Research Collection
Doctoral Thesis
Novel Approaches for Direct Electron Transfer of Redox
Enzymes and their Implementation in Biosensors and Biofuel Cells
Author(s):
Trifonov, Alexander Publication Date:
2020-03
Permanent Link:
https://doi.org/10.3929/ethz-b-000405205
Rights / License:
In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use.
ETH Library
DISS. ETH NO. 26597
NOVEL APPROACHES FOR DIRECT ELECTRON TRANSFER OF REDOX ENZYMES AND THEIR IMPLEMENTATION IN
BIOSENSORS AND BIOFUEL CELLS
A thesis submitted to attain the degree of DOCTOR OF SCIENCES of ETH ZURICH
(Dr. Sc. ETH Zurich)
presented by
ALEXANDER ADAM TRIFONOV
M.Sc. in Chemistry, The Hebrew University of Jerusalem
born on 04.04.1985 citizen of Israel
accepted on the recommendation of Prof. Dr. Andreas Stemmer, examiner Prof. Dr. Jeroen Anton van Bokhoven, co-examiner
Dr. Ran Tel-Vered, co-examiner
2020
Abstract
i
Enzyme-based biosensors are mainly used as diagnostic tools for the measurement of different substances such as sugars, alcohols, and human body metabolites. Specific electrode modifications allow the fabrication of biofuel cells that deliver on-demand electrical power from biomass for different applications, including self-powered biosensing. Despite their promising merits, enzyme-based bioelectronic products are still scarce on the market due to several reasons. These include a cost-inefficient production, poor stability of the biocatalysts operating outside their natural environments, and the frequent use of poisonous and/or leaking electron mediator molecules.
This thesis addresses two main challenges that bioelectronics face nowadays: the effective and sustainable electric wiring of enzymes to the electrode, and the lifetime extension of enzyme-based biofuel cells and biosensors.
Mediated electron transfer, the prevalent strategy to electrically link enzymes with electrodes, suffers from inefficiency, and naturally limits the biocompatibility as well as the theoretical power output of fuel cells.
Hence, alternatives are needed.
It has been shown that direct (mediatorless) electron transfer might provide the most efficient and sustainable enzyme-electrode communication pathway, allowing the design of biosensors of higher sensitivity, and biofuel cells with higher current densities and potential biases. Yet, the mediatorless wiring of enzymes is not an easy task and requires significant distance reduction between the enzymatic catalytic site and electrode on the nanometric level.
Abstract
ii
This work presents two novel techniques for enzymatic direct electrical wiring: an electrochemical and an enzymatic approach. Both methods rely on the pore-confined synthesis of metal nanoparticles in the vicinity of enzymatic catalytic sites as a strategy for shortening the donor- acceptor distance, facilitating heterogeneous electron transfer between enzyme and electrode. To this end, metal ions were loaded inside the pores of a mesoporous carbon nanoparticle matrix and capped with enzymatic units. While the electrochemical wiring requires application of electrical potential for nanoparticle formation, the enzymatic approach employs catalytic activity of the enzyme for metal nanoparticle creation, resulting in so-called self-wiring.
The two wiring methods presented, electrochemical and enzymatic, result in different wiring characteristics. Consequently, different catalytic efficiencies are obtained. The catalytic properties of the electrodes are thoroughly studied and compared. Also, a theoretical explanation of the observed characteristics is proposed and validated.
An additional problem, the solution of which could promote the broader introduction of bioelectronics in our daily life, is the short lifetime of enzyme-based bioelectronic devices. The lifetime of biocatalysts is dictated by relatively fast natural degradation that stems from a variety of interactions of amino acids with different compounds, the external environment, and the functional activity of the enzymes. These factors limit the usage of biosensors and biofuel cells to several tens of hours of operation or a few weeks of storage. Therefore, the development of strategies to significantly increase the operational duration of bioelectronic devices is one of the goals of this work.
We present a self-assembled fructose/oxygen all-direct electron transfer biofuel cell, which allows replacement of the exhausted biocatalysts by a
Abstract
iii
magnetically induced collection of enzyme-immobilized nanoparticles, and the introduction of a fresh batch of those. This reusable platform provides a practical engineering solution that ensures fast and straightforward cell reenergization, and allows continuous operation and power supply.
The architecture of the biofuel cell is based on differently modified glassy carbon electrodes, which interact exclusively with anodic or cathodic populations of enzyme-modified carbon-coated magnetic nanoparticles. The resulting biofuel cell demonstrates power outputs of ca. ~ 25µW·cm-2 and reproducible cycles of biocatalysts exchange.
Furthermore, this biofuel cell is shown to self-assemble and operate in grape juice as an example of a complex environment.
In addition, we demonstrate a method to prolong the catalytic activity of enzyme-based electrodes by coating them with carbon-coated magnetic nanoparticles as a protection layer. This approach proves to be useful for extending the biocatalysts’ lifetime and allows storage of the electrodes at room temperature for weeks, with only minor losses of catalytic activity after magnetically-stimulated removal of the protection layer.
Furthermore, this method allows us to use the electrodes with the protective coating on, granting additional stability to the enzymes while operating under proteolytic conditions.
We believe that the concepts introduced will promote a fundamental understanding of electrical wiring between enzymes and modified surfaces, and we hope that it will motivate a scientific exploration of novel approaches in order to find better solutions to daily sensing and energy concerns.
Zusammenfassung
iv
Enzymbasierte Biosensoren werden hauptsächlich als Diagnosewerkzeuge für die Messung verschiedener Substanzen verwendet, z.B. von Zuckern, Alkoholen und Metaboliten des menschlichen Körpers. Spezifische Elektrodenmodifikationen ermöglichen die Herstellung von Biobrennstoffzellen, die bei Bedarf Strom aus Biomasse für verschiedene Anwendungen liefern, einschliesslich der autarken Biosensorik. Trotz ihrer vielversprechenden Vorteile haben enzymbasierte bioelektronische Produkte aus mehreren Gründen den Marktdurchbruch noch nicht geschafft. Zu diesen Gründen gehören eine teure Herstellung, mangelnde Stabilität der Biokatalysatoren, welche ausserhalb ihrer natürlichen Umgebung arbeiten, und die häufige Verwendung von giftigen und/oder auslaufenden Mediatormolekülen.
Diese Dissertation befasst sich mit zwei Hauptherausforderungen, mit denen die Bioelektronik aktuell konfrontiert ist: der effektiven und nachhaltigen elektrischen Verkabelung von Enzymen mit der Elektrode, und der Verlängerung der Lebensdauer von Biobrennstoffzellen beziehungsweise Biosensoren auf Enzymbasis.
Der mediierte Elektronentransfer, die vorherrschende Strategie zur elektrischen Vernetzung von Enzymen mit Elektroden, leidet unter Ineffizienz und schränkt die Biokompatibilität sowie die theoretische Leistungsfähigkeit von Brennstoffzellen ein. Daher sind Alternativen erforderlich.
Es wurde gezeigt, dass der direkte (mediatorlose) Elektronentransfer den effizientesten und nachhaltigsten Kommunikationsweg zwischen Enzym
Zusammenfassung
v
und Elektrode darstellt. Dieser ermöglicht den Entwurf von Biosensoren mit höherer Empfindlichkeit und Biobrennstoffzellen mit höheren Stromdichten und vorteilhaften Überspannungen. Die mediatorlose Vernetzung von Enzymen ist jedoch keine leichte Aufgabe und erfordert eine signifikante Abstandsreduzierung zwischen dem katalytischen Zentrum und der Elektrode im Nanometerbereich.
In dieser Arbeit werden zwei neuartige Techniken für die enzymatische, direkte elektrische Verdrahtung vorgestellt: ein elektrochemischer und ein enzymatischer Ansatz. Beide Methoden beruhen auf der Synthese von Metallnanopartikeln, eingeschlossen in Poren, in der Nähe enzymatischer katalytischer Zentren, als Strategie zur Verkürzung des Elektronentransfers zwischen den beiden Reaktionspartnern über den Donor-Akzeptor-Abstand. Zu diesem Zweck wurden Metallionen in die Poren einer mesoporösen Kohlenstoffnanopartikelmatrix eingelagert und mit enzymatischen Einheiten gedeckelt. Während die elektrochemische Verdrahtung die Anlegung eines elektrischen Potentials zur Bildung von Nanopartikeln erfordert, nutzt der enzymatische Ansatz die katalytische Aktivität des Enzyms zur Erzeugung von Metallnanopartikeln, was zu einer sogenannten Selbstvernetzung führt.
Die beiden vorgestellten Vernetzungsmethoden, elektrochemisch und enzymatisch, führen zu unterschiedlichen Vernetzungseigenschaften.
Folglich werden unterschiedliche katalytische Wirkungsgrade erzielt.
Die katalytischen Eigenschaften der Elektroden werden sorgfältig untersucht und verglichen. Ausserdem wird eine theoretische Erklärung der beobachteten Merkmale vorgeschlagen und validiert.
Ein weiteres Problem, dessen Lösung einen breiteren Einzug der Bioelektronik in unseren Alltag fördern könnte, ist die kurze Lebensdauer enzymbasierter bioelektronischer Geräte. Die Lebensdauer
Zusammenfassung
vi
von Biokatalysatoren wird durch einen relativ schnellen natürlichen Abbau bestimmt. Dies ergibt sich aus einer Vielzahl von Wechselwirkungen von Aminosäuren mit anderen Verbindungen, der äusseren Umgebung und der funktionellen Aktivität der Enzyme. Diese Faktoren begrenzen die Verwendung von Biosensoren und Biobrennstoffzellen auf wenige Dutzend Betriebsstunden oder einige Wochen Lagerung. Daher ist die Entwicklung von Strategien zur signifikanten Verlängerung der Betriebsdauer von bioelektronischen Geräten eines der Ziele dieser Arbeit.
Wir präsentieren eine selbst oragnisierte, komplett mediatorlose Fructose/Sauerstoff-Biobrennstoffzelle. Diese Konstruktion ermöglicht es, die verbrauchten Biokatalysatoren mittels magnetischer Stimulation aufzusammeln und die enzymimmobilisierten Nanopartikel durch eine neue Charge zu ersetzen. Diese wiederverwendbare Plattform bietet eine praktische technische Lösung, die eine schnelle und unkomplizierte regenerierte Energieversorgung der Zellen sowie einen kontinuierlichen Betrieb und eine dauerhafte Stromversorgung gewährleistet.
Die Architektur der Biobrennstoffzelle basiert auf unterschiedlich modifizierten Glaskohlenstoffelektroden, die ausschliesslich mit anodischen oder kathodischen Populationen enzymmodifizierter, kohlenstoffbeschichteter, magnetischer Nanopartikel interagieren. Die resultierende Biobrennstoffzelle weist eine Leistung von ca. ~ 25µW · cm-2 und reproduzierbare Zyklen des Biokatalysatoren austauschs auf. Darüber hinaus wird gezeigt, dass sich diese Biobrennstoffzelle in Traubensaft, als Beispiel für eine komplexe Umgebung, selber organisiert und funktioniert.
Ausserdem zeigen wir eine Methode zur Verlängerung der katalytischen Aktivität enzymbasierter Elektroden, indem diese mit kohlenstoffbeschichteten, magnetischen Nanopartikeln als Schutzschicht
Zusammenfassung
vii
bedeckt werden. Dieser Ansatz erweist sich als nützlich, um die Lebensdauer der Biokatalysatoren zu verlängern und die Elektroden wochenlang bei Raumtemperatur zu lagern, wobei die katalytische Aktivität nach magnetisch stimulierter Entfernung der Schutzschicht nur geringfügig nachlässt. Darüber hinaus können wir mit dieser Methode die Elektroden mit der Schutzbeschichtung verwenden, um den Enzymen zusätzliche Stabilität zu verleihen, während sie unter proteolytischen Bedingungen arbeiten.
Wir glauben, dass die präsentierten Konzepte ein grundlegendes Verständnis der elektrischen Vernetzung zwischen Enzymen und modifizierten Oberflächen fördern werden, und wir hoffen, dass sie eine wissenschaftliche Erforschung neuartiger Ansätze anregen werden, um bessere Lösungen für alltägliche Sensor- und Energieprobleme zu finden.
Acknowledgments
viii
I would like to highly acknowledge my parents Irina and Victor Trifonov, my brother Yevgeny, my grandmother Valentina Berman, and the rest of my relatives for their love and support throughout my whole life.
In addition, I would like to thank:
• Prof. Dr. Andreas Stemmer for the opportunity to work under his supervision, for the guidance, support, and highly fruitful collaboration during my doctoral studies.
• Dr. Ran Tel-Vered, first of all, for his friendship, which I appreciate and cherish. For all the professional skills and critical thinking that he taught me throughout my academic and personal life.
• Dr. Katharina Herkendell for being a great friend and excellent colleague with whom I could discuss the practical problems during the laboratory work.
• Blerim Veselaj for friendship and technical support.
• The group members for being great colleagues and friends.
Table of contents
ix
Abstract ... i
Zusammenfassung ... iv
Acknowledgments ... xiii
Table of contents ... ix
Abbreviations ... xii
1. Introduction ... 1
1.1 Enzymes as effective catalysts ... 2
1.2 Principles of bioelectrocatalysis and heterogeneous electron transfer ... 5
1.3 Enzymatic electrochemical biosensors and biofuel cells ... 9
1.3.1 Mediated electron transfer ... 12
1.3.2 Direct electron transfer ... 14
1.3.3 Carbon-based conductive materials as hosting matrices for enzyme immobilization ... 17
1.3.4 Operational lifetime and shelf-time of enzyme-based bioelectronic compartments ... 19
1.4 Objectives of the thesis ... 20
2. Electrochemical analysis ... 23
2.1 Cyclic voltammetry ... 25
2.1.1 Homogeneous electron transfer processes and cyclic voltammetry in enzymatic catalytic reactions ... 31
2.2 Electrochemical impedance spectroscopy ... 35
Table of contents
x
3. Electrical wiring of redox enzymes ... 40
3.1 Induction of direct electron transfer on redox enzymes ... 40
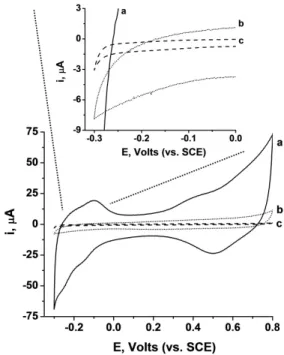
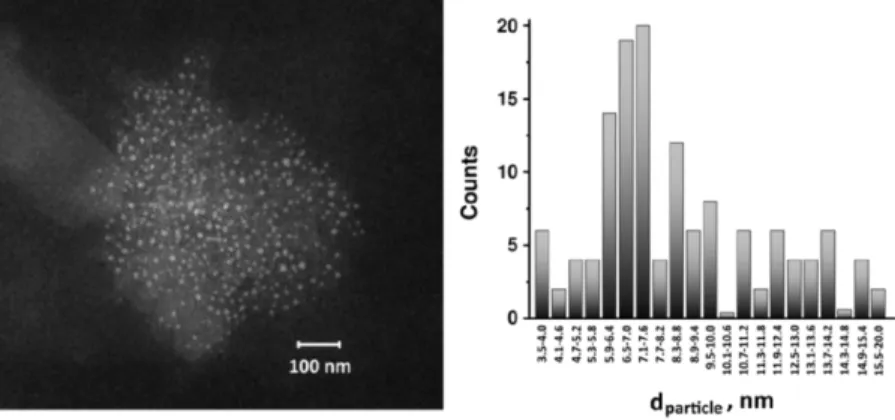
3.2 Induction of DET on HRP and GOx using the “outside-in” synthetic approach ... 44
3.2.1 Bioelectrocatalytic properties of HRP-AuNPs/MCNPs ... 51
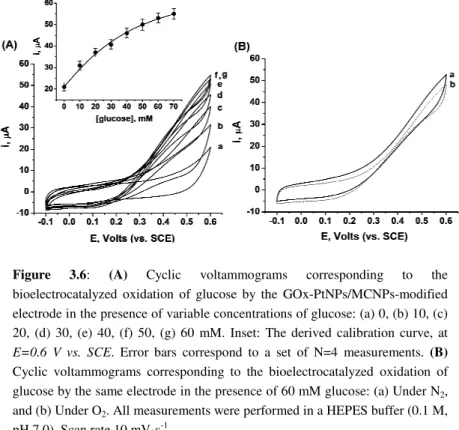
3.2.2 Bioelectrocatalytic properties of GOx-PtNPs/MCNPs ... 52
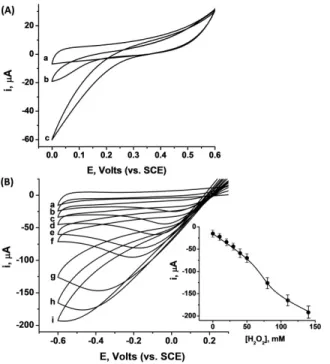
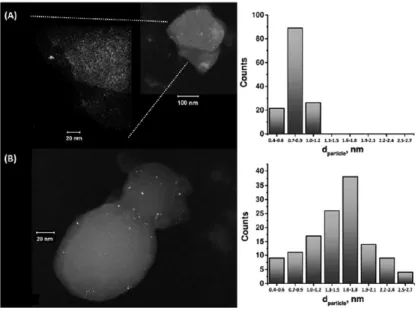
3.3 Induction of DET on GOx using the “inside-out” synthetic approach ………...……….54
3.4 Comparison of the PtNP@GOx/MCNPs, PtNP-GOx/MCNPs and FcMe-mediated assemblies ... 71
4. Strategies for the operation and lifetime extension of bioelectronic devices, using ccMNPs ... 78
4.1 All-DET-operated self-assembled biofuel cell ... 79
4.1.1 The self-assembled anode ... 82
4.1.2The self-assembled cathode ... 90
4.1.3 Self-assembled biofuel cell ... 96
4.2 Extension of the biocatalytic activity of the BOD-based cathode by ccMNPs protection ... 100
5. Summary and future perspectives ... 110
A. Appendix ... 114
A.1 Materials ... 114
A.2 Electrochemical measurements ... 115
A.3 Spectroscopic measurements ... 115
Table of contents
xi
A.4 Electrode modification with PtNP-GOx/MCNPs and AuNPs-
HRP/MCNPs ... 117
A.5 Electrode modification with enzymatically-grown PtNP@GOx/MCNPs ... 118
A.6 Electrodes modifications, ccMNPs modifications, and chemical synthesis for self-assembled BFC ... 119
A.7 Preparation of ccMNPs-protected BOD/SWCNTs cathode ... 122
Bibliography ... 123
Curriculum Vitae ... 132
Abbreviations
xii
ABTS2- - 2,2'-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) AlcDH - alcohol dehydrogenase
AlcOx - alcohol oxidase BFC - biofuel cell BOD - Bilirubin oxidase
BS3 - Bis(sulfosuccinimidyl)suberate Cat - catalase
ccMNPs - carbon-coated magnetic nanoparticles CE - counter electrode
CNTs - carbon nanotubes
CNTFs - carbon nanotube forests CV - Cyclic voltammetry
CytC - cytochrome c
DET - Direct electron transfer
EDC - 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride EIS - electrochemical impedance spectroscopy
FAD - Flavin adenine dinucleotide FcMe - Ferrocene methanol FMN - Flavine mononucleotide GC - glassy carbon
GDH - Glucose dehydrogenase GOx - Glucose oxidase
HEPES - 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid HRP - Horseradish peroxidase
Lac - laccase
MCNPs - Mesoporous carbon nanoparticles MES - 2-(N-morpholino)- ethanesulfonic acid MET - Mediated electron transfer
NAD - Nicotine adenine dinucleotide
Abbreviations
xiii NHS - N-Hydroxysuccinimide NMP - N-methyl pyrrolidone NPs - nanoparticles
OCV - open circuit voltage PEI - polyethyleneimine
PRR - proton reduction reaction PTS - pyrene tetrasulfonate PVDF - polyvinylidene fluoride RE - reference electrode
SCE - saturated calomel electrode SEM - scanning electron microscopy
STEM - scanning transmission electron microscopy
sulfo-EMCS - N-ε-maleimidocaproyl-oxysulfosuccinimide ester WE - working electrode
β-CD - β-cyclodextrin
1
1 Introduction
1
Bioelectronics is a rapidly evolving field that leagues biology and electronics together to facilitate the development of sensors, actuators, and energy conversion devices to meet human needs in a rapidly changing world. As the name hints, bioelectronics uses biological materials like nucleic acids (DNA), enzymes, antigens, or even living cells and the unique properties thereof for sensing applications,1-4 digital logic gates operations,5-7 filters,8,9 communication modules,10,11 nano/micromotors actuation,12-14 and energy harvesting.15-18
Biosensing is one of the hottest topics in bioelectronics due to the growing development of the medical, pharmaceutical, and food industries. For example, using the unique hybridization features of nucleic acids, a variety of portable optical19-21 and electrochemical22,23 biosensors (including self-amplified platforms,24,25 which allow the detection of single molecules) have been developed. These features were used for forensic and medical applications, providing a valid diagnosis of metabolites, 26-28 small molecules,29 genetic disorders,30,31 tissue matching,32,33 as well as detection of pathogens34,35 and heavy metals36,37 in food and water. Moreover, the specificity of enzymatic reactions enabled the development of biosensors for specific sugars,38 alcohols,39 pesticides,40 human body metabolites,41 heavy metals,42 utilizing different sensing platforms with immobilized proteins. So the unique architecture of biological materials, which has evolved over thousands of years, indeed serves humanity nowadays.
Another, but no less exciting direction in bio-inspired technologies is the development of alternative energy sources that ideally will enable people to use multi-component biomass oxidation43,44 for mass energy production. Yet, in light of some limitations that will be discussed later, a realistic goal at the present stage of technology development is energy production on small scales, enough to run low-powered devices45,46 for
Introduction
2
daily needs. In this sense, the conversion of biomass, which exists in abundance in nature, into clean energy by biofuel cells (BFC) has a substantial potential to support the popular trend of sustainability. Additionally, enzyme-based biofuel cells can be used as self-powered biosensors in the detection of a variety of substances, including carbohydrates and metabolites like glucose, fructose, ethanol, acetylcholine, ascorbic acid, or cholesterol.47-50 These cells monitor the levels of the substrates, correlating the power outputs with their concentration in absence of any other external power source. Enzymatic biofuel cells produce enough energy to power low voltage electronics (10-100 µW) and can be worn51 or implanted52 into the human body, enabling continuous monitoring of the metabolite or maintaining other functionality. This technology can ease the life of diabetics, for example, providing information about their glucose levels, avoiding a painful prick-based test.
This monograph discusses the fundamental aspects of both enzyme- based electrochemical biosensors and biofuel cells and provides solutions to two underlying problems in bioelectronics: the electrical communication between the biocatalysts and electrodes, and the lifetime extension of bioelectronic devices.
1.1 Enzymes as effective catalysts.
Enzymes belong to the group of biological macromolecules with dimensions in the range of 6-20 nm in diameter,53 usually proteins that catalyze chemical reactions and are responsible for most of the vital processes that take place in living organisms. Exceptional specificity and very high efficiency make enzymes the ultimate natural catalysts, capable of accelerating reaction rates as much as 1017-fold,54,55 and
Introduction
3
providing conversion rates in the range of 1.104-1.106 substrate molecules·(min·enz.unit)-1. The secret of such a high catalytic efficiency of enzymes originates from the unique three-dimensional structure of the active site. This site consists of a specific set of amino acids and cofactor molecules, whose combination determines the substrate and the catalytic reaction that takes place.
The uniqueness of enzymatic catalytic reactions56-58 is best appreciated when compared to chemical catalysis. For example, acid- or subcritical water-catalyzed hydrolysis59 of any amino acid sequence will cause degradation of every single peptide bond, producing individual amino acids. However, using the enzymatic catalysis of trypsin,60 the peptide bond cleavage will occur only on the carboxylic side of lysine and arginine amino acids, producing shorter sequences with specific ends.
The high specificity towards the substrate or the product makes enzymatic catalysis a potent tool, which can be used for specific chemical syntheses61,62 or very specific modifications.63-66
The Michaelis-Menten kinetic model67 suggests that the enzymatic catalytic reaction occurs in several steps, equation (1.1).
+ ⇌ · ⎯ + (1.1)
First, the substrate [S] binds to the active site of the enzyme [E] with a certain affinity, creating an enzyme-substrate [E.S] reversible complex with the forward rate constant kf, and the reverse rate constant kr. Next, the complex [E.S] undergoes the irreversible catalytic reaction, via intermediate species formation to get the enzyme [E] and the resulting product [P], with the catalytic rate constant kcat. Under equilibrium and steady-state approximation, considering free ligand and catalytic
Introduction
4
reaction irreversibility assumptions, the model is described by equation (1.2), where υ is the reaction rate, [E]0 = [E] + [E·S] , and KM = (kr+kcat)/kf.
= + (1.2)
The constant KM represents the substrate concentration at half of the maximum possible reaction rate and provides an inverse indication for the affinity towards the substrate. The turnover number, kcat describes the maximum number of substrate molecules converted into product per unit time. Another interesting value is kcat/KM which represents the catalytic efficiency of the enzyme. In the case of high catalytic efficiency, catalysis is limited by the diffusion rate of the substrate rather than by kcat, yet such enzymes are rare.68 Independent of constants mentioned above, the enzymatic reaction rate increases by elevation of the substrate concentration till it reaches the saturation due to kinetic (kcat), or diffusion limitations.
It should be noted that the Michaelis-Menten model is not perfect,69 yet describes enzymatic catalytic activity in good approximation. In reality, the second step of the reaction, ruled by kcat, is reversible; however, this fact can be neglected since the driving force of the reaction towards the product is much higher.
The specificity of an enzyme is determined by the conversion rate rather than by the affinity of the substrate toward the catalytic site.70 Often, an enzyme can catalyze chemical reactions of several different substrates.
This happens when their chemical structures are suitable to access and stay at the enzymatic active site. Yet, the turnover rate associated with the primary substrate will significantly exceed the values related to the conversion of the secondary substrates, providing a clear indication of
Introduction
5
the different molecules reacting. For example, the relative activity of glucose oxidase (GOx), known for catalyzing the oxidation of β-D- glucose, toward the catalytic oxidation of α-D-glucose is ca. 0.64%
compared to the primary substrate.
The exceptional catalytic and recognition properties of biological functional macromolecules, like enzymes, motivate scientists from different fields, such as biology, material science, organic chemistry, and interdisciplinary bioelectronics, to use these features for various applications, to expose the inherent potential, hidden in the unique, naturally evolved architecture, to explore new scientific horizons.
1.2 Principles of bioelectrocatalysis and heterogeneous electron transfer.
Like every other group of enzymes, oxidoreductases use cofactors, molecules or ions, which fulfill functions, like assistance in substrate orientation prior to catalytic reaction, catalytic reaction, or proper folding of the three-dimensional enzymatic structure. In general, cofactors can be divided into two groups: inorganic71 and organic.72 The inorganic cofactors are metal ions of different oxidation states, like Cun+, utilized by bilirubin oxidase (BOD) and Laccase (Lac), or metallo-organic complexes, like redox-active heme (a = C49H56O6N4Fe, b = C34H32O4N4Fe, c = C34H36O4N4S2Fe, and o = C49H58O5N4Fe), or Zn and Ni protoporphyrins (C34H32O4N4Zn and C34H32O4N4Ni, respectively), which are employed by horseradish peroxidase (HRP), cytochrome c (Cyt C), or Catalase (Cat).
Organic cofactors are redox-active molecules. Nicotinamide adenine dinucleotide/phosphate (NAD/P), flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD) and different quinones are frequently used
Introduction
6
by oxidative redox enzymes, like glucose oxidase (GOx), glucose dehydrogenase (GDH), fructose dehydrogenase (FDH), and alcohol oxidase (AlcOx). Sometimes, enzymes of the same functionality, but from different organisms can utilize different cofactors, for example, FAD-dependent GOx and FMN-dependent GOx.
To explain the principle of bioelectrocatalysis, let us look at the enzymatic oxidative catalytic reaction. During the catalytic reaction, which always occurs in a liquid phase, the substrate diffuses to the active site of the enzyme and undergoes catalytic oxidation. The cofactor serves as an intermediate acceptor of the electrons acquired from the oxidized substrate; therefore, in order to maintain continuous reaction flow, the cofactor has to be repeatedly regenerated (oxidized to its original oxidation state) by the final electron acceptor, Scheme 1.1.
In nature, oxygen and hydrogen peroxide commonly serve as the final electron acceptors in oxidative enzymatic reactions. The electrons obtained during the enzymatic catalytic reaction are transferred in the same liquid phase, so this type of reaction classifies as homogeneous electron transfer.
Scheme 1.1: Schematic illustration of enzymatic catalytic activity through continuous regeneration of the cofactor by oxygen.
Introduction
7
However, the operation principle of bioelectronic devices relies on heterogeneous electron transfer, because the electrons gained from the substrate oxidation must be transmitted to the electrode, i.e., through the interface of the two, in this case, liquid-solid phases, Scheme 1.2.
Scheme 1.2: Schematic illustration of heterogeneous electron transfer principle in bioelectronic devices.
The electrical current resulting from enzymatic catalytic activity can be directly used for powering electronic devices or further converted into the concentration of the substrate in the sample, using the respective calibration curve.
The latter pathway might be achieved upon application of electrochemical potential on the electrode, suitable for the regeneration (in this example, oxidation) of the enzymatic cofactor, using a potentiostat.
When such a pathway of electron transfer becomes available, electrical communication between the enzyme and the electrode is established.
Introduction
8
However, in reality, the creation of electrical communication between enzymes and electrodes is not obvious and might involve multiple chemical modifications and usage of specific materials. Therefore, the electrical wiring of enzymes to the electrode is one of the primary challenges of bioelectronics, which is spotlighted in this thesis.
An explanation for the lack of enzymatic electrical communication was given by Marcus et al., suggesting that the distance between the electrode and deeply buried catalytic site73 of the enzyme is too large and serves as a barrier for electron tunneling between the two reaction ends.
According to the Marcus theory,74 equation (1.3), the heterogeneous electron transfer kinetics, besides the empirical constant c, depends on the product of two exponential functions, which comprise several thermodynamic parameters, like, the free energy of reaction, !G0, the reorganization energy, λ0, the gas constant, R, and the temperature, T.
Additional important parameters are the tunneling barrier coefficient, β, which depends on the medium of the reaction, r0 the van der Waals radius, and r the donor-acceptor distance, the distance between the enzymatic active site and the electrode. Whereas the increase in the temperature in the presence of enzymatically modified surfaces is limited due to a risk of denaturation, shortening the gap between the electrode and the enzymatic cofactor (r, in our case) can significantly affect the heterogeneous electron transfer kinetics.
" = # · $%&( %')· $%(∆)
'*+'),
-./+' (1.3)
The dependence of the electron transfer rate on the gap between the cofactor and the electrode surface is highly significant. It can be calculated that even a 5 nm gap leads, under common experimental conditions, to a transfer rate equivalent to only 1e- per 100 years, thus
Introduction
9
indicating any possible application of such bioelectronic device as impractical. According to (1.3), the smaller the distance, the higher the electron transfer rate. Consequently, to establish a viable electrical connection between the enzyme and the electrode, the gap between the enzymatic active site and the collector surface must be significantly shortened.75-77
The efficiency of the electrical wiring of enzymes is reflected by heterogeneous ket, the electron transfer rate constant. The better heterogeneous wiring efficiency (and higher transfer rates), the system is expected to be less susceptible to homogeneous reactions involving the natural donors or acceptors of the enzymes in the solution phase. For example, the higher the heterogeneous ket of electrically wired GOx, the less O2-sensitive it is. This, in turn, allows the assembly of more accurate biosensors and membrane-less biofuel cells, where the anode is (almost) insensitive to oxygen, the primary substrate of the cathode.
1.3 Enzymatic electrochemical biosensors and biofuel cells.
Typical enzymatic electrochemical biosensors consist of two components: the electrode, which serves as a support and current collector, and the biorecognition element (an enzyme). The electrical wiring of the biocatalyst to the electrode is essential for the development of modern biosensors. Simple biosensors utilize enzymatic units of a single type,78-80 whereas advanced biosensors may employ various kinds of enzymes to detect multiple substrates.81-84
Over the past few decades, several strategies were developed for the electrical wiring of enzymes to electrodes. They can be divided into two
Introduction
10
sub-groups, based on the electron transfer pathway established between the donor and acceptor sides, namely, mediated electron transfer (MET) and direct electron transfer (DET), which will be further discussed.
Generally speaking, three generations of biosensors were developed.
The first-generation biosensors85 do not require electrical communication between the enzymes and the electrode, and the detection procedure combines two separate processes: the enzymatic catalytic O2-assisted oxidation of the substrate, accompanied by the byproduct formation (e.g., hydrogen peroxide) and its subsequent electrochemical oxidation on the electrode. However, such biosensors suffer from poor repeatability, integrability, and applicability due to restricted oxygen solubility in biological samples, high operation potential, required for effective hydrogen peroxide oxidation, which causes subsequent oxidation of interferences such as ascorbic and uric acids. These factors cannot be improved due to the working principle of such devices. Thus, the first-generation biosensors will be out of the scope of this thesis. Biosensors and biofuel cells of the second and third generations that require electrical wiring and operate via mediated or direct electron transfer, respectively, will be discussed.
Enzymatic biofuel cells86-91 (BFCs) are a specific type of fuel cell in which enzymes replace the noble metals as active catalytic elements, and the related enzymatic substrates serve as fuel and oxidizer. The operation principle is similar to conventional fuel cells, where a catalyst-decorated anode oxidizes the fuel, and the electrons gained during the process are passed in an external circuit towards the cathode, where a complementary reduction reaction takes place, avoiding the electrolyte barrier and producing the electric current. Scheme 1.3
Introduction
11
exemplifies the operation of glucose oxidase/bilirubin oxidase (GOx/BOD)-based enzymatic biofuel cell.
R load
glucose
gluconic acid
O2
H2O
i i = GOx
= BOD
e- e-
Scheme 1.3: Schematic illustration of the operation of the GOx/BOD-based biofuel cell.
The driving force for the generation of the electrical current is the continuous catalytic oxidation and reduction processes occurring on the anode and cathode, respectively.
The power output of BFC may be limited by the cathode or anode, depending on the turnover rate of the employed enzymes and efficiency of their electrical wiring. To maximize the power output of the cell, it should be operated under saturation conditions of both fuel and oxidizer.
Since the electrical current produced by the enzymatic oxidation reaction is proportional to the substrate concentration in the electrolyte, BFCs can also serve as the self-powered biosensors monitoring the fuel concentration, avoiding the usage of potentiostat.
Introduction
12
1.3.1 Mediated electron transfer (MET)
The second generation of biosensors and biofuel cells involves the usage of electron-transfer mediators (or relay units). Mediators are molecules that serve as a bridge between the two separated reaction sites (the enzyme and the current collector), which can be reduced and oxidized, shuttling the electrons from one reaction site to another, providing the desired electrical communication, Scheme 1.4.
Scheme 1.4: Principle of mediated electron transfer.
This electrical wiring strategy is widely implemented in bioelectronics.
However, not every redox-active molecule can mediate the electron transfer between the two reaction centers, and there are two factors that determine whether a particular redox-active unit can be coupled to the specific enzyme as its mediator:
a) The ability to penetrate and reach the enzymatic active site. This requirement is connected to the structure of the molecule and its specific properties like solubility, overall charge, and functional groups.
Introduction
13
b) The redox potential of the mediator has to be higher, in case of an oxidative, and lower in case of a reductive enzyme, compared to the redox potential of the enzymatic active site to facilitate regeneration of the cofactor, Scheme 1.5.
With regard to biofuel cell construction, the second requirement has an additional weight since the potential bias will be defined not by the difference of the redox potentials of the enzymatic cofactors, but by the potential difference of the cathodic and anodic mediators used.
Δ E Δ E
Scheme 1.5: Potential losses caused by the usage of electron mediators.
So, mediators always reduce the !E of the cell; thus, the power output according to equation (1.4).
"11 = 2 · ! (1.4)
To minimize energy losses, while building second-generation biofuel cells, one needs to choose the electron mediators wisely, with the redox potentials as close as possible to those of the enzymatic cofactors, although this is not always possible.
Introduction
14
Based on the principle of shuttling electrons via external mediators, several configurations have been developed for biosensor and biofuel cells construction. These include: an electrode-immobilized enzyme with freely diffusing mediators,92-94 mediators that are covalently bound to the electrode-immobilized enzyme,95-97 mediator-functionalized hydrogel-incorporated enzymes,98-101 reconstituted enzyme, linked via redox mediator102, or porous matrices with the pore-encapsulated mediators and enzyme caps.103,104 All configurations listed above have their own disadvantages, like the non-integrable architecture of freely diffusing mediators, limiting this platform to in-vitro measurements only, the destructive effect of covalently bonded mediators to the enzymatic shell, or leakage of entrapped poisonous mediators. The most successful architectures of electron mediated systems are the redox hydrogel- and polymer-incorporated biocatalysts, which are commonly used in commercially available biosensors.
1.3.2 Direct electron transfer (DET)
Enzymatic biosensors and biofuel cells based on DET are called third- generation devices. Direct electron transfer is considered to be the most efficient strategy for electron transfer. For example, GOx-based ferrocene methanol (FcMe)-mediated glucose biosensors exhibit ket values that usualy do not exceed ca. 900 s-1, whereas DET operated biosensors reach ket values up to ca. 5000 s-1.105 DET does not involve the usage of external, diffusional redox-active species. This communication methodology becomes possible when the distance between the enzymatic reaction center and the electrode is short enough to enable the electron transfer process (through tunneling). One way to decrease the r distance is by inducing conformational changes of the
Introduction
15
enzymes (e.g., BOD, Lac, or FDH) upon their deposition on the carbonaceous matrices, e.g., carbon nanotubes CNTs,106-108 carbon nanoparticles CNPs,109,110 and other carbonaceous materials.111-115 For example, hydrophobic interactions between certain regions of enzymes and electrodes may sometimes lead to non-destructive conformational changes in the three-dimensional enzymatic structure, shortening the distances between the reaction centers of enzymes and the electrodes, Scheme 1.6. Immobilization of enzymes on hybrid conductive materials which comprise metal nanoparticles,116-118 metal oxides,119,120 conductive polymers,121-123 and composite materials124,125 can also lead to direct electrical communication (e.g., GOx, AlcDH, and LOx).
Scheme 1.6: DET electrical communication establishment upon immobilization of redox enzyme on CNTs.
Both processes reduce the distance between the electrode and the enzymatic catalytic site, enabling effective electron tunneling in agreement with the Marcus theory.
In case of catalytic center of enzyme is very close or physically connected to the electrode (i.e., no barrier for electron tunneling), the observed onset potential of the catalytic reaction will be equal to E of the cofactor. However, a larger gap between the two ends will be expressed in overpotential (additional energy needed to overcome the energetic barrier for electron tunneling), and the catalytic onset potential will be higher for the oxidative and lower for the reductive enzymes.
Introduction
16
If we compare mediated and direct communication strategies, it is clear that DET is preferable over MET from several perspectives:
a) DET does not use any electron mediators that might leak and cause malfunction of the biosensor and the sample poisoning.
b) Mass transfer in DET systems is limited only by diffusion and not by additional layers required for enzyme immobilization, e.g., redox-active polymeric hosting matrix.
c) Since DET-based devices do not use relay units for electrical communication, no further losses of !E are caused in BFC applications.
d) ket is usually higher for DET communication, implying insensitivity towards oxygen, granting higher bioelectrocatalytic currents, which are beneficial for both energy harvesting applications and biosensing.
Considering these advantages of DET over MET, the global trend in bioelectronics in the last years is shifting toward direct electrical communication architecture and developing novel pathways to establish more powerful and more sensitive devices.
Introduction
17
1.3.3 Carbon-based conductive materials as hosting matrices for enzyme immobilization.
Immobilization of enzymes is one of the essential steps for establishing electrical communication in bioelectronic devices. Nowadays, four main strategies are used for enzymatic immobilization, including adsorption, covalent binding, affinity immobilization, or entrapment, as was previously exemplified.
Covalent attachment is an aggressive technique that frequently uses lysine (as a relatively common amino acid with good reactivity) as a point of covalent binding.126 The matrix side requires the presence of specific functional groups, for example, epoxide, which is stable, has good reactivity with lysine moieties and is easy to quench with primary amines after successful immobilization.127 Alternatively, one may use functional groups, like amine and carboxylic acid, to carry out the covalent binding using activation reagents, like water-soluble N-ε- maleimidocaproyl-oxysulfosuccinimide ester (sulfo-EMCS) or 1-(3- Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride and N- Hydroxysuccinimide (EDC/NHS) to carry out the reaction under mild conditions. However, a covalent linkage approach requires some experience since even minimal excess of the coupling reagent can cause complete deactivation of the enzyme.
Entrapment is a very common method for enzymatic immobilization and is frequently used for second-generation biosensors assembly.
Conductive polymers,128 sol-gels,129 and electrospun nanofibers130 are very friendly materials for this purpose where covalent (mainly click- chemistry) or non-covalent interactions prevent enzymatic detachment from the surface while the matrix protects the catalytic units from
Introduction
18
induced degradation. However, excessive thickness of the matrix may cause mass transfer problems, limiting the free diffusion of the substrate to the inner layers of the electrode and reducing efficiency of the device.
Affinity immobilization is based on enzymatic specificity and carried out either by decoration of the host matrix with affinity ligands towards a specific region of the enzyme or by conjugation of the enzyme to an entity with a certain affinity towards the host matrix. This method uses known affinity interactions, yet is not simple and usually requires several preparation steps to induce the self-assembly properties for immobilization. For example, affinity immobilization was used to electrically-contact apo-GOx via FAD-decorated SWCNTs.131
Physical adsorption is the least complicated technique since the immobilization process is based on hydrophobic132 or salt bridge133 interactions and practically occurs via self-assembly.134-137 Moreover, adsorption minimizes enzymatic motion, partially protecting the catalytic units from aggregation, additional interactions with hydrophobic interfaces, and proteolysis. Frequently this methodology is used for inducing DET communication. The immobilization can be done by drying the enzyme on the matrix or by dipping the matrix-containing electrode into an enzyme solution. Since most of the redox enzymes are water-soluble, their hydrophobic regions are mainly located in the inner parts of the three-dimensional structure. Thus, using conductive matrices with hydrophobic regions for enzyme immobilization, direct electrical communication can be achieved in some cases.
Owing to large surface area, high conductivity, high stability, and commercial availability, carbon-based materials like multi-walled or single-walled carbon nanotubes (MWCNTs or SWCNTs), carbon nanotube forests (CNTFs), carbon nanoparticles (CNPs), graphene,
Introduction
19
graphene oxide, ketjen black, or carbon-coated metal nanoparticles are widely used in bioelectronic applications.
1.3.4 Operational lifetime and shelf-time of enzyme- based bioelectronic compartments.
Employing enzymes as catalysts, bioelectronics faces the problem of a relatively short lifetime when enzymes are taken out of their native environment.138,139 Enzymes are dynamic structures that continuously bend and stretch during rest and activity, which accounts for one of the causes for their denaturation. The establishment of electrical communication requires mounting of the enzymes on different matrices as part of the fabrication process. Immobilization of the enzymes leads to their stabilization, minimizing free movements of the amino acid moieties and preventing non-covalent pathways of deactivation such as aggregation and precipitation. Additionally, covalent deactivation pathways are reduced as well, restricting amino acid chain interactions with reactive molecules.140 Moreover, other factors, like type of medium141-144 (aqueous or organic), immobilization method,145,146 temperature,147,148 pH,149,150 presence of particular inhibiting metal ions,151 all may affect the enzymatic structure, and as a result, the catalytic properties of the biocatalyst.
Additionally, the presence of proteolytic enzymes152 leads to rapid deactivation of biocatalysts, cutting them into inactive amino acid chains.
The lifetime of enzyme-based electrodes depends on the platform used (immobilization type, usage of mediators, protecting layers), but undoubtedly the main factor of electrodes’ deactivation is enzymatic
Introduction
20
degradation, and usually, operational functionality is limited to several days or weeks.
The lifetime extension of enzyme-based functional electrodes is considered one of the most critical topics. Undoubtedly, progress in this direction might help to elevate the interest of the market in bioelectronic devices and to attract appropriate attention to the utilization of enzyme- based accurate sensors and sustainable energy sources. The development of strategies for lifetime and shelf-time extension of bioelectronic products will allow producing more user-friendly devices and to make this field cost-effective and economically sound.
1.4 Objectives of the thesis
This doctoral thesis focuses on the development and investigation of novel synthetic approaches for electrical communication between enzymes and the carbonaceous hosting matrices by means of direct electron transfer (DET) with the goal of increasing the efficiency and broadening the spectrum of enzyme-based sensors and fuel cells.
Ultimately, the innovative concepts presented may lead to the development of lifetime extended, multi-integrable platforms for sensing (wearable or stationary) and energy harvesting, making bioelectronics cost-efficient, user-friendly, and more convenient.
Chapter 1 provides an introduction to bioelectronics and essential problems in the field. It supplies the necessary theoretical background regarding enzymes, principles of bioelectrocatalysis, and heterogeneous mediated and direct electron transfer as the communication pathways between the biocatalysts and electrodes.
Introduction
21
Chapter 2 describes the principles of electrochemical analysis and provides an introduction to two analytical techniques that have been used as the main characterization tools during the work, namely, cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS).
Chapter 3 deals with one of the fundamentally exciting and scientifically challenging tasks: enzyme-electrode communication. Here we demonstrate and discuss the electrochemical and the enzymatic synthetic approaches developed for direct electrical communication between redox enzymes, namely, glucose oxidase (GOx) and horseradish peroxidase (HRP) and electrodes. Both methods led to direct electrical communication, yet of different efficiency. The resulting electrodes were characterized, and their bioelectrocatalytic properties compared.
In addition, observing peculiar bioelectrocatalytic characteristics of enzymatically wired GOx, a mechanistic explanation of catalytic activity was suggested, supported by experimental data.
In Chapter 4, we demonstrate a novel approach to biofuel cell architecture, significantly simplifying its use and ensuring uninterrupted, on-demand power generation based on selective self- assembly of biocatalysts. The developed fructose/O2 biofuel cell is based on two ingredients: two chemically-modified glassy carbon electrodes, and two populations of chemically- and enzymatically (FDH and BOD)-modified carbon-coated magnetic nanoparticles (ccMNPs).
Each population of nanoparticles has a high affinity towards only one of the electrodes. Upon injection of the ccMNPs into the cell containing stationary electrodes, fully functional anode and cathode are formed within a few minutes, producing ca. 25 µW·cm-2 from fructose oxidation. We further demonstrate fast and convenient disassembly of
Introduction
22
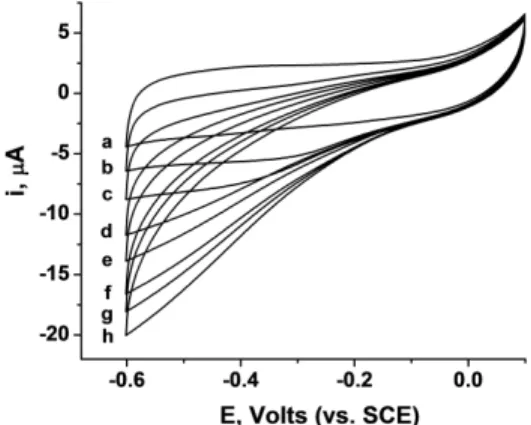
the cell by using an external magnetic field, allowing an exchange of biocatalysts on demand (for example upon exhaustion of biocatalyst units) by injection of a fresh batch of modified ccMNPs.
As a solution for long term storage of bioelectronic compartments, we implemented the ccMNPs for BOD/SWCNTs electrodes protection against natural degradation and proteolysis. The deposition of magnetic nanoparticles onto the electrode leads to the formation of a protective layer, which can be removed by an external magnetic field. We demonstrate that the ccMNPs-covered BOD-based electrode has superior stability retaining ca. 70% of its initial catalytic activity after 30 days of storage at room temperature whereas non-protected electrode does not exhibit any catalytic activity after the same time.
2 Electrochemical analysis
23
Electrochemical analytical techniques provide powerful tools to investigate electron transfer in chemical and biochemical reactions that occur at the solid-liquid interface, namely, on the electrode.
Voltammetry, one of the most convenient and powerful tools, is based on the observation of changes in electrical current upon application of variable potentials. For this purpose, a potentiostat is used as the primary instrument. In this work, cyclic voltammetry (CV) was used as the main analytical technique for the investigation of bioelectrochemical processes and determination of kinetic and thermodynamic parameters of the systems.
The electrochemical impedance spectroscopy (EIS) was used to complement this information with insights about mass transfer resistivity and processes that take place on the electrode’s surface.
This chapter provides a brief overview of the two electrochemical techniques.
In electrochemistry, the emergence of electrical current is related to chemical changes that take place in the system. In nature, molecules always interact and frequently oxidize and reduce each other; these processes involve electron transfer. Let us consider the following reaction, equation (2.1), where species An+ chemically oxidizes B in the solution. The driving force of this reaction originates in thermodynamics.
The lowest unoccupied molecular orbital (LUMO) of An+ has lower energy than the highest occupied molecular orbital (HOMO) of B, so the electron, preferring lower energies, migrates from B to An+, forming Bn+
and A. The electron transfer which occurs between the two species in the same phase, like in the discussed case, is called homogeneous electron transfer.
Electrochemical analysis
24 45*+ 6
⎯⎯⎯⎯ 4 + 65* (2.1)
However, when electrochemical oxidation takes place, electron transfer occurs between electrode and electrochemically active compound A, according to the reaction represented by equation (2.2).
4 ⇌ 45*+ 7$% = +0.19 (2.2)
The potentiostat, connected to the electrode, allows one to control the energy of the electrons on the HOMO/LUMO of the surface, equation (2.3), enabling oxidation or reduction of A/An+ at the liquid/solid interface (electrolyte/electrode surface) by setting the potential above and below E0 of A, respectively. The electron transfer on the interface of two different phases is called heterogeneous electron transfer.
!: = −7< (2.3)
In electrochemistry, the Nernst equation, (2.4) describes the relationship between the standard redox potential E0, temperature T, species activities (concentrations) [A] and [An+], and the actual potential at the electrode E, at equilibrium, where R is the gas constant, F the Faraday constant, and n the number of electrons involved in the electrochemical reaction. If the potential E applied to the electrode is identical to E0, at equilibrium, the concentration of [A] is equal to [An+]. By varying potentials, the ratio between [A] and [An+] is changed to satisfy the Nernst equation.
= −=>
7< · ?7 4
45* (2.4)
Electrochemical analysis
25
Considering E0 of equation (2.2) to be +0.1 V, and n = 1, at room temperature, upon application of E = +0.2 V, the ratio @
@AB = . CD , meaning that 98% of species A remains oxidized in the form of An+ at equilibrium.
2.1 Cyclic voltammetry
Cyclic voltammetry153 (CV) is one of the basic yet powerful techniques in electrochemistry. It is used for investigating redox reactions and (bio)catalytic responses of functional surfaces in liquid. CV measurements are carried out in electrolyte solution and employ a 3- electrode cell, composed of the working electrode (WE), reference electrode (RE), and counter (auxiliary) electrode (CE). During the measurement, the potential is applied between WE and RE, while the developed current flows between WE and CE, Scheme 2.1.
Scheme 2.1: Schematic illustration of a three-electrode electrochemical setup cell.
Electrochemical analysis
26
In principle, the electrochemical measurements could be carried out in a 2-electrode cell when counter and reference electrodes are a single wire.
However, the current flow can change the potential applied to the electrode and thus disturbs the reliability of the measurement.
Two basic requirements must be respected to assure reliability of the results:
(a) The potential of the RE should stay constant during the measurement;
hence, saturated calomel (SCE) or Ag/AgCl electrodes are widely used for this purpose in aqueous media.
(b) The surface area of the CE has to be larger than that of the WE to prevent limitation of electrical current due to insufficient capability of the CE to perform the complementary electrochemical reaction.
Cyclic voltammetry, as an electrochemical technique, investigates processes associated with heterogeneous electron transfer; however, processes involving homogeneous electron transfer associated with chemical reactions could also be indicated.
In CV, the potentiostat controls the applied potential, which is swept in a defined range, from E1 to E2 and back to E1, forming the potential circle. The user specifies the scan rate υF 5, with F 5 =GHG . In the presence of the electrochemically-active compound, e.g., A, whose E0 lies between E1 and E2, electrical current is developed upon scanning, indicating the oxidation or reduction process of the species, and the resulting current (Y axis) is plotted versus potential (X axis). Such a plot is called a voltammogram.
Let us consider A, from equation (2.2), is present in the electrolyte, and its redox potential lies in the range between E1 and E2. The potential is
Electrochemical analysis
27
swept from E1 to E2 ( 1 < 2), with =HC%HD
J%', Figure 2.1 (A). As the potential rises closer to the redox potential of A, an electrical current starts to grow, indicating the oxidation process on the electrode, and formation of An+. The electrochemical response is proportional to [A] in the system, expressed by the value of the anodic peak current, KL, Figure 2.1 (C). The resulting electrical current is called anodic and has positive values.
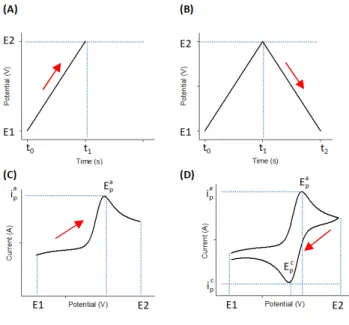
Figure 2.1: Diagrams of reversible redox couple in cyclic voltammetry measurement. (A) The profile of linear change of the potential in time (the forward scan); (B) The profile of complete cycle of potential application in a single scan; (C) The obtained linear voltammogram as a result of potential application in (A); (D) The complete cyclic voltammogram obtained as a result of potential application in (B).
Electrochemical analysis
28
Then, upon scanning from E2 to E1, the reverse process takes place, causing the reduction of An+ that have been accumulated in the vicinity of the electrode to A, through the development of a cathodic current of negative values (in agreement with the IUPAC convention). The full potential scan, Figure 2.1 (B), is reflected by the voltammogram. The electrical current appears as a duck-like wave, forming a typical voltammogram of the electrochemically reversible process (different from chemical reversibility). The midpoint between L M7N L (the potential values at the anodic and the cathodic peak currents) defines the redox potential of the species, Figure 2.1 (D).
The observed peaks, KL and KL, are created due to the depletion of [A]
and [An+] in the vicinity of the electrode, indicating the mass transport limitations since the electron transfer process is way faster than the diffusion of the species in the solution. Consequently, diffusional constraints of the redox species lead to the current drop, according to Fick’s second law, equation (2.5), where [A] is the species concentration, D[A] the diffusion coefficient of A, and x the distance from the electrode.
If convection is applied during the measurement (by direct mixing or by using rotating disc electrode), fresh molecules are regularly supplied to the electrode surface, achieving current values proportional to the concentration of the redox species in the solution and preventing the current drop and peak formation.
R 4
RS = T@ URC 4
RVC W (2.5)
Electrochemical analysis
29
The redox reaction is defined reversible if peak-to-peak separation equals to 59 mV per electron, L − L =YZ[\5 (derived from Nernst equation), meaning that electron transfer occurs easily. However, if electron transfer is hindered, for example, by an insulating layer on the surface, the peak-to-peak separation increases, accompanied by a less steep current growth, which provides an indication of an electrochemically semi (quasi)-reversible or irreversible reaction, Figure 2.2 (A), solid line.
Figure 2.2: (A) Comparison of the profiles of quasi-reversible (solid line) and reversible (dotted line) electrochemical redox reaction. (B) Illustration of non- Faradaic (solid line) and Faradaic (dotted line) currents.
It should be noted that the electrical current in cyclic voltammetry is produced as a result of two processes: redox reaction (Faradaic current), and electrode charging (non-Faradaic or charging current). The non- Faradaic current depends on the WE capacity and the scan rate applied.
In the absence of redox-active species, or if redox reactions do not occur in the chosen potential range, only non-Faradaic electrical current is developed, Figure 2.2 (B), solid line.