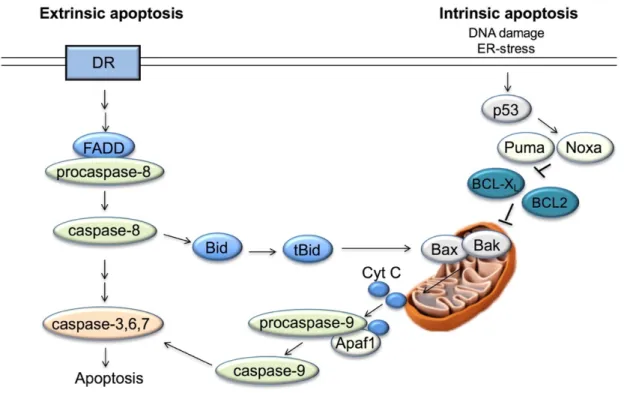
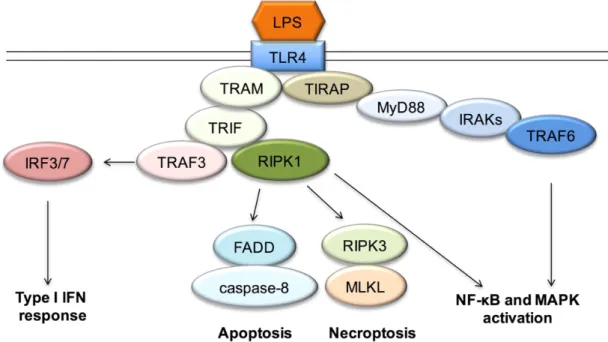
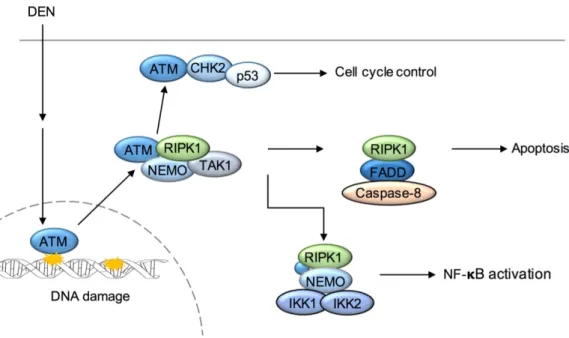
liver injury and cancer
127
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
Abbildung
+7
ÄHNLICHE DOKUMENTE
Improved understanding of the immune cell-mediated mechanisms involved in hepatocyte cell death could be beneficial for the development of common therapeutic strategies
In conclusion, from this current study, by using PrPC knockout mice, we showed a critical role of PrPC in the liver of aging mice by regulating the glucose/lipid metabolism, which
In a sensitivity analysis including only patients with liver imag- ing > 30 days before onset of COVID-19, imaging evidence of hepatic steatosis remained associated with
A New Flavonoid C-Glycoside from Solanum elaeagnifolium with Hepatoprotective and Curative Activities against Paracetamol- Induced Liver Injury in Mice..
There was a signifi cant (P < 0.05) decrease in the protein content in the livers of the paracetamol-treated group compared to the control group, while the groups treated
Taken together this study shows that the liver endothelial layer, mainly LSE Cs, represent a direct target of the cytotoxic effect of paracetamol and that activation of
Synergistic induction of cell death by TRAIL and chemotherapeutics has been described in different tumors cell Iines.6-8,14 Similarly, we have previously reported
Consequently, there is an urgent need for liver models that allow to screen drug candidates for their potential to induce idiosyncratic DILI in the early non- clinical
