Autophagie und Staphylococcus aureus Pathogen-Wirtszell-Interaktion
98
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
(39)
(40)
(41)
(42)
(43)
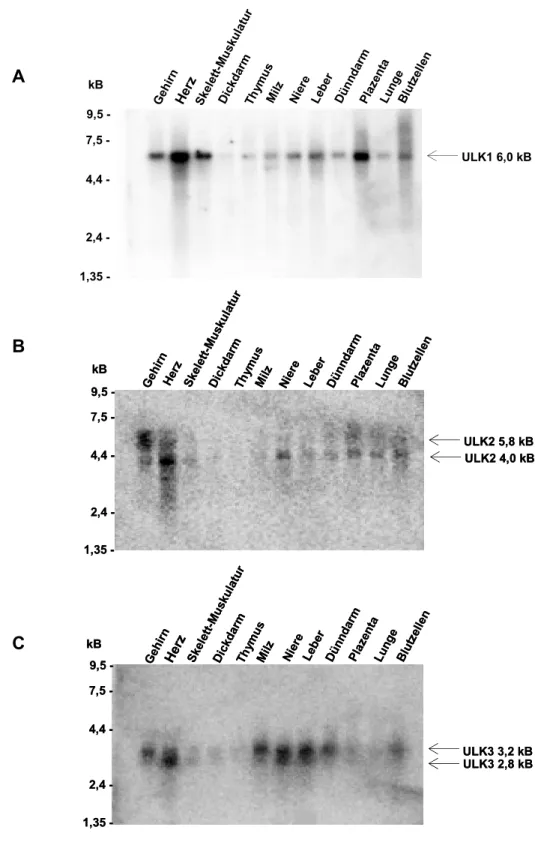
Abbildung
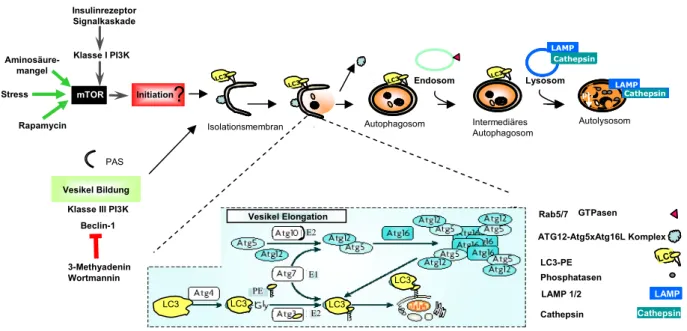
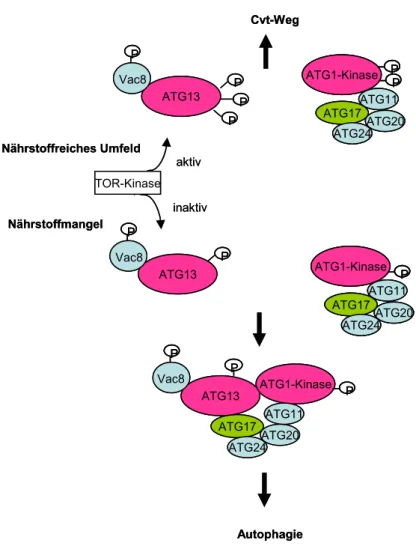

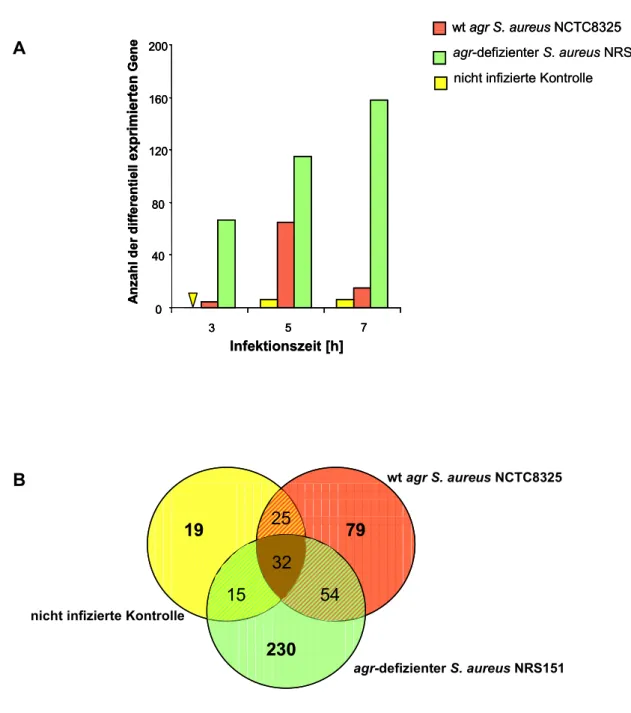
+7



ÄHNLICHE DOKUMENTE
• Proben mit Bronopol oder anderen Hemmstoffen können nicht verwendet werden, da diese eine Anreicherung, wie sie zum Nachweis gemacht wird, verunmöglicht. Die neue
aureus sind in einem Model zusammengefasst dargestellt (Abb.30). Nach dem Eintritt in die Wirtszelle befindet sich S. aureus in einem Rab7- positiven Phagoendosom. Kurz danach kommt
Die Mutation des ribosomalen Proteins L3 betraf zwar eine an der Peptidyltransferase-Reaktion unmittelbar beteiligte Komponente, wie im Fall der 23S-rRNA und der
Wie unter 'Isolierung neutrophiler Granulozyten aus venösem Blut' beschrieben, wurden neutrophile Granulozyten auf eine Konzentration von 5 x 10^6/ml gebracht mit dem
ii) Wie groß sind für diese neuen Preise die kompensierten Nachfragen beim ursprüng- lichen Nutzenniveau aus c)..
aureus starins isolated from 14 catheter asso- ciated infections with the ability to form slime layer based on te presence of the gene icaA, but also icaD.. Indeed, while icaA
Aus 27 Stuhlproben (12,8%) konnte Staphy- lococcus aureus als einziger fakultativ pathogener Keim angezüchtet werden (ausgeschlossen wurden Proben, in denen Salmonellen,
Aim 2: Exploration of antimicrobial and immunomodulatory effects of Gum arabic on human and bovine granulocytes against Staphylococcus aureus and Escherichia
