Research Collection
Doctoral Thesis
Development of split NanoLuc-based arrestin biosensors for both ligand- and light-activated GPCRs
Author(s):
Spillmann, Martin Publication Date:
2020
Permanent Link:
https://doi.org/10.3929/ethz-b-000457859
Rights / License:
In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use.
ETH Library
DISS ETH NO. 26922
Development of split NanoLuc-based arrestin biosensors for both ligand- and light-activated GPCRs
A thesis submitted to attain the degree of DOCTOR OF SCIENCE of ETH ZÜRICH
(Dr. sc. ETH Zürich)
presented by
Martin Spillmann
Master of Science in Immunology and Microbiology, ETH Zürich
born on 20.01.1992
citizen of Villnachern AG, Switzerland
accepted on the recommendation of Prof. Gebhard Schertler, examiner Dr. Philipp Berger, scientific co-supervisor Dr. Maria Waldhoer, scientific co-supervisor
Prof. Sabine Werner, co-examiner 2020
2
3
Funding
This work was supported by the NCCR Molecular Systems Engineering (MSE) to Prof. Gebhard F.X.
Schertler.
We would like to thank Robert Lucas (University of Manchester) for providing the human rhodopsin gene.
4
Acknowledgements
Here, I would like to thank all the people who helped me during this project. This work would not have been possible without the support, help and guidance of many. Science is always a
collaborative effort. I would like to offer my gratefulness to everyone that made a key difference and highlight his or her contribution and commitment during my time at PSI.
Gebhard Schertler was not only officially responsible for my PhD as my professor, but also gave valuable insights into my project and professional career. I also greatly appreciate his honest and kind attitude.
Philipp Berger acted as my supervisor and was key in planning experiments, analysing data, publishing my work, as well as practical support in the lab. From start to finish, his experience, knowledge and feedback were invaluable for this project.
Maria Waldhoer, as my co-supervisor, shared with me her experience in cellular assays. Her input was always insightful and broadened my horizon.
Elena Lesca introduced me to the fascinating world of opsins and I greatly enjoyed the
methodological and scientific challenge they posed, as well as the great discussions we had about work and life in general.
Aurélien Rizk and Susanne Roth showed great patience in explaining me, how math can help in better understanding biology. They also greatly supported me in the development of a mathematical model.
Mirjam Zimmermann and Nadine Dobberstein are fellow lab workers and collaborators. They
advised me in establishing the methods and protocols used in this study and supported me in the lab on countless occasions.
Maysam Mansouri developed the baculovirus system used to transduce multiprime plasmids. As a collegue, he answered each of my many questions and his know-how was immensely helpful.
5 Benoît Meger always found time for intense scientific discussions and burgers. I am also grateful for his technical and psychological support in the lab. I greatly enjoyed his company and admired his throughput in agar plates and SDS gels.
Nina Romantini provided me with key receptors and ligands used in this study. We also had many insightful scientific exchanges and shared an excitement for on fantasy conventions and literature (and dinosaurs).
Shahidul Alam developed GRK biosensors tested in this work. We had constructive talks about experimental procedures and interpretation of our data and I appreciated his welcoming attitude.
Additionally, three exceptional undergraduate students contributed to this work. Daniela Büttiker who further developed the mathematical model, and Larissa Thurner and Corinne Kaufmann who expanded our biosensor array and collected valuable data.
The people of LBR and LNB as well as the team of InterAx Biotech AG were creating a fun
environment, be it in the lab, on the ski slope, or while trying foreign cuisine. I hope they enjoyed it as much as I did.
Looking back, I have learned a lot - both scientifically and personally - in these three and a half years and I am grateful for this experience.
6
Summary
G-protein coupled receptors (GPCRs) are transmembrane proteins responsible for a vast variety of cellular functions including smell, vision and the detection of neurotransmitters or hormones. Their structure enables them to respond to outside stimuli and transmit these signals into the cell by binding various cytosolic proteins. Their eponymous binding partners are G proteins, who further relay the signal to second messengers like cAMP or Ca2+ ions.
Arrestins are cytosolic proteins, known to bind GPCRs. They act as multifunctional scaffold proteins involved in internalization, endosomal trafficking and signal modulation of GPCRs by binding various signalling components. The GPCR-arrestin interaction has proven to be useful for high-throughput screening of GPCR ligands. Vertebrates possess four arrestin genes: S-arrestin (SAG), arrestin-C (ARR3), β-arrestin-1 (ARRB1) and β-arrestin-2 (ARRB2). S-arrestin and arrestin-C are essential for human vision, while β‐arrestin-1 and β‐arrestin-2 are expressed more ubiquitously in a large range of tissues. GPCRs show different selectivity towards arrestins. It has been shown, that arrestin selectivity correlates with the interaction dynamics and the receptor’s trafficking behaviour. Currently, most studies focus on only one arrestin at a time due to their highly conserved structure and sequence. However, there is mounting evidence that each arrestin has unique functions.
Nanoluciferase (NanoLuc), a very bright, small luciferase is a promising new tool for molecular biology.
It has advantages in size, light intensity and is independent on cofactors like ATP or flavins. Further, it can be used in protein fragment complementation assays by splitting it into two subunits. The enzyme complementation is reversible allowing time resolved detection of protein-protein interaction dynamics and short-lived protein complexes.
In the first part of this study, an enzyme complementation reporter assay based on the split NanoLuc that allows time resolved observation of the recruitment of arrestins to GPCRs in living cells is presented. Two assay approaches were planned: 1) a direct variant, where both interaction partners are tagged with a split luciferase subunit and 2) an indirect variant that leaves the receptor unlabelled and instead localizes one luciferase subunit to the plasma membrane using a CAAX polyprenylation motif. Constructs were cloned for the β2-adrenergic receptor, the gastrin releasing peptide receptor and the somatostatin receptor type 2, as well as all four human arrestins. Different plasmid backbones were used in order to enable the use of biosensors in various applications. The finished constructs were first tested using transient transfection of HEK293 cells and stimulation of the cells with a respective agonist. A workflow for the assay was established.
7 Both assay types were tested, and arrestin selectivity as well as dose response parameters were compared. In both the direct and indirect assay, the clear preference of the β2AR for β-arrestin-2 could be shown. GRPR and SSTR2 showed no bias towards any arrestin and showed a much stronger interaction than β2AR in the direct assay. However, in the indirect assay, their recruitment was much weaker, possibly due to their trafficking behaviour. Concentration response experiments were performed for both assay types, showing that both assays produce comparable - but not identical - results. β2AR and SSTR2 showed no significant difference in their IC50 values between direct and indirect assay. GRPR showed a measurable shift in potency. Stable HEK293-derived cell lines were created expressing the CAAX-polyprenylated luciferase subunit as well as one luciferase-tagged arrestin. This allows the detection of arrestin recruitment by simple transfection of an unlabelled GPCR into the stable cell lines.
Using a Cre-recombinase reaction the aforementioned plasmids were combined into assembly vectors which are able to express several proteins from one vector. This allowed transfection and expression of the entire direct or indirect assay system from one plasmid. Further, two promoters of different strengths (cytomegalovirus and phosphoglycerate kinase) were used to vary the expression stoichiometry of the assay’s components. The different promoter strengths showed varying responses, indicating that overexpression of the assay’s components limits the dynamic window. The Cre- assembly plasmids were further introduced into a baculovirus. This makes the assay applicable for a variety of cell lines, including primary cell lines. Both the direct and indirect assay systems were successfully transduced into HEK293 cells using a single viral vector.
The effect of the GPCRs C-tail on arrestin binding was also analysed by creating C-tail truncated variants of β2AR and GRPR. For these mutants two main effects could be observed: Arrestin selectivity was altered compared to the wt receptors and similar between both mutants indicating that the C-tail is in part responsible for the differential recruitment of arrestins. Further the dynamics of the arrestin interactions were changed, with a stronger recruitment early after stimulation but reduced long-term interactions.
In the second part of this thesis, the assays developed in part 1 were applied to study light sensitive GPCRs, called opsins. Specifically, the binding behaviour of the rhodopsin 1 from the jumping spider Hasarius Adansonii (JSR) was compared to human rhodopsin (hRho). Both their arrestin binding as well as their recruitment of all seven human GRKs was measured. Little is known about JSR1’s cellular behaviour. The purified protein has been shown to be bistable and its photocycle has been well described. Here, we aim to study its signalling behaviour to investigate the possible application of JSR1 as an optogenetic tool.
8
Opsins differ from ligand-activated GPCRs in that they bind retinal, a vitamin A derivative, covalently to a lysine residue. Retinal is photosensitive and upon light absorption able to isomerize from 11-cis to all-trans-retinal, or vice versa. This isomerization activates the receptor.
The assay’s workflow needed to be adapted in order to measure opsin behaviour and activity. Retinal had to be added to the cells 24 hours before the measurement and the cells had to be kept in the dark during that time. The amount of retinal needed was evaluated using a concentration response curve.
Additionally, an illumination strategy needed to be developed to activate the opsins. This was done using either a laser or a xenon flash lamp combined with optical filters. Different wavelengths of light were tested and the amount of light needed for full saturation of the opsins was determined.
Unfortunately, the photocycle of JSR1 could not be observed by measuring arrestin recruitment, either because it does not occur in the cellular context or the interaction of JSR1 with arrestin is irreversible.
Recruitment of GRK could only be detected for GRK2 and GRK3 for both JSR1 and hRho. GRK1 binding to hRho could not be detected contrary to literature. This may be due to the GRK biosensors not changing their localization, as all except GRK2 and GRK3 are localized to the plasma membrane.
Therefore, no significant redistribution of them could be detected after stimulation.
9
Zusammenfassung
G Protein gekoppelte Rezeptoren (GPGR) sind Transmembran-Proteine und für eine grosse Vielfalt an zellulären Prozessen beteiligt, dazu gehören der Seh- und Geruchsinn sowie die Wahrnehmung von Neurotransmittern oder Hormonen. Ihre Struktur erlaubt es, Signale von ausserhalb der Zelle wahrzunehmen und diese durch das Binden von cytosolischen Proteinen weiterzuleiten. Ihr namensgebender Bindungspartner sind G Proteine, welche die Signale der GPGRs auf sekundäre zelluläre Botenstoffe wie cAMP und Ca2+ übertragen.
Arrestine sind cytosolische Proteine, die an GPGRs binden. Sie fungieren als multifunktionale Plattformen welche an Endozytose, der Steuerung von Endosomen, sowie dem modulieren von GPGR beteiligt sind. Dies erreichen sie durch die Interaktion mit verschiedensten Signalmolekülen [1-5]. Die Interaktion von GPGRs und Arrestinen wird sowohl in der Grundlagenforschung als auch in der Pharmazeutischen Industrie benutzt, um im Grossdurchsatz neue Medikamente, welche an GPRGs binden, zu entdecken [6-10]. Wirbeltiere verfügen über vier Arrestin gene: S-arrestin (SAG), Arrestin- C, β-Arrestin-1 (ARRB1) und β-Arrestin-2 (ARRB2) [11]. S-arrestin und Arrestin-C sind vor allem im Auge exprimiert und essentiell für den Sehsinn des Menschen, während β-Arrestin-1 und β-Arrestin-2 in verschiedensten Geweben zahlreiche Funktionen übernehmen [12]. GPGRs zeigen verschiedene Selektivitäts-Muster gegenüber den Arrestinen. Es wurde gezeigt, dass diese Selektivität mit der Art des Ablaufs und der Geschwindigkeit des Rezeptor-Transports korreliert. Studien zu dieser Interaktion testen meist nur ein Arrestin, da alle Arrestine eine ähnliche Struktur und Sequenz besitzen [13]. Es gibt jedoch eine wachsende Zahl an Hinweisen, die darauf hindeuten, dass jedes Arrestin einzigartige Funktionen erfüllt [14-16].
Die Nanoluciferase (NanoLuc) ist eine sehr helle, kleine Luciferase, welche ein vielversprechendes neues Instrument für die Zellforschung darstellt. Neben ihrer Grösse und Intensität ist ihre Aktivität auch unabhängig von Cofaktoren wie ATP. Sie kann zudem in zwei Untereinheiten gespalten werden, wodurch sie für Enzymfragment-Komplementierungs-Messungen benutzt werden kann. Die Komplementierung ist reversibel, was das Aufzeichnen von Proteininteraktionsdynamiken sowie kurzlebigen Proteinkomplexen erlaubt [17, 18]. Im ersten Teil dieser Arbeit, wird eine Enzymfragment- Komplementierungs-Messmethode basierend auf NanoLuc vorgestellt. Die Methode erlaubt die Detektion der Interaktion von Arrestinen mit GPGRs in Echtzeit und in lebenden Zellen. Zwei verschiedene Messstrategien wurden entwickelt. 1) In der direkten Variante werden beide Bindungspartner mit einer Untereinheit der Luciferase verbunden. 2) In der indirekten Variante wird der Rezeptor unverändert eingesetzt, stattdessen wird eine Luciferase-Untereinheit mit Hilfe eines CAAX Polyprenyl-Motifs an die Zellmembran angebracht. Konstrukte wurden für den β2-adrenergen
10
receptor (β2AR), den Gastrin freisetzenden Peptid Rezeptor (GRPR) und den Somatostatin Rezeptor Typ 2 (SSTR2), sowie alle vier menschliche Arrestine entwickelt. Die Konstrukte wurden in verschieden Plasmide eingebaut, damit die Methode mit verschiedenen biochemischen Applikationen kompatibel ist. Die fertigen Plasmide wurden zuerst mithilfe transienter Transfektion in HEK293 Zellen getestet.
Stimulationen wurden mit entsprechenden Liganden durchgeführt. Ein Arbeitsablauf für die Methode wurde etabliert.
Beide Varianten wurden getestet und auf ihre Arrestin-Selektivität sowie pharmakologische Eigenschaften verglichen. In beiden Varianten wurde eine klare Präferenez von β2AR für β-Arrestin-2 festgestellt. GRPR und SSTR2 zeigten hingegen keine solche Präferenz und eine viel stärkere Interaktion mit allen Arrestinen in der direkten Variante. In der indirekten Variante zeigten sie hingegen eine wesentlich schwächere Bindung, möglicherweise aufgrund ihres zellulären Transport- Verhaltens. Konzentrations-Wirkungs-Kurven wurden für beide Varianten und alle drei Rezeptoren gemessen. sowohl β2AR und SSTR2 zeigten sehr ähnliche IC50-Werte in beiden Messvarianten. GRPR zeigte einen signifikanten Unterschied. Stabile Zelllinien basierend auf HEK293 wurden entwickelt, welche sowohl eine Luciferase Untereinheit an einem CAAX Polyprenyl-Motif sowie ein Arrestin mit der anderen Untereinheit konstant exprimieren. Dies Zellen erlauben die Messung der Arrestin Bindung durch einfache Transfektion eines unveränderten Gens in die Zellen.
Mithilfe einer Cre-Rekombinations-Reaktion kombinierten wir unsere Plasmide zu Vektoren, welche mehrere Proteine von einem Plasmid exprimieren können. Dies erlaubt es, alle für die Messmethode notwendigen Biosensoren von einem einzigen Plasmid zu exprimieren. Wir verwendeten zusätzlich zwei verschiedene Promotoren (Cytomegalovirus and Phosphoglycerat Kinase) von unterschiedlicher Stärke. Dies erlaubt es, die Expression unserer Proteine zu variieren. Alle möglichen Kombinationen von GRPR mit β-Arrestin-1 oder β-Arrestin-2 mit den zwei Promotoren wurden geklont und getestet.
Die verschiedenen Vektoren zeigten, dass übermässige Expression unserer Messkomponenten unser Messfenster verkleinert. Die Vektoren aus der Cre-Reaktion wurden zusätzlich in einen Baculovirus- Vektor eingeführt. Dies erlaubt die Applikation unserer Messmethode in diversen Zelltypen, insbesondere Primärzellen. Die Komponenten für die direkte sowie die indirekte Messmethode wurden erfolgreich mittels eines einzelnen Virus in HEK293 Zellen transduziert.
Der Einfluss des C-Terminus eines GPGR auf die Interaktion von Arrestin wurde untersucht indem Mutanten von B2AR und GRPR, welche über keinen C-Terminus verfügen, erstellt wurden. Zwei Effekte konnten dabei beobachtet werden: 1) Die Arrestin-Selektivität wurde im Vergleich zum Wildtyp verändert. Dies lässt schlussfolgern, dass der C-Terminus eine Rolle bei der unterschiedlichen Rekrutierung der Arrestine spielt.
11 Im Zweiten Teil dieser Arbeit wurde die in Teil eins entwickelte Methode auf lichtempfindliche GPGRs, auch genannt Opsine, angewandt. Das Signalverhalten von Rhodopsin 1 der Springspinne Hasarius Adansonii (JSR1) und menschlichem Rhodopsin (hRho) wurde verglichen. Deren Binding zu sowohl Arrestinen als auch G protein-gekoppelte Rezeptor Kinasen (GRKs) wurde untersucht. Es ist wenig über JSR1’s Signalverhalten bekannt. Es wurde jedoch mit gereinigtem Protein gezeigt, da es bistabil ist und der Photozyklus des aufgereinigten Proteins bekannt ist. Das Verhalten von JSR1 wurde analysiert im Hinblick auf eine mögliche Anwendung als optogenetisches Werkzeug.
Opsine unterscheiden sich von Ligand-aktivierten GPGRs indem sie Retinal, ein Vitamin A Derivat, kovalent an einer Lysin Seitenkette binden. Retinal ist lichtempfindlich und isomerisiert unter Beleuchtung von 11-cis zu all-trans-retinal, oder umgekehrt. Es ist diese Reaktion, welche den Rezeptor aktiviert.
Die Messmethode musste an die Ansprüche der Opsine angepasst werden. Retinal wurde zu transfizierten Zellen 24 Stunden vor der Messung zugegeben und die Zellen wurden von dem Zeitpunkt an vor Licht geschützt. Die benötigte Menge an Retinal wurde mithilfe Dosis-Wirkungs-Kurven bestimmt. Zusätzlich wurde eine Stimulierung mit Licht benötigt. Dies wurde entweder durch Laser oder einer Xenon-Blitz-Lampe und optischen Filtern erreicht. Verschiedene Wellenlängen an Licht wurden getestet und die Lichtmenge ermittelt, die für eine vollständige Stimulation benötigt wird.
Leider konnte der Photozyklus von JSR1 nicht direkt beobachtet werden. Zusätzlich zur Interaktion mit Arrestinen wurde auch die Interaktion mit G protein-gekoppelten Kinasen ermittelt. GRK2 und GRK3 zeigten signifikante Interaktion mit JSR1 und hRho nach der Lichtstimulation.
12
Table of Contents
Part 1 Establishment of arrestins as biosensors for GPCRs using a split nano-luciferase assay ... 14
Introduction ... 15
G protein coupled receptors (GPCRs) ... 15
Structure ... 16
β2-adreneric receptor ... 17
Gastrin-releasing peptide receptor ... 17
Somatostatin receptor type 2 ... 17
Ligands Types ... 17
Cytosolic Binding partners of GPCRs ... 19
Arrestins ... 20
Biased signalling ... 28
Luciferases... 29
PCA- and BRET-based assays ... 31
Baculoviral Vectors ... 33
Results and Discussion ... 34
Goal ... 34
Development of arrestin biosensors ... 34
Direct Assay ... 35
Indirect Assay ... 37
Assay comparison and verification ... 39
Gene Dose experiments ... 40
Generation of baculovirus constructs ... 44
Effect of the GPCR C-tail on arrestin binding ... 45
Conclusion ... 49
Part 2 Signalling of opsins ... 50
Introduction ... 51
Retinal ... 51
Light perception in Humans ... 52
Rhodopsin signalling ... 52
Bistable vs. monostable opsins ... 53
Vision of the jumping spider Hasarius Adansoni ... 55
Results and Discussion ... 56
Goal ... 56
13
Assay adjustments ... 56
Arrestins binding to JSR1 and hRho ... 57
Retinal controls ... 58
Saturation curves ... 60
JSR1 Photocycle... 62
GRK recruitment ... 63
Conclusion ... 66
Part 3 Mathematical Modelling ... 67
Introduction ... 68
ODE models ... 68
Parameter estimation ... 69
Results and Discussion ... 70
Conclusion ... 72
Future Perspectives / Outlook ... 73
Output ... 74
Teaching responsibilities ... 74
Materials and Methods ... 75
Molecular Biology ... 75
Cell culture ... 75
Stable cell lines ... 75
Split Luciferase Assay ... 76
Baculovirus generation ... 77
Statistical analysis ... 78
References ... 79
Appendix ... 89
Plasmids used ... 90
14
Part 1 Establishment of arrestins as biosensors for GPCRs using a split NanoLuc assay
Contributions:
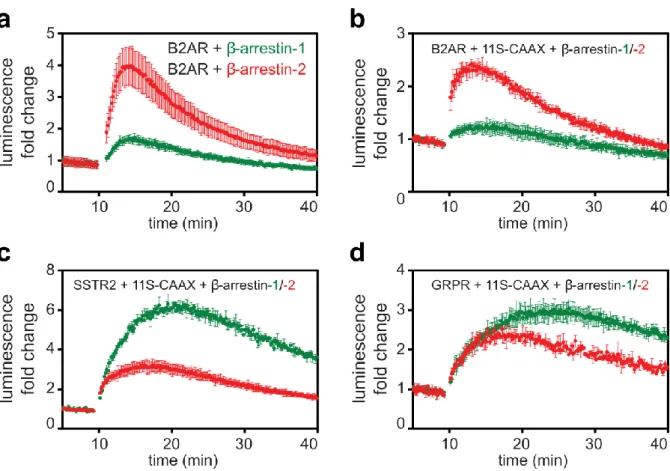
Larissa Thurner, Corinne Kaufmann and Dr. Philipp Berger cloned plasmids. Compounds for GRPR and SSTR2 were provided by Dr. Martin Behe. Dr. Benoît Meger provided arrestin mutants, and Dr.
Mirjam Zimmermann performed comparative BRET assays. Dr. Maria Waldhoer, Prof. Gebhard Schertler, and Dr. Philipp Berger supervised the project.
15
Introduction
G protein coupled receptors (GPCRs)
GPCRs are a major family of membrane proteins that transmit signals from the outside of the cell into the cytoplasm. GPCRs create three of our senses: sight, scent and taste as well as allowing cell to cell communication by acting as receptors for a slew of hormones and neurotransmitters throughout the body. This heavy involvement in countless physiological processes makes GPCRs involved in a plethora of diseases and conditions including pain, blindness and neurological disorders. As such, they are extremely important pharmaceutical targets. GPCRs are currently the largest target family of clinically used drugs, and present a huge potential for future treatments. Many non-olfactory GPCRs have not yet been explored in clinical trials and have a broad untapped therapeutic potential, particularly in genetic and immune system disorders [19]. Despite their well-conserved basic structure, GPCRs evolved into an extremely diverse family, with over 800 unique receptors expressed in the human body [20]. They detect a huge variety of molecules, with each receptor being highly specific to its selected targets.
GPCRs can be classified in several ways. The most common classification is based on sequence homology and function, dividing them into six classes: Class A—rhodopsin-like receptors, Class B—
secretin family, Class C—metabotropic glutamate receptors, Class D—fungal mating pheromone receptors, Class E— cyclic AMP receptors, and Class F—frizzled (FZD) and smoothened (SMO) receptors[21, 22].
Another classification by Oakely et al [23] is based on how receptors interact with arrestins -their cytosolic binding partners. This creates two categories: class A and B. Class A GPCRs interact only transiently with arrestins and those interactions are generally restricted to the plasma membrane.
Additionally, class A GPCRs have a strong preference for β-arrestin-2 showing only marginal recruitment of other arrestins. In contrast, class B GPCRs strongly interact with S-arrestin, β-arrestin- 1 and β-arrestin-2. Their interactions are sustained during internalization and trafficking of the receptor into endosomes. Examples of class A receptors include the β2-adrenergic receptor, μ opioid receptor, dopamine receptor D1 and endothelin receptor type A. Examples of class A receptors include the type-2 angiotensin II receptor, vasopressin receptor 2 and neurotensin receptor 1.
16
Structure
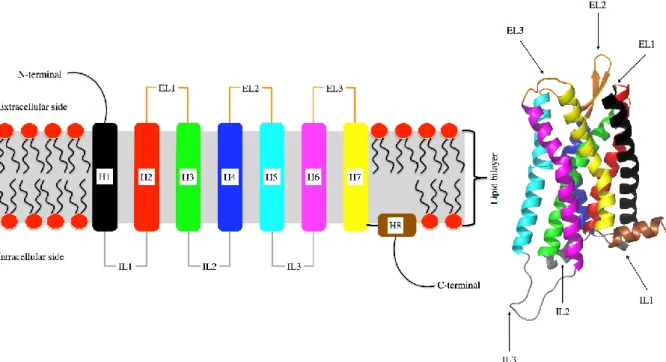
All GPCRs are composed of a bundle of seven transmembrane helixes (H1 – H7, Fig. 1) connected by flexible loops. All helixes span the membrane bilayer. The extracellular side of a receptor consists of three flexible loops as well as the N-terminus. The intracellular side also contains three loops and the flexible C-terminus, also called C-tail. A short intracellular helix can be found at the start of the C-tail (H8, Fig. 1). The helix bundle forms a cavity, usually opened towards the extracellular side, that acts as ligand binding site. The cytoplasmic loops and C-tail are involved in the interaction of cytosolic binding partners like G proteins, arrestins or GRKs.
The first crystal structure of a GPCR that was available was bovine rhodopsin [24]. Later, the structures of many other GPCRs, including pharmacologically important ones, were elucidated [25-27]. These structures not only helped in gaining a mechanistic understanding of GPCR functions, but also enabled the rational design of ligands based on the target GPCRs binding pocket in so-called virtual screens [28]. More recently, developments in time-resolved crystallography allow the observation of receptor dynamics at high resolution [29].
Figure 1. GPCR Structure Left: schematic of a G protein coupled receptor in its membrane environment showing the connectivity of helixes (H1-H8), extra- and intracellular loops (EL/IL). Right: 3D ribbon structure of µ-opioid receptor, the C-tail is not shown. Helices and loops are coloured identically in both images. Image adapted from: Fossépré et al (2015) [30].
17 β2-adreneric receptor
The β2-adreneric receptor (B2AR) is one the best studied GPCRs often used as a model system. Its physiological functions are manifold, but mostly related to muscle function. It causes the relaxation of smooth muscle cells in the gastrointestinal tract, the bronchi, uterus and eye making it involved in digestion, respiration, labour and vision[31, 32]. The β2AR is also triggering many of the physiological effects of the fight-or-flight response [33].Further, is expressed in the pancreas where it modulates the secretion of insulin and glucagon[34]. β2AR’s native ligand is epinephrine (adrenaline), but many artificial ligands have been developed and are commonly used compounds in the clinics. Drugs targeting the β2AR are used to treat asthma, chronic obstructive pulmonary disease and abnormal heart rhythms (beta-blockers).
Gastrin-releasing peptide receptor
The gastrin-releasing peptide receptor is expressed in the pancreas, stomach and colon. It causes the release of hormones such as glucagon, insulin and -as its name implies- gastrin. GRPR activity also releases neurotransmitters, which stimulate smooth muscle cells regulating gastrointestinal motility.
Further GRPR is involved in several neurological functions such as the circadian cycle, memory modulation, stress, anxiety and fear[35]. GRPR is overexpressed in many breast and prostate cancers making it a potential radiotherapy target[36, 37]. An analogue of the GRPRs namesake ligand called bombesin, originally isolated from the skin of the toad Bombina bombina, is studied as a potential radioligand [38].
Somatostatin receptor type 2
Similar to the GRPR, the somatostatin receptor type 2 is expressed in the pancreas, as well as in the kidneys and the cerebrum. SSTR2 reduces the secretion of pancreatic enzymes in the pancreas upon binding of its ligand somatostatin [39]. In the brain, it mediates neuronal migration and axon outgrowth [40]. Further is has also been shown to be overexpressed in a large number of tumours, making it a potential radiotherapy target [41].
Ligands Types
Ligands typically bind a GPCR in a pocket that is opened towards the extracellular side of the receptor, inducing a conformational change. This change is relayed through the transmembrane helices to the intracellular side of the receptor. There, signal transmission is continued by various cytosolic proteins binding the intracellular side of the receptor. The signalling outcome is determined by the ligand, each inducing a specific receptor conformation. Ligands are grouped into categories, depending on the signalling output they create [42].
18
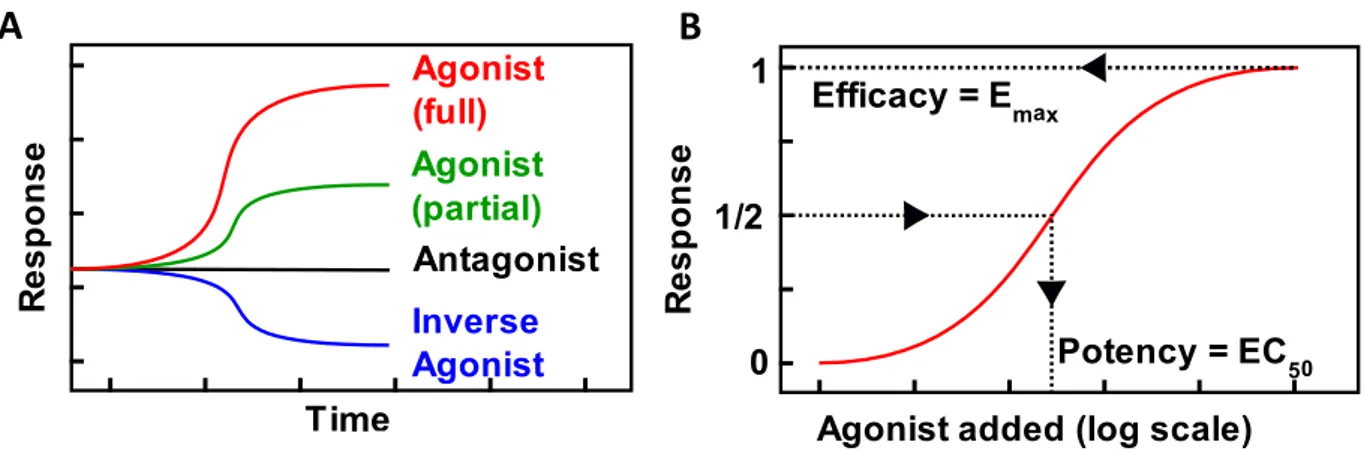
A B
Figure 2. A) Schematic representation of time courses showing a receptor’s response over time depending on the type of ligand added. B) Schematic concentration response, showing efficacy as the maximum response generated by a ligand and potency indicating the ligand concentration at which half the maximum response is generated.
➢ An agonist activates a GPCR, causing the generation of downstream signalling events. The extent of this activation is measured by the ligand’s efficacy. A very efficacious or “full” agonist creates the maximal amount of signalling, i.e. induces a maximally activated receptor structure. A less efficacious or “partial” agonist creates a weaker response.
➢ An antagonist, has no effects on signalling, but occupies the ligand binding pocket of the GPCR, preventing other ligands from interacting with the receptor.
➢ An inverse agonist reduces the constitutive activity of a GPCR. Certain receptors are continuously creating a signalling response even with no ligand bound. In such cases, an inverse agonist is able to induce a receptor conformation that is less active, thereby reducing the signalling output of the receptor.
Efficacy is independent of the affinity of a receptor towards a ligand. Instead, it is a measure of the affinity of the ligand bound receptor towards downstream effector molecules. Efficacy is often represented as Emax value, indicating the maximum signal that may be generated with a given ligand.
Potency on the other hand, indicates how much of a ligand is needed to induce a certain response, it is often quantified by the EC50 value. Potency is directly proportional to both the affinity of a ligand to a receptor and the ligand bound receptor to the effector. Potency and efficacy are independent from each other and differ for each ligand – receptor – effector system.
19 Cytosolic Binding partners of GPCRs
GPCRs have three many cytosolic binding partners, the most prominent among them are G proteins, arrestins and G protein coupled receptor kinases (GRKs) [43-45].
G proteins
G proteins are the eponymous interaction partners of GPCRs and are the main inducers of the GPCRs downstream response. They are heterotrimeric proteins consisting of an α- β- and γ-subunit. G proteins bind the cytosolic cavity created by the active state of the GPCR with the α-subunit determining the specificity of the G protein heterotrimer to different GPCRs, enabling them to cause a large diversity of cellular signalling effects. The active GPCR acts on the G protein α-subunit as a nucleotide exchange factor. Binding to a GPCR causes the G protein to open its nucleotide-binding pocket, releasing GDP and binding GTP. The G protein then dissociates from the receptor, allowing the receptor to bind another G protein molecule, leading to a first amplification in signal. The active G protein then splits into its α- and βγ-subunit. Each subunit activates its own downstream effectors, usually enzymes or ion channels. The activation of a single G protein therefore causes movement of many ions or molecular change in many molecules, leading to a second signal amplification.
Humans possess four classes of α-subunits that determine the G protein complex's signalling output.
The four classes of α-subunits are: Gαq, Gαi, Gαs and Gα12 each family causing a different downstream effect when activated by a GPCR.
➢ Gαs binds to adenylyl cyclases activating the production of cyclic AMP from ATP.
➢ Gαi also binds to adenylyl cyclases but inhibits their activity, decreasing cAMP levels.
➢ Gαq stimulates phospholipase C , which cleaves phosphatidyl inositol bisphosphate into inositol-trisphosphate and diacylglycerol, two important second messengers.
➢ Gα12 is involved in cell cytoskeleton control via Rho GTPase signalling [46, 47].
In mammals, 5 Gβ and 11 Gγ subunit genes have so far been discovered [48]. The βγ-subunit of a G protein can be comprised of any combinations of a Gβ and Gγ subtype and the specifc combination can influence the effect of the βγ-subunit on downstream effectors. Additionally, the βγ-subunit also affects the structure and behaviour of the Gα subuit [49].
GRKs
GRKs phosphorylate serine and threonine, but not tyrosine, residues of GPCRs C-tails and cytoplasmic loops[50]. Not all GRKs act on the same residues, resulting in different phosphorylation patterns on
20
the GPCR dependent on the GRKs recruited. It has been shown, that phosphorylation of the GPCR, especially its C-tail, is important for arrestin binding (see Arrestin Binding).
There are seven GRKs in humans, GRK1 and 7 are highly expressed in the photoreceptor cells of the eye, phosphorylating rod and cone opsins respectively. GRK 4 is only expressed in the testes. The remaining four GRKs are ubiquitously expressed. GRK2 and 3 were discovered to interact with the β2AR and were initially referred to as β2AR kinases. All GRKs localize to the plasma membrane. GRK2 and 3, bind the G protein βγ-subunit after G protein activation, recruiting them to the plasma membrane. GRK 4 and 6 are anchored at the plasma membrane via a palmitoylation while GRK1 and 7 are prenylated [51]. GRK5 has a unique amphipathic helix that inserts into the plasma membrane.
Figure 3. Table and graphic overview of the seven GRKs found in humans, ordered after the localization and their type of membrane association[52].
Arrestins
Arrestins have been named for their ability to arrest, or stop, G protein signalling [53]. Further research on them has uncovered many additional functions [1-5]. Mammals possess four arrestins, S-arrestin (SAG), arrestin-C (ARR3), β-arrestin-1 (ARRB1) and β-arrestin-2 (ARRB2) [11]. S-arrestin and arrestin-C are also called visual arrestins, as they are expressed mostly in photoreceptor cells of the retina, where they are responsible for the desensitization of rhodopsin and cone opsins. β-arrestin-1 and β-arrestin- 2 are ubiquitously expressed throughout the body and are therefore involved in the desensitization of a wide range of GPCRs (See Figure 4a). The GPCR-arrestin interaction has been utilized to study receptor activity caused by ligand binding, in both basic research and the pharmaceutical industry and has been proven to be useful for high-throughput screening of GPCR ligands [6-10].
21 Figure 4. A) Graphic overview of the four human arrestins, showing the tissues they are prevalently expressed in, as well as their main GPCR binding partner B) overview of receptor desensitization and endosomal trafficking depending on the GPCRs arrestin recruitment type. Image adapted from Kendall et al (2009) [54].
Structure
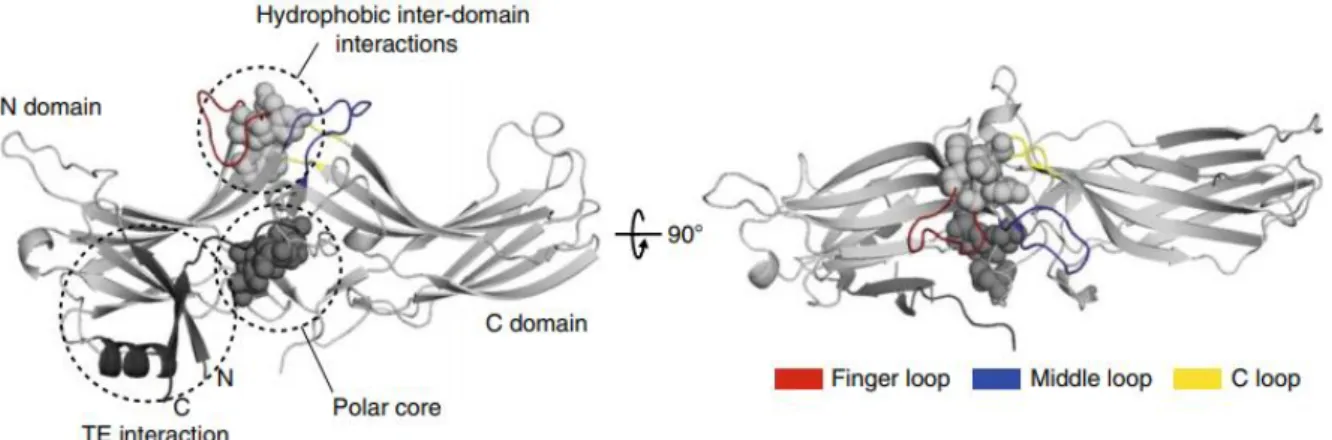
All arrestins share a common basic structure consisting of two domains. Each domain is composed of a β-barrel sandwich. The two domains are called N and C domain.
The N-domain harbours the three-element interaction. It is composed of the first β-strand of arrestin, the only α-helix of the protein as well as arrestin’s C-tail that is folded over the N-domain. The C- domain contains the C-edge composed of several hydrophobic residues that insert into the plasma membrane. It also contains a receptor-binding element that seems to be important for receptor selectivity [55]. The C-tail of arrestin contains several important binding sites for downstream effectors involved in ERK signalling or the endocytic pathway.
The interface where the two domains interact forms two important regions. The central crest, a series of flexible loops which function as the main interaction surface of arrestin with GPCRs. The most prominent loop is the finger loop, that binds the GPCR in the same binding pocket as the α-subunit of G proteins. The other two loops contained in the central crest, the middle and C-loop, both interact with the cytosolic side of GPCRs. The other region is the polar core of arrestin that is composed of five charged residues forming salt bridges connecting the N and C-domain.
22
Figure 5. Structure of arrestin in its inactive state. The three-element interaction, polar core, and hydrophobic inter-domain interactions are highlighted; left: side view; right: top view (PDB 1CF1).
Image adapted from Sente et al (2018) [56].
S-Arrestin
(alternative names: 48 kDa protein, arrestin-1, retinal S-antigen)
S-arrestin is -at least structurally- the best studied member of the arrestin family. Its structure in complex with rhodopsin has been solved and is used as a template to understand the interaction of other arrestins [57, 58]. It is mainly expressed in the retina in rod photoreceptor cells, where it is responsible for the desensitization of rhodopsin. It was the first arrestin to be discovered as the causing agent of an autoimmune disease of the eye called uveitis. Therefore, it was named S-antigen. It was further referred to as 48kDa protein, referring to its molecular weight. Its biological function, the desensitization of active opsin signalling, was later discovered. Upon activation of a photoreceptor cell, S-arrestin is translocated to the rod cell’s outer segments where it turns off the transducin signalling started by active rhodopsin. Transducin is a member of the Gi G protein family specific for active opsins. S-arrestin selectively binds to light activated, GRK1 phosphorylated rhodopsin (Rho*-P) and is essential for the proper electrophysiological function of rod photoreceptor cells[59]. S-arrestin -/- knockout mice suffer from retinal degeneration even if they are never exposed to light.
Arrestin-C
(alternative names: arrestin-3, arrestin-4, cone arrestin, X-arrestin)
Arrestin-C is the least studied member of the arrestin family, as it is only expressed in cone photoreceptors and pinealocytes. Despite its similar function it is not closely related to S-arrestin. In fact, it shares around 58% sequence identity with both β-arrestins-2 and S-arrestin. Therefore, the grouping of the two visual arrestins is misleading as they differ as much from each other as from the
23 β-arrestins. It also shows an intermediate receptor selectivity, not being as specialized as S-arrestin but not as promiscuous as β-arrestin-2[60]. Arrestin-C binds cone-opsins with high affinity but also shows recruitment to non-opsin receptors. It is mainly expressed in M and L-cones and with a reduced level in S-cones. It has been shown to be able to bind to photoactivated cone opsins and be able to partially rescue S-arrestin deficiency when expressed in mouse rod cells [61]. In mouse cone cells, it is co-expressed with S-arrestin but at a 50x lover expression level. S-arrestin can fulfil the same roles as arrestin-C, however it cannot rescue all functions of arrestin-C in cones, where arrestin-C seems to be responsible for fast desensitization of cone opsins. Conversely arrestin-C cannot rescue all functions of S-arrestin in the rods.
Apart from opsins, arrestin-C has been shown to interact with c-Jun N-terminal kinase and E3 ubiquitin ligases which act as scaffolds in receptor trafficking. Its non-opsin interactions and functions in pinealocytes are virtually unexplored.
β-arrestins
Both β-arrestins are expressed in a wide variety of tissues and are responsible for the desensitization and regulation of a vast number of receptors, with numerous functions.
This is facilitated by β-arrestin’s relative promiscuity, as they are able to adopt an active conformation more readily than the more specific S-arrestin[62].
β-arrestins have been considered to be functionally redundant due to their high sequence identity and similar expression pattern [13]. Additionally, they may substitute each other in single knockout mice.
Even after Kohout et al. [13] showed that GPCRs differ in their selectivity for β-arrestins, they were mostly seen as interchangeable and redundant, evidenced by the fact that both β-arrestin-1 and 2 knockout mice are viable. However, their respective phenotypes differed. Among other effects, β- arrestin-1 knockout mice showed increased baseline locomotion and mice lacking β-arrestin-2 showed a lack of morphine-induced respiratory suppression [63, 64]. More recent studies have further hinted that the redundancy of β-arrestins may b e oversimplification and the two proteins do indeed have unique functions. In fact, they can show opposite regulation of different pathways [14-16].
β-arrestin-1
(alternative names: β-arrestin, arrestin-1, arrestin-2)
β-arrestin-1 is specific for Class B GPCRs with only weak binding to class A GPCRs. Class B GPCRs are usually degraded and not recycled back to the plasma membrane, indicating that β-arrestin-1 is involved in receptor degradation. Studies with the δ-opioid receptor have confirmed that [65]. It has
24
numerous interactions with proteins involved in internalization, trafficking and signalling. β-arrestin-1 is overexpressed in joints affected by arthritis and seems to enhance inflammation. It binds and scaffolds to p38 mitogen-activated protein kinases (MAPK) causing activation of 7-p38 MAPK [66, 67].
β-arrestin-2
(alternative name: arrestin-3)
The most promiscuous member of the arrestin family as it binds both class A and B GPCRs with high affinity. Structural studies and cellular assays further revealed that β-arrestin-2 is capable of pre- engaging inactive receptors at the plasma membrane [68]. For the δ-opioid receptor β-arrestin-2 knockout mice showed unaltered internalization, but a delayed resensitization, indicating that β- arrestin-2 is involved in endosomal trafficking and recycling of receptors to the plasma membrane.
This is in line with the observation that class A GPCRs that preferably recruit β-arrestin-2 are often recycled to the membrane instead of degraded [65]. β-arrestin-2 seems to have a protective function in arthritis, by reducing the inflammatory response, and its expression is increased in the affected joint tissue [69].
Alternative splicing variants
There are further arrestin variants found in the human body, created by alternative splicing of the four genes mentioned above:
p44 an S-arrestin variant lacking its C-tail. This causes the form to be pre-activated with the three- element interaction destabilized, eliminating the need for the receptor’s C-tail to bind. Therefore, p44 is able to bind unphosphorylated, light-activated rhodopsin [70]. p44 is expressed in rod cells alongside S-arrestin where it is able to desensitize rhodopsin faster than the full-length variant and contributes to G protein signalling shutoff [71].
β-arrestin-1-B is lacking 8 amino acids due to alternative splicing[72]. It was found to be expressed in human leucocytes, heart, lung, liver and smooth muscle cells[73]. In rat, tissue specific expression of β-arrestin-1-B could be shown in peripheral organs, whereas it was lacking in the central nervous system. Understanding the biological significance of this tissue‐specific splicing pattern awaits further investigation.
25 Arrestin Binding
Arrestins accomplish receptor desensitization - arresting of the G protein pathway- by outcompeting the G protein for its binding site on the cytoplasmic side of the receptor.
Arrestins exist in both an active and inactive form. Typically, binding to an active GPCR causes the conversion from the inactive to the active state. The active form is also able to recruit other effector proteins like ERK1/2 and C-Raf [74] [75].
For the binding of arrestin to an active GPCR, two binding modes have been described in structural studies: One features the GPCR’s C-tail that becomes phosphorylated by GRKs shortly after ligand binding. Arrestin’s N-domain interacts with the flexible C-tail of the receptor. This leaves the transmembrane bundle of the receptor core free allowing other effector molecules like G proteins to bind. This is referred to as “tail-interaction”.
The second more fully engaged mode includes in addition to the tail-interaction a second binding interface. This additional interface is composed of the finger loop of arrestin interacting with the transmembrane core of the receptor. At this site arrestins are in direct competition with G proteins for binding to the GPCR. This binding mode is known to be necessary for receptor desensitization by inhibiting G proteincoupling and referred to as “core-interaction” [76, 77].
Previous studies have shown that either core or tail binding mode is sufficient to recruit arrestin, indicating that neither binding site is essential for binding. [78, 79][103, 129, 130]. The biological function of these two sites are not well understood. Recently a study found that tail binding of arrestin is sufficient to trigger arrestin-dependent signaling pathways and internalization [102]. It was also shown that exchanging the C-tail of B2AR with V2R (a prototypical class B GPCR) changes the B2AR’s behavior to a mimic that of a class B GPCR. This indicates that the tail interaction is important for receptor trafficking and arrestin selectivity, but so far, the differential effects of the different interaction modes are not well understood.
Based on these two binding modes and further structural information available about arrestin in its active and inactive form, the theory of arrestin binding has emerged. There are two hallmark steps in arrestin activation. First the three-element interaction is disrupted causing the release of arrestin’s own C-tail from the N-domain. This release allows the GPCRs C-tail to bind instead, with positive charges on the surface of arrestin stabilizing the interaction with the negatively charged phosphates featured on the GPCRs C-tail. Second, the disruption of the polar core allows a 20° rotation of arrestin’s two domains towards each other. Further structural changes occur especially in the central crest: The
26
finger loop forms an α-helix and inserts itself into the cavity formed by the GPCR’s transmembrane bundle, leading to direct competition with G proteins [56, 80].
The interaction of the phosphates on the receptor’s C-tail with positive charges on arrestin’s N-domain can combinatorically change the conformation of other motifs, like the finger loop, leading to a set of distinct conformations that may lead to distinct functional outcomes[76].
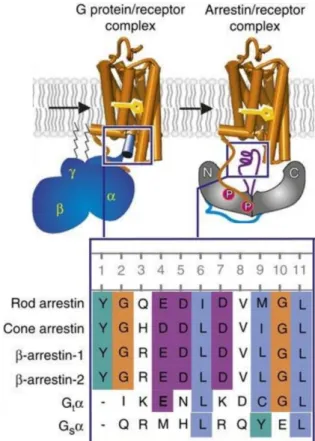
Figure 6. Overview of rhodopsin binding of G proteins and arrestins. Left: Coupling to the G protein.
Right: Arrestin binding to the phosphorylated receptor. Inset (bottom): Sequence alignment of a proposed common sequence motif: rod photoreceptor arrestin (Arr1; residues 68–77), cone photoreceptor arrestin (Arr4; 63–72), β-arrestin-1 (Arr2; 64–73), β-arrestin-2 (Arr3; 65–74), wildtype GtαCT (340–349), Gsα (385–394). Figure adapted from Szczepek et al (2014) [81].
27 Arrestin functions
Arrestin in its active conformation is acting as a scaffold, recruiting a variety of proteins to the receptor- arrestin complex [82]. The main differences between the four human arrestins are found in the C- terminus. β-arrestin-1 and β-arrestin-2 each contain a clathrin and AP2 binding site whereas such a site is lacking in visual arrestins [83]. The C-tail of arrestin is only released and accessible for binding after activation. Therefore, these effectors are only recruited to active GPCR-arrestin complexes [84].
Both clathrin and AP2 are important in the assembly of clathrin coated pits, leading to internalization of the GPCR-arrestin complex [85]. In the visual system, internalization is not required as the opsins in rod and cone cells do not internalize after activation, but are instead digested by the retinal pigment epithelium (discussed more in detail in part 2 of this thesis).
Arrestins also recruit signalling molecules like MAP kinase modules like ERK1/2 JNK and p38. The best studied downstream effect of arrestin is its effect on ERK. Arrestin has been shown to interact with cRAF-1, ERK2 and MEK1, bringing together all components required to trigger ERK on the arrestin scaffold[86]. Further, arrestin recruits E3 ubiquitin ligases, acting as adaptor for the ubiquitination of GPCRs, thereby guiding their degradation [1]. Ligases and arrestins are interdependent: a lack of arrestin reduces ubiquitination of GPCRs and a lack of ligase activity decreases arrestin-mediated endocytosis and MAP kinase signalling [87].
Lastly, arrestin has been shown to interact with tubulin, inositol-6-phosphate and calmodulin, an exhaustive discussion of all these interactions is beyond this thesis, the reader is directed to an excellent review regarding the interactome of arrestins from Peterson and Luttrell "The diverse roles of arrestin scaffolds in g protein–coupled receptor signalling (Peterson, Yuri K. Luttrell, Louis M. [11])".
While it is clear, that arrestins modulate the receptor’s response, it is passionately debated whether arrestins should be classified as signalling molecules, or merely scaffolds with no signalling activity of their own. Several studies [88-90] have proclaimed arrestin dependent (and G protein independent) signalling while other studies have shown a complete absence of arrestin signalling in cells lacking functional G proteins [91]. While this issue may be seen as a technicality, many side-effects of drugs are caused due to their target receptor activating both the arrestin and G protein pathways. Therefore, this distinction has a profound pharmacological impact, especially for drug design.
28
Biased signalling
Arrestins and G proteins cause different downstream effects but originate from the same input: an active GPCR. By competing for the cytoplasmic binding pocket of the receptor, they create a balance between their respective signalling pathways, determined by the specific conformation of the receptor. Mutant receptors have been developed that are unable to trigger G protein signalling while retaining the ability to recruit arrestin [92, 93]. Additionally, siRNA and PKC inhibitor studies tried to dissect the signalling of the Angiotensin II receptor type 1 and showed that arrestin and G protein induced ERK1/2 phosphorylation are independent and additive[94]. In the meantime, the signalling of the µ-opioid receptor has been dissected, showing that its G protein pathway causes analgesia (pain relief), while its arrestin pathway causes respiratory depression and seems responsible for the addictive potential of opioids. Structural studies then revealed, that a receptor can assume many different conformations depending on the ligand provided, resulting in different affinities for different cytosolic binding partners.
The interplay between arrestins and G proteins is central in a receptor's signalling output and therefore on the effect a given drug has on the patient. These findings have become a basis of many new pharmaceuticals, aiming to bias a receptor towards a specific pathway. This concept of pluridimensional efficacy is called biased signalling, meaning that a certain receptor conformation is biased towards binding one downstream partner over the other. An ideally biased ligand would induce a receptor conformation that activates a desired pathway, while simultaneously lacking other undesired interactions, thereby avoiding side effects of conventional ligands.
One key problem when creating biased ligands is that it requires quantitative measurements of several different pathways under the same conditions, making the development process more laborious and raising requirements on cellular assays. Regardless, biased ligands are currently developed for many GPCRs including the μ- and ĸ-opioid receptor, the dopamine d2 receptor and chemokine receptors [95].
Biased signalling is one of the most promising, but also ambitious drug development endeavours, offering the possibility to design an entirely new class of therapeutic agents [96].
29 Luciferases
Luciferases are enzymes capable of producing bioluminescence: the production of light from a living organism. Bioluminescence emerged independently approximately 30 times over the course of evolution in various organisms from unrelated phyla [97-99]. The emission of light might not have been the original function of luciferases. As all luciferases use oxygen to reduce a substrate, their antioxidant properties suggest a detoxification function during the oxygen revolution 2.4 billion years ago [100]. Bioluminescence has a variety of biological functions in different organisms, and is used in predation, camouflage and communication [97]. 80% of all known light-emitting metazoan genera are in deep sea environments where light is sparse. Under these circumstances, bioluminescence is a very advantageous trait [101].
All luciferases produce light by oxidizing a substrate, called luciferin. Luciferin is a generic term for light-emitting compounds. Their products called oxyluciferins are initially produced with some of their electrons in a higher energy state. These electrons then quickly adopt a lower energy level by emitting the excess energy as light. The wavelength of light created depends on the luciferin used and the chemical environment created by the enzyme [102-104]. Some luciferases require additional cofactors like ATP or flavins to function.
Figure 7. Chemical structure of D-Luciferin
The luciferases of the firefly family Lampyridae and the click beetle genus Pyrophorus use D-luciferin (also called firefly or beetle luciferin) as their substrate. These enzymes have evolved from fatty acyl- CoA synthetases involved in the beta-oxidation of fatty acids. These enzymes catalyse a two-step reaction: The first step requires ATP to activate D-luciferin, releasing pyrophosphate. In a second step, O2 attacks the luciferin-AMP complex releasing the AMP and creating oxyluciferin, CO2 and light [105].
The emitted light spectra typically ranges in colour from green to red.
Coelenterazine (CLZN) is mainly used by many marine organisms including the coral Renilla reniformis, the shrimp Oplophorus gracilirostris or the jellyfish Aequorea victoria. Other families using CLZN for bioluminescence include Gaussia, Metridia, Pleuromamma, Lucicutia, Heterorhabdus and Heterostylites [106]. This reaction only requires the luciferin and O2 releasing oxyluciferin and CO2 with no cofactors required. Furthermore, marine bioluminescence differs from the terrestrial in that the predominant light emission colour is blue instead of green [107].
30
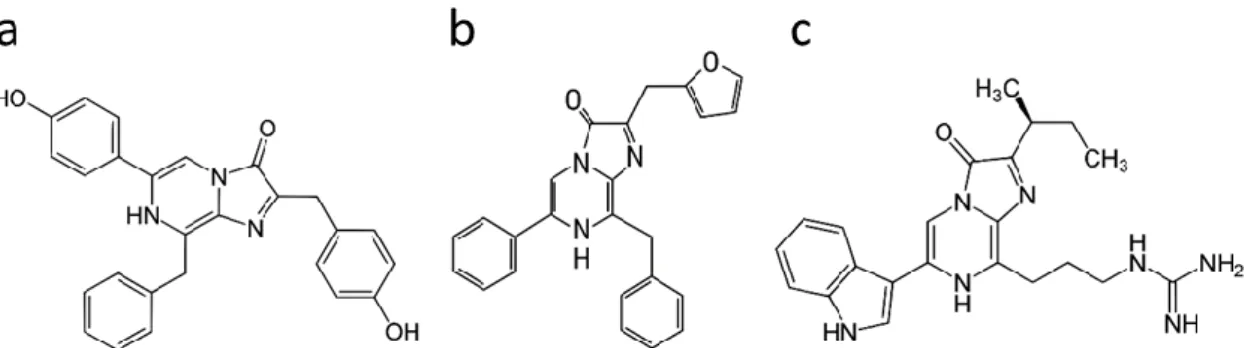
Figure 8. Chemical structures of related luciferins: a) coelenterazine, b) furimazine and c) vargulin.
Nanoluciferase (NanoLuc/Nluc) is an enzyme optimized for biosensor assays based on the Oplophorus gracilirostris luciferase. It can use a CLZN analogue called furimazine to produce light (Fig. 8).
Additionally, the ketone group of furimazine can be protected by esterification, making it more stable in the cellular environment. Endogenous esterases then slowly cleave the ester creating a slow and steady supply of furimazine during the assay [108]. Nluc bioluminescence output is sensitive in a pH- range from 5 to 8, a characteristic which has been used to study GPCR internalisation [109].
Cypridina hilgendorfii, a marine ostracod, uses a luciferin called vargulin. This luciferase uses a similar mechanism to CLZN-based enzymes. Vargulin itself is also similar in structure to CLZN with the same pyrazine-imidazole ring structure at its centre.
Figure 9. Chemical structures of a) 3-hydroxy hispidin (fungal luciferin), and b) Bacterial luciferin (decanal)
Fungi are also able to create bioluminescence also known as foxfire. The ability is common among the genus Armillaria [110]. The fungal luciferin is 3-hydroxyhispidin and is synthesized from hispidin.
Fungal luciferases process it into caffeylpyruvic acid, releasing CO2 and light in the process. It is the first complete pathway from eukaryotes that can be genetically encoded. This allows the creation of autonomously bioluminescent eukaryotes by expression of three transgenes. This might enable applications where tissues or entire organisms report on their physiological state by light emission or even the integration of glowing plants into city infrastructure [111].
Bacterial chemiluminescence is created by the lux operon, encoding both the luciferase and a fatty- acid reductase complex. The complex creates a long-chain aliphatic aldehyde necessary for the
31 reaction. The actual luciferin of the enzyme is a reduced flavin mononucleotide cofactor (FMNH2). The enzyme first oxidizes the cofactor, which then reacts with the aldehyde. The resulting product then decays into a fatty acid, an oxidised cofactor (FNM), water and light [112]. This reaction is useful, as all its components are encoded in a single operon, making it independent of exogenous luciferin. An enhanced version further enables long-term visualization of bacterial viability and behaviour on a single-cell level [113].
There are many lesser-known luciferase systems using different luciferins and chemical mechanisms in dinoflagellates, molluscs (genus latia) and annelida (earthworms, genus diplocardia and fridericia) [106].
After the first luciferase genes were cloned, they were quickly used as reporter systems in a multitude of research areas in life science and medicine. Coelenterazine-using luciferases like Renilla or Gaussia luciferase (Rluc or Gluc respectively) have various biomedical applications, including in vivo animal models. Several variants of Rluc have been engineered for increased brightness or a red shifted emission spectrum[114, 115]. This makes them useful for in vivo reporter studies as red light penetrates tissue more easily [116]. Firefly luciferase (Fluc) or other D-Luciferin-using luciferases are frequently employed in detection systems for contaminations in drinking water, various food poisoning agents and even tumour diagnostics [117-120]. Because Fluc and Rluc use different substrates with no cross-reactivity and different emission spectra the two luciferases be used in a sequential dual reporter assay [121].
The bacterial lux luminescence operon has been used for the detection of pollutants in water like heavy metals and phenols as well as the detection of pathogens in vivo [122-125].
PCA- and BRET-based assays
Protein fragment complementation assays (PCAs) are a useful method for the detection of protein- protein interactions. They are based on a monomeric enzyme that has been split into two fragments.
Each fragment is inactive on its own, but when the two fragments are brought into close proximity, they can reform the monomeric enzyme and regain their catalytic activity. The enzymatic activity is then detected either directly or indirectly, allowing the measurement of the interaction. This approach has been successful used in basic research as well as drug discovery [126].
32
Many different enzymes have been employed as split reporters. Split β-lactamases or murein dihydrofolate reductase can be used to detect protein interactions [127]. Split Cre-recombinase allows inducible, time controled DNA recombination [128, 129].
Split fluorescence reporters such as GFP or dihydrofolate reductase offer the advantage that they not only detect protein interactions but also determine their localization inside a cell. Their output is also quantifiable, as it scales linearly with enzymatic activity. Split luciferases, currently offer the most information. As their activity can also be quantified, and localized in the cell, but their output can further be detected in real time, making time resolved measurements possible [130]. Such PCAs can be useful for measurements of cellular signalling activity [131]. Additionally, multiplexed PCAs with two competing proteins have been shown to simultaneously compare both β-arrestin types [132].
Various split luciferases have been engineered. Their properties vary, offering different advantages and disadvantages. Fluc is relatively large (61kDa) with a weak luminescence requiring ATP [133]. Click beete luciferases (Bluc) are similar in size but much brighter. Additionally, they allow a dual colour output as the N-terminal fragment of Bluc determines the emission spectra. Therefore, two different N-terminal fragments can be combined with a C-terminal fragment in the same assay, with a different colour of light produced depending on the fragments combined. Rluc is a much smaller luciferase (36 kDa) and provides a similar light intensity as Fluc [134]. Gluc is even smaller (19.9 kDa), while providing a light intensity over 100 times higher than Fluc [135, 136]. Finally, an optimized version of NanoLuc is the smallest luciferase available yet (19.1 kDa) and provides the same light intensity as Gluc [137].
In conclusion, luciferases currently provide one of the best choices for the analysis of PCA assays, by detecting presence, location, dynamics of an interaction [17, 18].
BRET is an alternative to PCA based assays and can also be used to study interactions in live cells, cell lysates and purified proteins in real time [138]. Like FRET, BRET uses the Förster resonance energy transfer to detect the interaction of two proteins. Instead of a fluorophore that is excited by an external light source, the donor in BRET is a luciferase that produces light by its chemical reaction. This light is then transferred as a virtual photon to a nearby fluorescent acceptor that emits the light back in a different wavelength. The ratio between the light emitted by the luciferase and the light emitted by the donor acts as an indication of their interaction. The key advantage of BRET is that no external light source is needed. This avoids phototoxicity and reduces bleaching of the chromophores, however an exogenous luciferin must be provided. BRET has been used to study GPCR behaviour in mammalian cells, for example their homo- and heterotopic oligomerization of GPCRs [139-143]. In recent years, it became feasible to measure recruitment of G proteins and arrestin with BRET- and PCA-based assays
33 [143-147]. This is the most direct way to measure ligand-induced conformational changes and is therefore an excellent strategy to compare different ligands [148].
Baculoviral Vectors
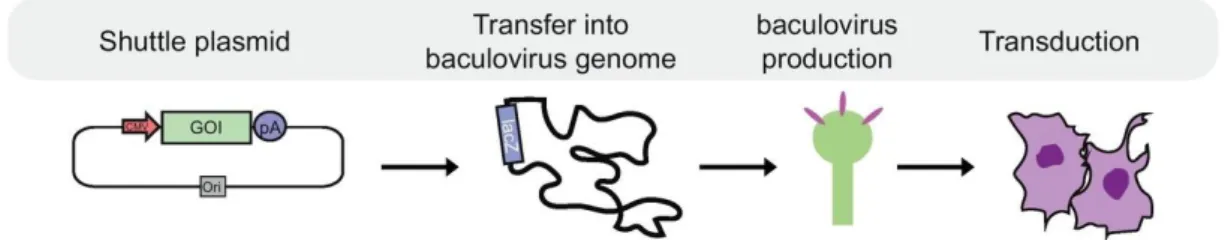
Figure 10. Schematic workflow of baculovirus production. A plasmid containing genes of interest is first transferred into a baculoviral genome using a Tn7 transposon. Then the bacmid is transferred into insect cells to produce a stock of viroids that can be used to transduce mammalian cells. Image adapted from Mansouri et. al 2018[149].
To express foreign genes in a cell, DNA or RNA encoding that gene must be introduced into the cytoplasm of the cell. This can be done by transfection where a chemical, usually a positively charged polymer, is used to bind to the DNA that enables uptake of the DNA via endocytosis [150].
However, the number of cell types that can be transiently transfected is limited. Especially primary cells can typically not be transfected efficiently. In order to express reporter genes in these types of cells, viral vectors can be used. Viruses are naturally able to deliver genetic cargo into mammalian cells, this ability can be used to express genes of interest. Additionally, viruses achieve a higher efficiency of transduction than transient transfection, successfully infecting up to 100% of cells in a culture.
To insert a gene of interest into a viral genome it needs to be flanked by sequences recognized by viral proteins, directing it to be packaged into viral particles (virions). Different viral vectors have been developed to infect mammalian cells, in this study, a baculovirus-based system was used.
Baculoviruses have the advantage of allowing for a very large genetic cargo (> 50kbp) enabling the delivery of several reporter genes within the same viroid. They do not integrate into the host cell’s genome and cannot replicate in mammalian cells, making them safe to work with requiring only biosafety level 1 [149].
Baculoviruses are produced and replicated in insect cells which are easy to culture allowing for an efficient production of a large number of virus particles.
34
Results and Discussion
Goal
The goal of part 1 of this thesis was the development and validation of biosensors detecting arrestin recruitment to GPCRs. Two different assay approaches were developed and compared. A complete analysis of the arrestin specificity of three different GPCRs: β2AR, GRPR and SSTR2 in a time resolved manner was performed. The developed biosensors were introduced into multigene expression constructs as well as viral vectors. Further, the effect of the GPCR’s C-tail on arrestin binding was analysed.
Development of arrestin biosensors
In this study, split NanoLuc biosensors based on a published principle were developed and compared [151]. This strategy was expanded by using all four known human arrestins (S-arrestin, arrestin-C, β- arrestin-1 and β-arrestin-2). They were N-terminally fused with the small subunit of the split luciferase
“114”. A flexible, 16 amino acids long serine-glycine linker was used to connect the arrestins N- terminus with the luciferase subunit.
To complement the arrestin probes different GPCRs were C-terminally tagged with the large subunit of the split luciferase “11S”. A 16 amino acids long linker was used to connect the two fusion proteins.
To avoid modifying the GPCRs C-tail, the 11S was fused with a C-terminal prenylation signal called CAAX. This localizes the luciferase subunit to the cytosolic side of the plasma membrane using a hydrophobic polyprenyl chain. A similar setup for BRET sensors was used by Carl W. White [152]. The term CAAX represents the amino acids sequence recognized for prenylation. C stands for cysteine, the thiol group of cysteine acts as the acceptor for the polyprenyl chain. Followed by any two aliphatic amino acids, valine and isoleucine were used in this case. X may be any amino acid, methionine was used for constructs in this study. The exact sequences used can be found in the supplementary of this work.
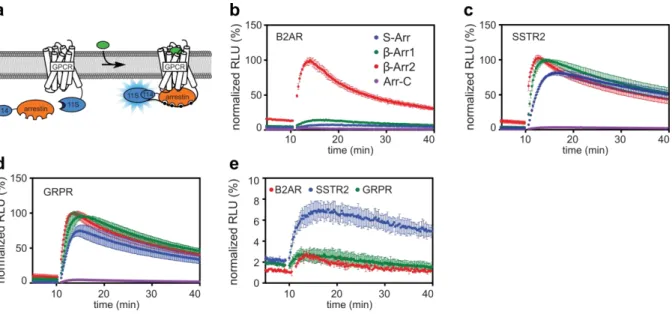
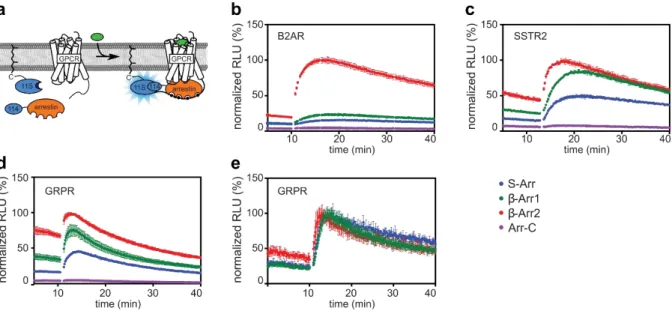
To evaluate the proper function of the biosensors all plasmids using transient transfection of HEK293 cells were tested. 114-tagged arrestin was transfected with either a 11S-tagged GPCR, or an untagged GPCR and 11S-CAAX. Furimazine, the substrate for the luciferase, was added to the cells. Light production of the cells was recorded before and after their stimulation with a respective agonist for the transfected GPCR. As the GPCR is activated by an agonist, its binding affinity to arrestin increases.
When arrestin is recruited, its 114 tag binds the 11S subunit attached to either a tagged GPCR or the CAAX membrane anchor. The complementation of the NanoLuc fragments causes an increase in light
![Figure 3. Table and graphic overview of the seven GRKs found in humans, ordered after the localization and their type of membrane association [52]](https://thumb-eu.123doks.com/thumbv2/1library_info/5480186.1684614/21.892.108.545.410.674/figure-table-graphic-overview-ordered-localization-membrane-association.webp)