse und Diskussion: Darstellung von SiFA-Poly(2-oxazolin)-basierte Radiopharmaka ... 94
6.2.1 Darstellung eines SiFA-Homopolymers ... 94
6.2.2 Darstellung von SiFA-funktionalisierten Nanopartikeln auf Basis von amphiphilen Alkin-Blockcopolymeren ... 97
6.2.3 Darstellung von SiFA-funktionalisierten Nanopartikeln auf Basis von amphiphilen Acrylat-Blockcopolymeren ...108
6.2.4 Darstellung von SiFA-kernfunktionalisierten Nanopartikeln ...117
6.2.5 Zusammenfassung ...121
7. Darstellung von Nanopartikel-Multimeren...122
7.1 Anisotrope Partikel ...122
7.2 Ergebnisse und Diskussion: Darstellung von Nanopartikel-Multimeren über Bis-Terpyridin-Eisen(II)-Komplexe ...125
7.2.1 Synthese des Terpyridin-Motivs ...126
7.2.2 Polymersynthese und Charakterisierung ...127
7.2.3 Nanopartikelsynthese und Charakterisierung ...129
7.2.4 Funktionalisierung der Nanopartikel mit dem Terpyridin-Motivs ...130
7.2.5 Komplexierung der Nanopartikel ...133
7.2.6 Zusammenfassung ...138
8. Zusammenfassung und Ausblick ...139
9. Experimenteller Teil ...149
9.1 Allgemeine Arbeitstechniken und Geräte ...149
9.2 Präparative Vorschriften ...152
9.2.1 Aufreinigung von kommerziell erhältlichen Substanzen...152
9.2.2 Synthesevorschriften der Monomere ...153
9.2.3 Synthesevorschrift der Chelat-Liganden ...157
9.2.4 Synthesevorschriften der Blockcopolymere ...159
9.2.5 Allgemeine Vorschrift der Nanopartikelsynthese über die Mikroemulsionspolymerisation mit dem Azo-Initiator AIBN ...170
9.2.6 Allgemeine Vorschrift der Nanopartikelsynthese über die Mikroemulsionspolymerisation mit dem UV-Initiator 2-Propanthiol ...170
9.2.7 Synthesevorschrift zur Staudinger-Reaktion von P5 ...171
INHALTSVERZEICHNIS
III
9.2.8 Allgemeine Synthesevorschriften der Entschützungen ... 171
9.2.9 Allgemeine Synthesevorschrift zur Gadolinium(III)-Komplexierung der Polymere P8b, P8d, P10 und des Nanopartikels NP15 ... 172
9.2.10 Allgemeine Synthesevorschrift der nanopartikelanalogen tpy-Kopplung an den Nanopartikeln NP28-32 ... 172
9.2.11 Allgemeine Synthesevorschrift der Eisen(II)-Komplexierung der Nanopartikel NP28tpy-32tpy ... 173
10. Literaturverzeichnis ... 174
11. Anhang ... 184
11.1 Abkürzungs- und Symbolverzeichnis ... 184
11.2 Abbildungsverzeichnis ... 186
11.3 Schemataverzeichnis ... 192
11.4 Tabellenverzeichnis ... 194
11.5 NMR-Spektren... 196
11.5.1 Monomer-Spektren ... 196
11.5.2 Chelat-Spektren... 199
11.5.3 Polymer-Spektren... 201
11.6 Eidesstattliche Versicherung ... 214
MOTIVATION
1
1.
Die Erkennung, Behandlung und Heilung von Krebs, (neuro)degenerativen und Stoffwechselerkrankungen liegen im Fokus von verschiedensten Forschungsfeldern und gelten nicht nur für die Chemie, sondern für alle Naturwissenschaftsbereiche, als große Herausforderungen.
Die Visualisierung von Krankheitsherden und deren selektive Unterscheidung vom gesunden Gewebe durch geeignete Diagnostika sind heutzutage nur zwei der Forschungsfelder mit denen sich die Naturwissenschaftler eingehend beschäftigen.
Zudem sind die Aufklärung von Wirkmechanismen und die Optimierung der bestehenden Methoden weitere Hauptaufgaben der heutigen Wissenschaft. Ein großes Ziel für die Zukunft ist die patientenspezifische Therapie durch begleitende Echtzeit- Diagnostik, welche die heutigen diagnostischen und therapeutischen Standards revolutionieren würde.[1]
In der vorliegenden Arbeit wird die Synthese und Untersuchung von (multi)funktionalen Nanopartikeln basierend auf Poly(2-oxazolin)en für den biomedizinischen Bereich beschrieben und erläutert.
Nach Implementierung einer reproduzierbaren Nanopartikelsynthese auf Basis eines biokompatiblen Polymersystems und deren Funktionalisierung mit biorelevanten Molekülen, lag der weitere Fokus auf der Darstellung von Nanopartikeln für die Diagnostik. Dabei sollte die Immobilisierung eines Kontrastmittels für die Magnetresonanztomographie (MRT) bzw. eines Radiopharmaka-Vorläufers für die Positronen-Emissions-Tomographie (PET) auf makromolekulare Strukturen erfolgen.
Zunächst werden in der Einleitung die Besonderheiten der unterschiedlichen Nanopartikel-Typen und deren physikalischen Eigenschaften beschrieben. Zudem werden die Anforderungen dieser Systeme für den Einsatz in der Biomedizin erläutert.
Des Weiteren werden die Grundlagen des gewählten Polymersystems beschrieben.
EINLEITUNG
2
2.
Nanopartikel können als Objekte in einem Größenbereich von 1100 nm definiert werden.[2] Aufgrund ihrer unterschiedlichen Eigenschaften im Vergleich zu Atomen, Molekülen und Feststoffen ist das Interesse an ihnen in den letzten Jahren enorm gestiegen. Angesichts ihres großen Oberflächen-zu-Volumen-Verhältnisses resultieren einzigartige physikalische Eigenschaften, wie verändertes optisches Verhalten und spezifisches Leitvermögen. Diese Eigenschaften macht sie unter anderem interessant für den Elektroniksektor.[3] In diesem Bereich finden vor allem anorganische Nanopartikel auf Basis von Gold[4], Palladium[5], Silicium[6] und Eisenoxid[7] Verwendung. Ein ebenso interessantes Gebiet stellt der Einsatz von Nanopartikeln in der Biomedizin dar.
Der gezielte Transport von Wirkstoffen und Diagostika kann durch nanopartikuläre Trägersysteme ermöglicht werden. Auch hier finden anorganische Partikel Verwendung, wobei im Weiteren lediglich auf organische Nanopartikel eingegangen wird. In Abbildung 1 sind die typischen Vertreter der organischen Nanopartikel- Systeme dargestellt. Neben Dendrimeren[8] und Mizellen[9], gehören auch Liposome[10]
bzw. Polymersome[11] zu möglichen Trägersystemen für den Wirkstofftransport und finden auch Anwendung in der Diagnostik.
Abbildung 1: Schematische Abbildungen der Strukturen von organischen Nanopartikeln, a) Dendrimer mit dem Kern (rot), 1. Generation (blau), 2. Generation (violett), 3. Generation (schwarz);
b) Liposom/Polymersom mit hydrophiler Kopfgruppe (blau) und hydrophoben Schwanz (schwarz);
c) Mizelle mit hydrophiler Kopfgruppe (blau) und hydrophoben Schwanz (schwarz).
Dendrimere sind mit einer Größe von 110 nm die kleinsten Vertreter der polymeren Nanopartikel. Über einen multifunktionalen Kern (rot) wird durch eine repetitive Synthese ein hochverzweigtes Makromolekül mit definierter Größe und Anzahl funktioneller Gruppen / Generation aufgebaut.[12] Liposome bestehen aus Phospholipiden, welche Doppelmembranen aufbauen und somit Wasser einschließen können. Diese geschlossenen Vesikel können durch Ausbildung von mehreren Schichten Größen von einigen Nanometern bis hin zu wenigen Mikrometern aufweisen.
EINLEITUNG
3 Im Gegensatz dazu bestehen die Doppelschichten der Polymersome aus Di-/Triblock- copolymeren. Dies ermöglicht im Vergleich zu Liposomen den Einbau von funktionellen Gruppen.[13] Mizellen hingegen sind sphärische Anordnungen amphiphiler Blockcopolymere, welche sich oberhalb der kritischen Mizellbildungskonzentration (CMC) bilden.[14]
Polymere Nanopartikel eröffnen durch ihre hohe Flexibilität die einfache Modifikation auf molekularer Ebene. Durch Veränderung der entsprechenden Monomer- bzw.
Polymereinheit können nicht nur aktive, funktionelle Gruppen in ein Nanosystem eingeführt werden, sondern auch verschiedene Architekturen aufgebaut und unterschiedliche übergeordnete Strukturen ermöglicht werden. In den letzten Jahren rückte die Darstellung und Entwicklung von multifunktionalen Nanomaterialien mit definierter chemischer Zusammensetzung für den Einsatz in der Biomedizin nicht nur in den Fokus der akademischen Forschung, sondern auch der industriellen Anwendung. In Tabelle 1 sind einige Beispiele kommerzieller Anwendungen von organischen Nanopartikeln im Bereich der Medizin aufgelistet. Dabei werden Liposome von einigen Firmen als Wirkstofftransport-Systeme zur Solubilisierung von hydrophoben Arzneimitteln in der Behandlung von Krebs und Pilzinfektionen, aber auch als Trägersystem für Impfstoffe gegen Influenza und Hepatitis A, eingesetzt. Die Firma Starpharma[15] fokussiert sich hingegen bei der Behandlung von Krebs auf den Einsatz von Dendrimeren als Trägersysteme für den gezielten Wirkstofftransport.
Tabelle 1: Beispiele von kommerziellen Anwendungen von Nanopartikeln.[16]
Nanopartikel-
Komponente Anwendung Indikation Firma
Liposome Wirkstofftransport Krebs Liplasoma Pharma (Lyngby, Dänemark)
Schering-Plough Corp.
(Kenilworth, NJ, USA) Wirkstofftransport Impfstoff für Influenza,
Hepatitis A
Berna Biotech AG (Basel, Schweiz)
Wirkstofftransport Pilzinfektion Enzon (Bridgewater, NJ, USA) Dendrimere Therapeutikum HIV, Krebs,
Entzündungen Starpharma (Melbourne, Australien)
Vor allem die Möglichkeit polymere Nanomaterialien kontrolliert aufzubauen und zu funktionalisieren, ermöglicht neue Ansätze in der Therapie und Diagnostik. Aus diesem Grund werden zunächst einige Strategien zur Nanopartikeldarstellung beschrieben. Im darauffolgenden Abschnitt werden dann die Anforderungen für Nanomaterialien für den Einsatz in der Biomedizin erläutert.
EINLEITUNG
4
2.1 Methoden zur Nanopartikel-Synthese
In der Vergangenheit hat das Themengebiet der Nanopartikel und deren Darstellung das Interesse vieler Forschungsgruppen geweckt. Über die letzten Jahrzehnte wurden verschiedene Darstellungsmethoden hin zu organischen Nanopartikeln entwickelt. Es werden im Allgemeinen zwei Strategien unterschieden: Dipersionen bestehend aus vorgeformten Polymeren (1) und die direkte Polymerisation von Monomeren (2). Bei beiden können unterschiedliche Prozesse und Methoden zur Einstellung von Größe, Form und chemischen Eigenschaften heran gezogen werden. Zu den klassischen Methoden der Nanopartikeldarstellung über vorgeformte Polymere gehören unter Anderem das Aussalzen, die Nanoausfällung und das Dialyseverfahren. Weiter wird in diesem Kapitel auch die Kern-/Schalevernetzung von amphiphilen Polymeren betrachtet. Die Emulsionsverfahren (Mini-, Mikro-, Makro-Emulsion) sowie die Grenzflächenpolymerisation gehören zu der Darstellungsmethode über die direkte Polymerisation von Monomeren.[17] Die Wahl der Darstellungsmethode bezieht sich auf das verwendete Polymersystem und die gewünschte Größe, sowie die spätere Anwendung. Im Folgenden wird auf einige der oben genannten Darstellungsverfahren eingegangen.
2.1.1 Dispersionen aus vorgeformten Polymeren
In diesem Kapitel werden die Darstellungsmethoden über vorgeformte Polymere beschrieben und erläutert. Dazu zählen unter Anderem der Aussalzungsprozess, die Nanoausfällung und die Kern-/Schalevernetzung.
Aussalzen
Anfang der 1990er Jahre veröffentlichte Bindschaedler et al. eine Nanopartikelsynthese über einen modifizierten Emulsionsansatz, welcher als Schlüsselprinzip einen Aussalzungsprozess beinhaltet.[18] Ein großer Vorteil dieser Methode ist, dass auf große Mengen organisches Lösungsmittel und auf die Zugabe von oberflächenaktiven Substanzen verzichtet werden kann. In einem typischen Darstellungsprozess wird das vorgeformte Polymer in einem Gemisch aus Wasser und einem sich in Wasser löslichem Lösungsmittel (meist Aceton) dispergiert. Durch anschließende Zugabe von einer hochkonzentrierten wässrigen Salz-Lösung, meist bestehend aus Magnesium- oder Calciumchlorid, wird die Löslichkeit des organischen Lösungsmittels in der wässrigen Phase herabgesetzt. Dadurch ist das organische Lösungsmittel nicht mehr in Wasser löslich ist und eine Dispersion entsteht. Das Polymer ist im Organischen besser löslich, wodurch es in die organischen Tröpfchen gedrängt wird. Durch Verdünnen der
EINLEITUNG
5 Mischung mit einem Überschuss an Wasser kommt es zur Ausfällung des Polymers.
Durch die rapide Konzentrationsänderung wird das organische Lösungsmittel wieder in Wasser löslich. Die Wanderung des organischen Lösungsmittels in die wässrige Phase bewirkt den Ausfällungsprozess des Polymers als Nanopartikel. Neben Aceton, werden auch Ethanol oder Tetrahydrofuran (THF) als organische Lösungsmittel genutzt.[19] Die verwendeten Polymere basieren meist auf Polylactiden oder Polyacrylaten. Durch diese Methode können Partikel im Größenbereich 100 250 nm hergestellt werden.[20]
Poly(2-oxazolin)-basierte Nanopartikel, dargestellt über das Aussalz-Verfahren, sind nicht in der Literatur bekannt. Güner et al. verwendeten den Aussalzungsprozess lediglich zur Darstellung von Poly(2-ethyl-2-oxazolin)-Faser. Die Löslichkeit des hydrophilen Homopolymers wurde jeweils durch die Zugabe von Natriumacetat- und Natriumthiocyanat-Lösungen in Wasser so herabgesetzt, dass sich Fasern mit einem Durchschnittsdurchmesser von 2.7 ± 0.5 µm ausbildeten.[21]
Nanoausfällung
Das Prinzip der Nanoausfällung basiert auf der Grenzflächendeposition eines Polymers nach Verdrängung eines semipolaren Lösungsmittels, welches in einem unpolaren Lösungsmittel vorliegt.[22] Bei der Fällung kommt es durch intensives Vermischen aus drei Komponenten zu einer Übersättigung im Reaktionsraum. Dieser besteht aus dem Polymer, einem selektivem Lösungsmittel, in welchem das Polymer löslich ist, und einem nicht-selektivem. Zur Induzierung dieses Stoff-übergangs von einer Phase in die andere muss eine thermodynamische Triebkraft aufgeprägt werden, hier die übersättigte Lösung an Polymer. Nach Erreichen der Übersättigung werden mit Hilfe homogener und heterogener Keimbildung nanokristalline Partikel erzeugt. Nach Bildung der Primärpartikel beginnt ein diffusionskontrolliertes Partikelwachstum. Eine Nanoausfällung wird somit durch drei Mikroprozesse bestimmt: Keimbildung (1), Partikelwachstum (2) und Reifung der gebildeten Nanopartikel (3). Meist werden als organische Lösungsmittel, die in Wasser löslich sind, wie Aceton oder Ethanol gewählt.[23] Zudem sollten diese leicht verdampfen, um die Übersättigung zu erzeugen.
Bei dem nicht-selektive Lösungsmittel handelt es sich meist um ein Gemisch aus Wasser mit einem Tensid. In Nanoausfällungen kommen unterschiedliche Polymere zum Einsatz, vor allem biodegradierbare Polymersysteme, wie Polyester.[24] Durch diesen Prozess können Nanopartikel mit einer engen Verteilung in einem Größenbereich von 60 600 nm dargestellt werden.[25]
Kempe et al. synthetisierten 200 800 nm große Poly(2-oxazolin)-Partikel über die Nanoausfällung. Dabei verwendeten sie Copolymere bestehend aus dem hydrophilen 2-Ethyl-2-oxazolin (EOx) und dem hydophoben 2-Decyl-10-en-2-oxazolin (DOx), welche endständig mit einem Fluoreszenzfarbstoff funktionalisiert wurden. Als
EINLEITUNG
8
2.1.2 Polymerisationen von Monomeren
In diesem Kapiel wird auf die Nanopartikeldarstellung über die direkte Polymerisation von Monomeren eingegangen. Hierbei wird zunächst kurz auf die Grenzflächenpolymerisation eingegangen, anschließend werden die Emulsionspolymerisationen eingehend betrachtet.
Grenzflächenpolymerisation
Bei der Grenzflächenpolymerisation handelt es sich um eine fest etablierte Nanopartikelherstellung. Dabei wird eine Stufenpolymerisation von zwei reaktiven Monomeren ausgelöst, welche in zwei nicht miteinander mischbaren Phasen gelöst sind.[39] Die eigentliche Polymerisation findet somit nur an der Grenzfläche der beiden Phasen statt. Dabei kann durch Wahl der Polymerisationsart die Gestalt und Form der Nanopartikel eingestellt werden.[40] Hohle Nanopartikel können über Polyadditionen oder auch Polykondensationen dargestellt werden.[41] Nanokapseln, welche einen ölhaltigen Kern besitzen, werden über eine Grenzflächenpolymerisation in einem Wasser/Öl-System erhalten.[42] Dabei kann die Bildung von Nanokapseln durch Zugabe von Acteon oder Acetonitril zur wässrigen Phase unterstützt werden. Die erhaltenen Nanosysteme besitzen eine Größe von 20 350 nm.[43] Es sind keine Grenzflächenpolymerisationen mit dem hier verwendeten Poly(2-oxazolin)-System bekannt.
Emulsionspolymerisation
Im Allgemeinen versteht man unter einer Emulsion ein feinverteiltes Gemisch aus zwei nicht mischbaren Flüssigkeiten. Dabei unterscheidet man zwischen Makro-, Mini- und Mikroemulsionen. Eine Flüssigkeit bildet in der anderen kleine Tröpfchen aus. Die Tröpfchen-bildende Phase nennt man disperse Phase und die Phase in der die Tröpfchen schwimmen wird kontinuierliche Phase genannt (Abbildung 3)
a) Makroemulsion
Ein typisches Beispiel einer Makroemulsion (> 1 µm) sind Öl-Tröpfchen in Wasser.
Generell sind Emulsionen thermodynamisch instabil, da die disperse Phase bestrebt ist zusammen zu fließen. Diesen Vorgang nennt man Koaleszenz. Eine weitere Destabilisierung kann das System über die Ostwald-Reifung erfahren. Hier wird der Konzentrationsunterschied verschieden großer Tropfen ausgeglichen, indem ein Materiestrom von den kleinen zu den großen Tröpfchen fließt.
EINLEITUNG
10
nanopartikelanaloge Reaktionen auf der Oberfläche der 140 195 nm großen Nanopartikel durchgeführt werden.[49]
Amato et al. zeigten eine weitere Methode der Oberflächenmodifizierung. Über eine UV-induzierte Photopolymerisation von Thiolen und Alkenen in einer Miniemulsion stellten sie Nanopartikel im Größenbereich von 100 nm her. Durch die Variation der Menge an Thiol- bzw. Alken-Komponente konnte eine Thiol- bzw. Alken- funktionalisierte Schale erhalten werden. Diese Oberfläche konnte in entsprechenden nanopartikelanalogen Reaktionen weiter mit verschiedenen Farbstoffen umgesetzt werden.[50]
c) Mikroemulsion
Eine weitere Strategie im Bereich der Emuslionspolymerisationen ist die Mikroemulsion. Diese beschreiben Dispersionen, bestehend aus einem Gemisch aus hydrophober Flüssigkeit und einem Tensid in Wasser. Die Grenzflächenspannung ist aufgrund der hohen Tensidkonzentration fast gleich null. Hierdurch entsteht ein thermodynamisch stabiles System, welches Tröpfchen von 1 100 nm ausbildet.[44]
Somit ist durch die Mikroemulsionspräparation die komplette Monomer-Oberfläche mit dem eingesetzten Tensid bedeckt. Dies ist einer der Gründe warum diese Technik so attraktiv für die Stabilisierung von übergeordneten Strukturen geworden ist. 1977 veröffentlichten Riess und Nervo die ersten Versuche der Kombination von Polymeren und dem Mikroemulsionsansatzes.[51] 1985 beschrieben Candau, Leong, Fitch die Kinetik des Polymerisationsprozesses von mit Monomeren geschwollenen Mizellen über ein Model. Die Polymerisation wird hierbei radikalisch in der wässrigen Phase gestartet und die Monomere werden von der unpolymerisierten Mizelle durch das Wasser in den wachsenen Partikel transportiert. Dieses Verfahren führt zu Partikeln im Größenbereich von 1 100 nm und leeren Mizellen. In den darauffolgenden Jahren wurde weiter intensiv geforscht und der (Konzentrations)einfluss von unterschiedlichen Tensiden[52], Initiatoren[53] und Monomeren[54] auf die Partikelsynthese untersucht.[55]
Neben der radikalischen Polymerisation wurden auch kontrolliert radikalische Polymerisationsarten, wie die NMP[56] (engl.: Nitroxide-mediated Polymerization, ATRP[57] und die RAFT[58] (engl.: Reversible AdditionFragmentation Chain Transfer), in der Darstellung von Nanopartikeln über den Mikroemulsionsprozess eingesetzt. In der Mikroemulsion können allgemein Oberflächen-funktionalisierte Partikel durch die Copolymerisation von zwei hydrophoben Monomeren dargestellt werden. Dabei sollte eines dieser Monomere weniger hydrophob sein und sich an der Grenzfläche orientieren.[59] Antonietti et al. stellten durch den Einsatz von Lecithin als Tensid biokompatible Latex-Partikel auf Basis von Styrol und m-Diisopropenylbenzol
EINLEITUNG
11 her. Durch Oberflächen-aktive Proteine konnten sie die 10 20 nm großen Partikel auf der Schale funktionalisieren (ein Protein pro Partikel).[60]
Nachteil bei dieser Methode ist der vorhandene Restanteil an Tensid in der Nanopartikelprobe, welcher vor weiterer Anwendung, z.B. im Medzinbereich, über Ultrazentrifugation, Ultrafiltration oder Dialyse entfernt werden muss. Daneben wurden Untersuchungen angestrebt gewünschte Funktionalitäten über entsprechende amphiphile, bifunktionale Blockcopolymere ein zu bringen. Diese dienen somit als Makromonomere bzw. Makroinitiatoren im Mikroemulsionsprozess und werden kovalent am Partikel gebunden. Damit ist keine weitere Aufreinigung notwendig.[61]
Die bisher bekannten Systeme, in welchem Blockcopolymere als Makromonomere eingesetzt wurden, wurden von . veröffentlicht. Sie setzten Poly(N- acetylethylenimin)e mit polymerisierbaren Zimtalkohol-Endgruppen[62] und Maleinsäure-terminierten Poly(N-acetylethylenimin)e[63] als Co-Makromonomere neben dem eigentlichem Tensid (SDS) in einer Mikroemulsion ein und erhielten Acrylat- basierte Nanopartikel im Größenbereich von 40 100 nm.
Mitte der 1990er beschrieb Kobayashi et al. den Einsatz von vinyl-modifizierten Poly(2-oxazolin)en als Makromonomere in der Darstellung von monodispersen polymeren Partikeln im Mikrometer-Bereich auf Basis von Styrol[64] bzw. Acrylaten[65]. Zur Herstellung der polymeren Partikel wurde die Dispersionspolymerisation verwendet, wobei das Makromonomer zusätzlich als Stabilisator in einer wässrigen Alkohollösung diente. Weiter wurde der Einfluss der Makromonomerkonzentration von 1-5 Gew.-% auf die resultierende Partikelgröße untersucht. Es zeigte sich eine Abnahme des Durchmessers mit steigendem Poly(2-oxazolin)-Gehalt und eine gleichzeitige Verbesserung der Monodispersität.
Somit gibt es bis jetzt keine bekannte Nanopartikelsynthese, welches Poly(2- oxazolin)en als Makromonomere in einer Mikroemulsion einsetzt.
2.2 Anforderungen an polymere Nanopartikel in der Biomedizin
1975 löste Ringsdorf mit seiner Forschung auf dem Gebiet der Polymer-basierten Wirkstoff-Konjugate großes Interesse bei Chemiker und Pharmazeuten aus, Makromoleküle in der Biomedizin einzusetzen. Er zeigte damit entgegen der damaligen Skepsis, dass pharmakologisch aktive, makromolekulare Strukturen Vorteile im Bezug auf Löslichkeit, Toxizität und Bioverteilung im Körper besitzen.[66] Auch Jahre später zeigt sich, dass nicht nur Polymer-Wirkstoffkonjugate, sondern auch makromolekulare Komponeneten im Nanometerbereich in der Biomedizin als Grundgerüst für diagnostische und therapeutische Zwecke verwendet werden können. Aufgrund ihrerEINLEITUNG
13 Mechanismus der Erkennung und Entfernung von Fremdsubstanzen im Körper durch das Immunsystems. Aber auch das Ausscheiden über die Niere (MWCO-Größe von 10 nm), über die Milz (Fenestrations-Poren < 500 nm) und über die Leber sollte vermieden werden. Ein wichtiger Aspekt ist die sogenannte Tarnkappeneigenschaft
stealth-Eigenschaft) solcher Nanosysteme. Diese verhindert die Absorption von Plasmaproteinen und die Erkennung durch das mononukleär-phagozytierende System (MPS). Das MPS ist Teil des Immunsystems und erkennt fremdartige Stoffe im Körper, um sie dann aus dem Körper zu entfernen.[68] Ein erfolgreicher Ansatz, die Erkennung des Immunsystems zu vermeiden, ist das dichte Umhüllen des Nanosystems mit flexiblen, neutralen, hydrophilen Polymerketten. Eine Beschichtung mit unterschiedlich langen PEG-Ketten zeigte zum Beispiel einen starken Einfluss auf die Blutzirkulationszeit und die Beseitigung der Nanopartikel aus dem Körper.[69] Die Oberflächenladung der Partikel beeinflusst dabei stark die Toxizität und die Bioverteilung. Eine zu hohe positive Ladung bewirkt eine schnelle Entfernung durch das MPS, was eine Verstopfung der Blutgefäße zur Folge haben kann. Dennoch ist eine leicht positive Ladung der Oberfläche nützlich bei der Zellaufnahme über den EPR- Effekt (engl. enhanced permeability and retention = ilität und
. Die Einstellung der Oberflächenladung für die ideale Zellaufnahme stellt eine große Herausforderung dar.[70] Auch die Größe und Form der Nanopartikel hat einen Einfluss darauf, ob die Opsonisierung eingeleitet wird, eine Ausscheidung über die Niere erfolgt oder ob eine Zellaufnahme stattfindet. Sind die Partikel kleiner als 10 nm, erfolgt oft eine schnelle Ausscheidung über die Niere Zwischen 10 200 nm liegt die optimale Größe, um über den EPR-Effekt in die Zelle aufgenommen zu werden. Auch beschädigte Zellwände, wie es bei Tumorzellen der Fall ist, besitzen maximal 200 nm große Poren.[71] Bei größeren Partikeln (> 200 nm) steigt wiederum die Toxizität an, da hieraus eine Verstopfung der Blutgefäße resultieren kann. Neben der Größe, spielt auch die Morphologie der Partikel eine Rolle bei der Zellaufnahme.
Die sphärische Form ist die meist genutzte bzw. synthetisierte Form polymerer Nanopartikel, weil diese im Vergleich zu zylindrischen Formen eine bessere Aufnahme zeigen.[72] Ein weiterer wichtiger Aspekt ist die Stabilität von polymeren Nanopartikeln.
Nanopartikel aus Aggregaten, welche lediglich über entropische oder elektrostatische Wechselwirkungen stabilisiert werden, können ihre Strukturen in vivo durch äußere Einflüsse, wie pH-Wert- und Temperatur-Änderungen, oder Verdünnungseffekte verlieren. Mizellen zum Beispiel liegen unterhalb der kritischen Mizellbildungskonzentration (CMC) als einzelne Polymerketten vor.[73] Die Unterschreitung der CMC kann durch die Verdünnung im Blut ausgelöst werden.[74]
Aus diesem Grund ist eine Stabilisierung der eingesetzten Nanomaterialien sinnvoll und meist notwendig. In dem Kapitel 2.1 wurden einige gängige Synthesen zur Nanopartikelstabilisierung beschrieben und erläutert.
EINLEITUNG
14
2.3 Verwendetes Polymersystem
In dieser Arbeit wurde das Polymersystem basierend auf 2-Oxazolinen verwendet.
Dieses bietet vor allem für den Einsatz in der Biomedizin zahlreiche Vorteile. Poly(2- oxazolin)e gelten im Allgemeinen als nicht-toxisch und biokompatibel. Gerade die hydrophilen Poly(2-oxazolin)e weisen geringe Interaktionen mit dem menschlichen Serumproteinen auf. Somit werden sie als Alternative[75] zum Polyethylenglycol (PEG) gesehen, welches zurzeit in der Medizin und Kosmetik das meist eingesetzte Polymersystem ist.[76] Weiter weisen Poly(2-oxazolin)e strukturelle Ähnlichkeit zu Polypeptiden auf (Abbildung 5) und werden daher zu den sogenannten Pseudopeptide gezählt.[77]
Abbildung 5: Strukturausschnitte eines Poly(2-oxazolin)s (links) und eines Polypeptids (rechts).
Weitere Vorteile sind die zahlreichen Modifikations-Möglichkeiten des Polymersystems, welche damit den großen Nachteil des PEG-Systems ausgleicht.[78] Im Gegensatz zu PEG können viele Modifikationen am Monomer, dem Terminierungsreagenz und dem Initiator erfolgen, wodurch eine Menge von Funktionalitäten in das Polymer eingebracht werden können.[79] Ein weiterer Vorteil ist der lebende Charakter dieser Polymerisation. Hierdurch kann der Polymerisationsgrad eingestellt und eine definierte Polymerstruktur mit niedriger Polydispersität aufgebaut werden.[80] Jedoch zeigt das System den großen Nachteil, dass die Polymere nicht bioabbaubar sind.[81]
In den folgenden Abschnitten wird zunächst der Mechanismus der kationischen Ring- öffnenden Polymerisation der 2-Oxazoline beschrieben und weiter auf die Möglichkeiten der Funktionalisierung über das Monomer, den Initiator und das Terminierungsreagenz eingegangen.
2.3.1 Die kationische Polymerisation von 2-Oxazolinen
Die Polymerisation von 2-Oxazolinen gehört zu der Klasse der lebenden Polymerisationsarten, die 1956 von Szwarc definiert wurde.[82] Mitte der 1960er wurde die Poly(2-oxazolin)-Synthese von vier Forschungsgruppen unabhängig voneinander beschrieben.[83] Die Polymerisation läuft nach dem kationischen ringöffnenden Mechanismus ab. In Schema 2 wird die Polymerisation mechanistisch betrachtet. Diese
EINLEITUNG
15 verläuft über eine ionisch-zyklische Zwischenstufe und wird in drei Schritte gegliedert:
Initiierung, Kettenwachstum und Terminierung.
Schema 2: Mechanismus der kationischen Polymerisation von 2-Oxazolinen.
Gestartet wird die Polymerisation durch den nukleophilen Angriff des Stickstoffatoms eines 2-Oxazolins auf das elektrophile Zentrum des Initiators. Als Initiatoren können starke Brønsted-Säuren (z.B. H2SO4), Lewis-Säuren (AlCl3, BF3), Sulfonsäureester (MeOTs, MeOTf) oder Benzylhalogenide dienen. Es bildet sich eine kationische, zyklische Spezies, die mit einer offenkettigen, ladungsneutralen Spezies im thermodynamischen Gleichgewicht liegt. Bei einem idealen Verlauf dieser Polymerisationsart wird vorausgesetzt, dass ki >> kp ist. Dies bedeutet, dass die Initiierung schnell und vollständig verläuft. Somit kann von einem gleichmäßigen Kettenwachstum ausgegangen werden. Auf diese Weise lassen sich Polymere mit definierter Molmasse, Zusammensetzung und enger Molmassenverteilung herstellen.[84]
Im nächsten Schritt erfolgt das Kettenwachstum durch wiederholte Anlagerung weiterer Monomereinheiten. Nach Verbrauch aller Monomermoleküle bleibt das Makrokation aktiv, somit kann durch Zugabe neuer Monomere die Polymerkette weiter wachsen.
Dies wird dazu genutzt Blockcopolymere mit Monomerblöcken aufzubauen, welche unterschiedliche chemische Eigenschaften besitzen können. Die Isomerisierung der Iminoethergruppe zur energetisch günstigeren Amidgruppe trägt wesentlich zur guten Polymerisierbarkeit der Monomere bei. Die Terminierung des Systems wird durch die gezielte Zugabe eines Nukleophils (z.B. Piperidin, Piperazin, Wasser) erreicht.
Trotz des ideal-vorausgesetzten Mechanismus, entdeckten 1975 Litt et al. mögliche Neben- und Abbruchreaktionen.[85] Besonders beim Aufbau von längeren Ketten und bei hohen Temperaturen werden Nebenreaktionen der Oxazolinpolymerisation
EINLEITUNG
16
beschrieben. In Schema 3 sind die Folgen von möglichen Übertragungsreaktionen aufgeführt.
Schema 3: Übertragungsreaktion bei der Polymerisation von 2-Oxazolinen: a) Umwandlung eines Kettenendes (A) in eine Enamin-Ether-Endgruppe (B) und eine neue wachsende Spezies (C), b) Reaktion der Enamin-Ether-Spezies (B) mit einer aktiven Polymerkette zu hochmolekularen verzweigten Polymeren, c) Entstehung niedermolekularer Ketten.
Dabei wird ein Proton von einem lebenden Polymerkettenende unter Ausbildung einer Enamingruppe auf eine Monomereinheit transferiert (A). Diese Enaminfunktion kann mit dem lebenden Kettenende eines zweiten Polymers reagieren (B), wodurch sich verzweigte Makromoleküle bilden können. Das entstandene Oxazolinium kann nun als Initiator reagieren und somit eine neue Kette starten (C).[86]
2.3.2 2-Oxazolin-Monomere
2-Oxazoline (Dihydrooxazole) sind fünfgliedrige Heterozyklen mit einem Sauerstoff- und Stickstoffatom in 1,3-Stellung zueinander. In Abbildung 6 ist die allgemeine Strukturformel dargestellt. Über die Alkyl-Kettenlänge in Position 2 kann die Eigenschaft des entsprechenden Homopolymers generiert werden. Handelt es sich beim Rest R um eine Methyl- bzw. Ethylgruppe, ist das Homopolymer gut wasserlöslich. Die Wasserlöslichkeit nimmt jedoch mit länger werdender Alkylkette stetig ab.[87]
Abbildung 6: Allgemeine Strukturformel eines 2-Oxazolins und die Löslichkeitseigenschaften des Homopolymers in Abhängigkeit der Monomer-Seitenkette.
EINLEITUNG
18
Unter anderem können aromatische Systeme, sowie (endständige) Doppel- und Dreifachbindungen über entsprechende Monomere in das Polymer eingebracht werden.
Zudem werden Amine, Alkohole und Thiole in geschützter Form einpolymerisiert und anschließend polymeranalog entschützt.
Weitere Möglichkeiten, Funktionalitäten in ein Polymer einzubringen, ergeben sich durch die Derivatisierung des Initiators bzw. des Terminationsreagenzes.
2.3.3 Endgruppenfunktionalisierung
Durch den Einsatz von funktionalisierten Initiatoren bzw. Terminierungsreagenzen können funktionelle Gruppen endständig ins Polymer eingebracht werden. Dabei bietet der Einsatz von funktionalisierten Initiatoren den Vorteil, dass von einer hohen Funktionalisierungsdichte ausgegangen werden kann.
Initiatoren
Bei der Initiierung der kationischen Polymerisation von 2-Oxazolinen handelt es formal um eine SN2-Reaktion. Aus diesem Grund muss der Initiator eine gute Abgangsgruppe besitzen, welche als stabiles Gegenion während der Polymerisation vorhanden bleibt. Es konnte gezeigt werden, dass neben Benzylhalogeniden, Triflate besonders geeignet erscheinen.[90] Gerade das Triflat-Ion ist durch seine Resonanzstrukturen äußerst stabil und die Reaktion läuft schon bei Raumtemperatur quantitativ ab. Dadurch ergibt sich eine große Vielfalt an möglichen Initiatoren. In Abbildung 9 ist zum einen das oft verwendete Methyltriflat (a) und ein funktionalisierter Initiator auf Triflat-Basis dargestellt (b). Auch das simultane Starten von zwei Polymerketten kann durch die Wahl eines geeigneten Initiators erfolgen (c).
Abbildung 9: Beispiele für mögliche (funktionalisierte) Initiatoren, a) Methyltriflat, b) Pent-4-ynyl- triflat[91] -Dibromo-p-xylol[92].
Terminierungsreagenzen
Die Polymerisation von 2-Oxazolinen kann aufgrund ihres lebenden kationischen Mechanismus gezielt durch starke nukleophile Reagenzien beendet werden. In Schema 4 ist eine Auswahl von verschiedenen Nukleophilen dargestellt. Dabei können nicht nur
EINLEITUNG
19 Thiole (1), Amine (2), Natriumazid (4) und deprotonierte Carbonsäuren (5) zur Terminierung verwendet werden, auch Spuren von Wasser (3) führen zum Abbruch der Polymerisation. Aus diesem Grund ist eine wasserfreie Durchführung wichtig für eine erfolgreiche Polymerisation. Um eine Funktionalisierung über die Termination zu erreichen, muss der Rest R2 entsprechend modifiziert werden oder im Falle der Azidendgruppe (4) kann direkt eine funktionelle Gruppe erhalten.
Schema 4: Eine Auswahl an Terminationsreaktionen von Poly-(2-oxazolin)en über (1) Thiole, (2) Amine[93], (3) Wasser[94], (4) Natriumazid[95] und (5) deprotonierte Carbonsäuren[96].
Eine Endgruppenfunktionalisierung über einen geeigneten Initiator hat den Vorteil, dass die funktionelle Gruppe zu 100% an der Polymerkette vorhanden ist, wohingegen bei einer Terminierungsreaktion nicht von einer 100%igen Funktionalisierung ausgegangen werden kann. Nuyken et al.[93] testeten unterschiedliche Nukleophile auf ihre Terminationseigenschaft und analysierten die Kinetik der Terminierungen über
1H-NMR-Spektroskopie. Dabei wurde gezeigt, dass ein höherer pKS-Wert des Nukleophils eine schnellere Terminierung erreicht.
AUFGABENSTELLUNG UND ZIELSETZUNG
21 Daran schließt sich im Kapitel 5 und 6 die Synthesen von DO3A- bzw. SiFA- modifizierter Polymermizellen und Nanopartikel auf der Basis amphiphilier Poly(2- oxazolin)e an. Diese können eine mögliche Anwendung in der Magnetresonanz- Tomographie (MRT) bzw. Positronen-Emissions-Tomographie (PET) finden.
Im letzten Ergebniskapitel 7 sollte dann untersucht werden, wie sich polymere Nanopartikel über kontrollierte Metall-Ligand Wechselwikungen zu größeren Aggregaten assemblieren lassen.
NANOPARTIKELSYNTHESE MIT HILFE DER MIKROEMULSION
22
4.
Heutzutage werden die meisten industriellen Polymerisationen in dispergierten Systemen durchgeführt. Polymerdispersionen und Polymere, die in Dispersion hergestellt wurden, kommen in verschiedenen Bereichen, wie Kosmetika, Farben, Klebstoffe, Mikroelektronik und der Medizin, zum Einsatz. In der Forschung sind die Miniemulsions- und Mikroemulsionspolymerisation von großer Bedeutung, da über diese Partikel im Bereich von 10 100 nm produziert werden können. Im Kapitel 2.1 wurde auf die beiden Techniken im Zusammenhang mit amphiphilen Polymeren als Makromonomer bzw. Initiatoren eingegangen. Im Rahmen dieser Arbeit wurde die Nanopartikelsynthese mit Hilfe der Mikroemulsion durchgeführt.
Zielsetzung
Ziel dieses Kapitels war die reproduzierbare Darstellung von polymeren Nanopartikeln im Bereich von 20 100 nm mittels der Mikroemulsionspolymerisation und amphiphiler Poly(2-oxazolin)-Makromonomere. Diese Partikel sollten neben biokompatiblen Eigenschaften, auch die Möglichkeit einer nanopartikelanalogen Oberflächenfunktionalisierung besitzen. Daher sollten auch Modifikationen mit bioreleventen Molekülen über Kopplungreaktionen durchgeführt werden, um die mögliche Anwendung dieser Partikel in der Biomedizin zu zeigen.
Es wurde zunächst das bekannte Alkin-basierte System von Engelhardt et. al. auf die Mikroemulsionspolymerisation (Mikroemulison I) übertragen. Weiter wurde die Größe der enstehenden Nanopartikel in Abhängigkeit der Zusammensetzung des polymeren Amphiphils, Sonifkations- und Polymerisationszeit und der eingesetzten Monomer- sowie Polymerkonzentration untersucht. Im Weiteren wurde das verwendete Polymersystem weiter entwickelt und so optimiert, dass eine bessere Kompatibilität der funktionellen Gruppen der polymeren Amphiphile und des eingesetzten Monomers (1,6-Hexandioldimethacrylat) gewährleistet wurde. Dies sollte durch den Austausch der Alkin-Funktionalität durch Acrylatgruppen erreicht werden (Mikroemulsion II). Zudem sollten drei unterschiedlich große Nanopartikel mit Hilfe des zweiten Mikroemulsionsansatzes und Amin-endfunktionalisierten Polymeren dargestellt werden und anschließend auf der Oberfläche mit bioaktiven Molekülen modifiziert werden.
NANOPARTIKELSYNTHESE MIT HILFE DER MIKROEMULSION
23
4.1 Ergebnisse und Diskussion Mikroemulsion I
4.1.1 Synthese von Alkin-funktionalisierten amphiphilen Poly(2- oxazolin)en
In diesem Abschnitt wurden zunächst zwei amphiphile, Alkin-basierte Poly(2- oxazolin)e mit unterschiedlicher Zusammensetzung synthetisiert und charakterisiert.
Diese wurden im Anschluss zur Nanopartikelsynthese in der Mikroemulsionspolymerisation als amphiphile Makromonomere eingesetzt. Hierbei wurde der Einfluss der Polymer- und Monomerkonzentration, der Reaktionszeit und der Homogenierungsdauer auf die Partikelgröße untersucht.
Monomersynthesen und Charakterisierung
Um die nötigen Polymere über die kationische Polymerisation aufzubauen, mussten zunächst die Monomere 2-Heptyl-2-oxazolin (HOx) und 2-Pent-4-inyl-2-oxazolin (PenOx) dargestellt werden. In Schema 6 sind die beiden Strukturen der gewünschten Monomere aufgezeigt.
Schema 6: Darstellung der hydrophoben Monomere a) 2-Heptyl-2-oxazolin (HOx) und b) 2-Pent-4-inyl- 2-oxazolin (PenOx).
Das hydrophobe Monomer 2 (HOx) wurde über eine einstufige Reaktion ausgehend von n-Heptylcyanid mit Ethanolamin nach Witte und Seelinger[97] mit einer Ausbeute von 44 % und das PenOx über drei Stufen nach Luxenhofer[98] mit einer Gesamtausbeute von 85 % dargestellt. Für Letzteres wurde ausgegangen von
NANOPARTIKELSYNTHESE MIT HILFE DER MIKROEMULSION
24
5-Hexinsäure (3), welche mittels Thionylchlorid chloriert wurde. Das entstehende Säurechlorid 4 reagierte anschließend mit 2-Chloroethylamin Hydrochlorid zum entsprechenden Amid 5. Im letzten Schritt wurde unter basischen Bedingungen der Ring zum 2-Oxazolin 6 geschlossen. Um sicherzustellen, dass die dargestellten Monomere wasserfrei vorliegen, wurden beide Monomere über CaH2 getrocknet und über Molsieb gelagert. Das für den hydrophilen Teil des Polymers benötigten 2-Methyl- 2-oxazolin (MOx) ist kommerziell erhältlich. Eine Aufreinigung fand ebenfalls über die Destillation über CaH2 und anschließende Lagerung über Molsieb statt.
Polymersynthesen und Charakterisierung
Ausgehend von den erhaltenen Monomeren wurden die beiden Polymere P1 und P2 durch die in Schema 7 dargestellte Polymerisation synthetisiert. Über den Einsatz von doppelt so vielen Äquivalenten an den hydrophoben Monomeren HOx und PenOx und 1,5 Mal mehr MOx bei der Polymerisation von P2 zu P1 sollten zwei Polymere mit unterschiedlicher Zusammensetzung hergestellt werden. Zuerst wurde der hydrophobe Block aufgebaut. Dabei wurden HOx und PenOx zusammen in Acetonitril mit trockenem Methyltriflat initiiert. Nach Verbrauch der hydrophoben Monomere wurde der hydrophile Block durch Zugabe von MOx synthetisiert. Die Polymere wurden dann mit einer basischen Methanollösung terminiert. Hierdurch wurde eine Hydroxyl- Endgruppe am hydrophilen Teil des Polymers erhalten.
Schema 7: Allgemeine Polymerisation der amphiphilen Blockcopolymere P1 und P2.
Die Polymere wurden mittels Dialyse gegen Wasser aufgereinigt (MWCO=1000).
Dabei wurden die Polymere für mindestens 24 h in Wasser gerührt. Nach Gefriertrocknung folgten eine Fällung der Polymere in kaltem Diethylether und eine Trocknung am Hochvakuum. Somit konnte P1 in einer Ausbeute von 63 % und P2 in einer Ausbeute von 33 % erhalten werden. Anschließend wurden beide Polymere 1H- NMR-spektroskopisch auf ihre Zusammensetzung untersucht. In Abbildung 10(A) ist beispielhaft das 1H-NMR-Spektrum von P1 mit Zuordnung der Signale dargestellt.
Über das Referenzsignal des Initiators bei einer chemischen Verschiebung von
NANOPARTIKELSYNTHESE MIT HILFE DER MIKROEMULSION
26
Tabelle 2: Analytische Daten der Polymere P1 und P2.
# HOxa PenOxa MOxa Mna
/ g· mol-1
Mnb
/
g· mol-1
b dhc /nm
P1 2(4) 3(4) 20(20) 2530 3300 1.19 8.8 ± 0.4
P2 7(8) 8(8) 29(30) 4780 6930 1.09 22.4 ± 3.7
a) Blockcopolymerzusammensetzung und Molmasse wurde über 1H-NMR-Spektroskopie bestimmt, die theoretisch eingesetzten Äquivalente sind in Klammern angegeben; b) Molmasse Mn und Dispersitätsindex wurden über GPC-Analyse bestimmt (PMMA Standards, DMF+5 g/LLiBr; c) Der hydrodynamische Durchmesser (dh) wurde über DLS Messungen einer 1 mM Polymerlösung in Wasser bestimmt.
4.1.2 Nanopartikelsynthese mittels der Mikroemulsionspolymerisation und amphiphiler, Alkin-funktionalisierten Poly(2-oxazolin)-Makro- monomere
In der hier beschriebenen Mikroemulsionspolymerisation zur Darstellung der Nanopartikel werden die hergestellten amphiphilen Blockcopolymere P1 und P2 sowohl als Amphiphile als auch als Makromonomere eingesetzt. Durch Einlagerung eines hydrophoben Monomers (1,6-Hexandioldiacrylat, HDDA) in den Kern, soll neben der Quellung der Mizelle, ein vernetzter Kern entstehen. Hierbei fungiert die Alkingruppe des hydrophoben Blocks ebenfalls als funktionelle Gruppe für die Kernvernetzung und ermöglicht so eine kovalente Bindung des Polyacrylats im Kern zum umhüllenden Polymer.
Im Vordergrund stand eine reproduzierbare Methode zu entwickeln, die eine einfache Variation der Partikelgröße ermöglicht. Hierfür wurden zunächst zwei verschiedene Initiatorsysteme betrachtet. Anlehnend an Engelhardt et al.[38] sollte die Mikroemulsionspolymerisation über den Photoinitiator 2-Propanthiol gestartet werden, analog wurde dieselbe Partikelsynthese mit dem Azoinitiator Azobis(isobutylnitril) (AIBN) durchgeführt. Beide Systeme sollten im Anschluss auf ihre Durchführbarkeit und Reproduzierbarkeit verglichen werden.
In Schema 8 ist der Mikroemulsionsprozess schematisch dargestellt. Zuerst wird das amphiphile Poly(2-oxazolin) mit einer Konzentration von c = 1 mmol/L in Wasser vorgelöst. Damit wurde eine Konzentration oberhalb der CMC gewählt (A). Zu dieser Mizell-Lösung wurden unterschiedliche Mengen an HDDA, 5 Gew.-% Heptadekan und 0,06 eq. 2-Propanthiol bzw. 0,1 Gew.-% AIBN gegeben. Anschließend wurde die Mikroemulsion für 30 Minuten mit Argon entgast und eine Minute mittels Ultraschall behandelt (B). Somit wird eine homogene Einlagerung des hydrophoben Vernetzermonomers HDDA im Mizellkern gewährt (C).[47,101]
NANOPARTIKELSYNTHESE MIT HILFE DER MIKROEMULSION
29 Methanol. Die möglichen Reaktionen sind in Schema 9 dargestellt. Unter (1) ist der Mechanismus ausgehend über die UV-Initiierung gezeigt. Ein Thiolderivat kann unter UV-Bestrahlung aktiviert werden und sowohl die Polymerisation von Acrylaten[104] (a) und Alkinen[105] (b) starten. Die dann entstehenden Radikale sind in der Lage sowohl Homopolymere aufzubauen als auch Copolymerisationen mit der jeweils anderen Funktionalität einzugehen. Am Ende wird eine kovalente Verbindung zwischen der Alkingruppe des eingesetzten Polymers und der Acrylatfunktion des eingelagerten Vernetzers HDDA herbeigeführt.
Im Fall des Azoinitiators AIBN erfolgt die Aktivierung Dabei zerfällt das Molekül in zwei Isobutyronitril-Radikale und molekularer Stickstoff entweicht. Unter dem Punkt (2a) ist die Initiierung des Acrylats gezeigt, was wiederum eine Alkinfunktion einpolymerisieren kann.[106] Anders der Fall (2b), hier gibt es keine bekannte Literatur zu. Dennoch zeigt der Blick in die Literatur, dass bei (kontrolliert) radikalischen Polymerisationen Alkinfunktionalitäten geschützt vorliegen und erst anschließend polymeranalog entschützt werden.[107] Dies bedeutet, dass die Kernvernetzung schnell über die Polymerisation des Acrylats gestartet wird und die Einpolymerisation der Alkingruppen des Polymers langsam erfolgt. Dadurch entsteht eine kovalente Bindung zwischen dem eingelagerten Diacrylats und dem Polymer und somit kann ein stabiler wasserlöslicher Nanopartikel aufgebaut werden.
Aufgrund der einfachen Handhabbarkeit des AIBN-Systems und der hohen Sauerstoffempfindlichkeit des Thiol-Systems wurden im Folgenden weiterführende Parameterstudien der Mikroemulsionspolymerisation nur auf Basis der AIBN- Initiierung untersucht.
Neben der Variation der HDDA-Menge wurde nun auch der Polymerisationsgrad betrachtet. Zudem zeigte die Literatur, dass auch die Sonifizierungsdauer einen großen Einfluss nicht nur auf die Homogenität, sondern auch auf die Partikelgröße hat.[101]
Zudem sollte die Polymerisationsdauer betrachtet werden.
Einfluss der Vernetzerkonzentration HDDA auf die Partikelgröße
Wie schon im ersten Experiment (Tabelle 3) gezeigt worden ist, hat die Menge des eingesetzten Diacrylats HDDA einen wesentlichen Einfluss auf die resultierende Partikelgröße. Basierend auf dem AIBN-Initiationssystems wurde ein breiteres Spektrum von Polymer P1 zu Vernetzer HDDA untersucht (0-1600 Gew.-%). Die Polymerkonzentration wurde bei c = 1 mmol/L, die Sonifizierungsdauer bei t = 1 min und die Polymerisationsdauer bei 65 °C über Nacht konstant gehalten.
NANOPARTIKELSYNTHESE MIT HILFE DER MIKROEMULSION
30
0 200 400 600 800 1500 1600
0 10 20 30 40 50
dh/nm
Gew.-% (HDDA)
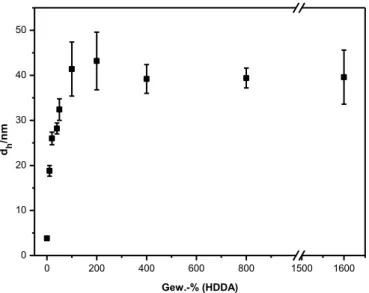
Abbildung 11: Einfluss der Vernetzerkonzentration HDDA auf die Partikelgröße ausgehend von P1 (c= 1 mmol/L, Ultraschallbehandlung: t = 1 min, Reaktionsbedingungen: T = 65 °C, über Nacht, hydrodynamische Durchmesser dh wurde über DLS-Messungen einer methanolischen Nanopartikel- Lösung (c = 1 mg/mL) bestimmt).
In Abbildung 11 sind die über die DLS gemessenen hydrodynamischen Durchmesser gegen die Gewichtsanteile an HDDA aufgetragen. Von 0 bis 200 Gew.-% steigt der Durchmesser der Partikel stetig an bis diese eine Größe von ungefähr 40 nm erreichen.
Mit der Stagnation der Partikelgröße ab 200 Gew.-% wird die limitierte Aufnahmekapazität der Mizellen bestätigt. Weiter kann beobachtet werden, dass sich bei der Aufreinigung der Partikel über die Zentrifugation mehr unlöslicher Feststoff absetzt je mehr HDDA eingesetzt wird. Es scheint, dass es sich hierbei um durchpolymerisiertes Diacrylat handelt, dass wasserunlöslich ist und deshalb ausfällt.
Antonietti-Wu -Auftragung überein. Diese geht detailliert auf die Korrelation zwischen der Partikelgröße und der eingesetzten Monomer-Menge ein.[52,108]
Einfluss der Polymerkonzentration auf die Partikelgröße
Im Folgenden sollte nun der Einfluss der Polymerkonzentration auf die Partikelgröße untersucht werden. Ein großer Vorteil von höher einsetzbaren Konzentrationen ist die größere Menge an Partikeln, die produziert werden kann. Hierfür wurden Polymerlösungen ausgehend von P1 mit c = 1, 5, 10 mmol/L hergestellt und einem Vernetzeranteil von 200 Gew.-% gewählt. Dieser, sowie die Sonifizierungsdauer von t = 1 min und die Polymerisationsdauer von 65 °C über Nacht wurden konstant gehalten.
NANOPARTIKELSYNTHESE MIT HILFE DER MIKROEMULSION
31
1mM 5mM 10mM
0 10 20 30 40 50
dh/nm
Konzentration (P1) Methanol
Wasser
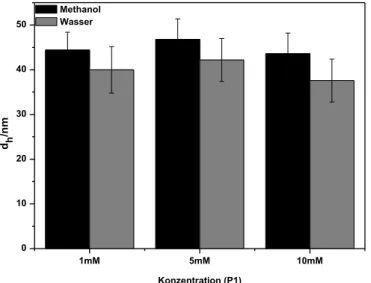
Abbildung 12: Einfluss der Polymerkonzentration P1 auf die Partikelgröße (HDDA-Menge: 200 Gew.-%, Ultraschallbehandlung: t = 1 min, Reaktionsbedingungen: T = 65 °C, über Nacht, hydrodynamische Durchmesser dh wurde über DLS-Messungen einer methanolischen Nanopartikel-Lösung (c = 1 mg/mL) bestimmt).
In Abbildung 12 sind die Partikelgrößen in Wasser und Methanol in Abhängigkeit der Polymerkonzentration dargestellt. Es zeigte sich, dass der Partikeldurchmesser unabhängig von der eingesetzten Menge an Polymer ist und jeweils bei 1, 5 und 10 mmol/L eine Partikelgröße von um die 40 nm erreicht werden konnte.
Einfluss der Ultraschalldauer auf die Partikelgröße
Weiter wurde der Einfluss der Sonifizierungsdauer auf die Partikelgröße untersucht. In diesem Versuchsansatz wurden jeweils Polymerlösungen von P1 und P2 mit c = 1 mmol/L und einem Vernetzeranteil von 100 Gew.-% verwendet. Die Reaktionsbedingungen blieben mit 65 °C über Nacht auch konstant. Die Sonifizierungsdauer variierte von 10 s bis hin zu 30 min. In Abbildung 13 ist die Sonifizierungszeit gegen die Partikelgröße in Methanol aufgetragen. Auch hier ist eine anfängliche Vergrößerung des Durchmessers mit längerer Ultraschallbehandlung zu beobachten, bis die Größe der Partikel ab einem bestimmten Zeitpunkt nicht weiter ansteigt. Bei P1 wird das Größenmaximum bei 3 min mit rund 35 nm und bei P2 bei 5 min mit ca. 70 nm erreicht. Die Mizellgröße von P1 liegt bei nur 8,8 ± 0,4 nm und ist damit dreimal so klein wie von P2 mit 22,4 ± 3,7 nm. Aus diesem Grund ist die maximale Einlagerung von HDDA in P1 eher erreicht. Weiter wurden auch nur halb so große Nanopartikel erhalten.[109]
NANOPARTIKELSYNTHESE MIT HILFE DER MIKROEMULSION
35
4.2 Ergebnisse und Diskussion Mikroemulsion II
4.2.1 Synthese von Acrylat-funktionalisierten amphiphilen Poly(2- oxazolin)en
In diesem Abschnitt soll die eingangs betrachtete Mikroemulsion I basierend auf Alkin- funktionalisierten, amphiphilen Poly(2-oxazolin)en für das AIBN-Initiationssystem angepasst und optimiert werden. Dabei sollte zunächst eine Acrylatfunktionalität an die Stelle des Alkins in die polymeren Amphiphile eingebaut werden. Dies sollte über die Synthese eines neuen Acrylat-funktionalisierten 2-Oxazolinmonomers erfolgen.
Anschließend sollte das Monomer in der Polymerisation von 2-Oxazolinen evaluiert werden, bevor die Polymere in der Mikroemulsion eingesetzt werden. Hierbei wurden die gewonnenen Erfahrungen über den Einfluss der Vernetzerkonzentration gezielt genutzt, um drei unterschiedlich große Nanopartikel zu erhalten.
Monomersynthese und Charakterisierung
Die im hydrophoben Block einpolymerisierte Alkinfunktion sollte durch ein Acrylat- modifiziertes Monomer ersetzt werden, um eine bessere Kompatibilität und Umsetzung des polymeren Amphiphils mit dem eingesetzten Diacrylats (HDDMA) in der Mikroemulsionspolymerisation zu gewährleisten.
In Schema 11 ist das Reaktionsschema zur Darstellung des neuen Monomers 2-(5-Pentyl-[(1,2,3-triazol)-4-yl-methacrylat)]-oxazolin (12) dargestellt.
Schema 11: Reaktionsschema zur Darstellung von 2-(5-Pentyl-[(1,2,3-triazol)-4-yl-methacrylat)]- oxazolin (12).
NANOPARTIKELSYNTHESE MIT HILFE DER MIKROEMULSION
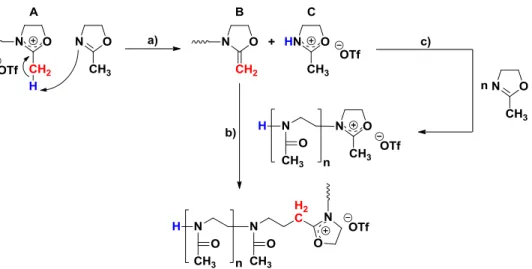
37 Spektrum zusätzlich eine Esterschwingungsbande bei 1715 cm-1, welche der Acrylatgruppe zugeordnet werden kann.
Abbildung 18: FT-IR-Spektren vom 2-(5-Azidopentyl)-2-oxazolin (11, oben) und von 2-(5-Pentyl- [(1,2,3-triazol)-4-yl-methacrylat)]-oxazolin (12, unten).
Im nächsten Abschnitt soll nun das neue Monomer 2-(5-Pentyl-[(1,2,3-triazol)-4-yl- methacrylat)]-oxazolin (12) in der Herstellung von amphiphilen Poly(2-oxazolin)en eingesetzt werden.
Polymersynthesen und Charakterisierung
Das neue Acrylat-funktionalisierte Monomer 12 wurde anschließend in der Synthese von drei Polymeren mit unterschiedlicher Zusammensetzung des hydrophoben Teils eingesetzt. In Schema 12 ist das Reaktionsschema zu den Polymersynthesen der Polymere P3-P5 dargestellt. Bei P3 handelt es sich um ein Triblockcopolymer. Hier sollte das Monomer 12 zunächst den ersten Block bilden, als zweite hydrophobe Komponente wurde wieder HOx eingesetzt. Zum Aufbau der Amphiphilie wurde sequentiell MOx einpolymerisiert. Das Polymer P4 unterscheidet sich lediglich in der Reihenfolge der Monomere im hydrophoben Teil, hier wurde erst HOx, gefolgt von Monomer 12 und anschließend MOx polymerisiert. Bei P5 handelt es sich um ein Blockcopolymer, dessen hydrophober Block sich statistisch aus HOx und Monomer 5 zusammensetzt. Initiiert wurden die Polymere P3-P5 mit Methyltriflat und die Termination fand bei den Polymeren P3 und P4 über Luftsauerstoff statt. Eine Terminierung über eine basische Methanollösung bei 60 °C über Nacht war nicht möglich, da unter diesen Bedingungen die Acrylatgruppe abgespalten wurde.[112]

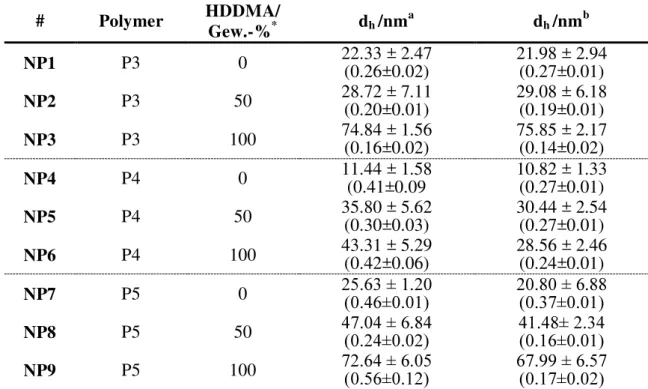
![Tabelle 1: Beispiele von kommerziellen Anwendungen von Nanopartikeln. [16]](https://thumb-eu.123doks.com/thumbv2/1library_info/3629247.1502177/21.892.147.789.690.934/tabelle-beispiele-von-kommerziellen-anwendungen-von-nanopartikeln.webp)
![Abbildung 18: FT-IR-Spektren vom 2-(5-Azidopentyl)-2-oxazolin (11, oben) und von 2-(5-Pentyl- 2-(5-Pentyl-[(1,2,3-triazol)-4-yl-methacrylat)]-oxazolin (12, unten)](https://thumb-eu.123doks.com/thumbv2/1library_info/3629247.1502177/55.892.200.734.179.530/abbildung-spektren-azidopentyl-oxazolin-pentyl-pentyl-methacrylat-oxazolin.webp)