Magnetic Hybrid Materials based on Co/C Nanoparticles and their Use in Catalysis
122
0
0
Volltext
(2)
(3)
(4)(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(17)
(18)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
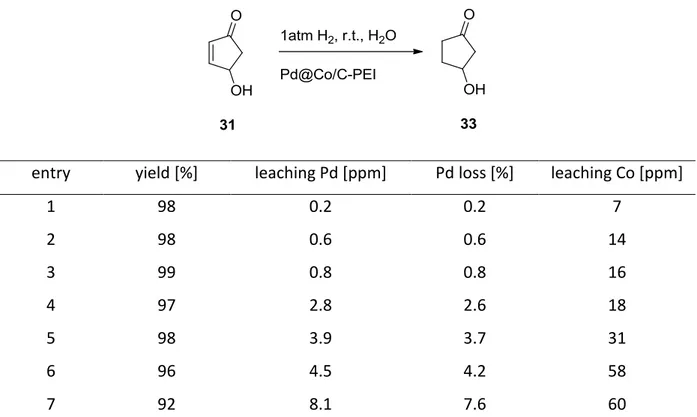
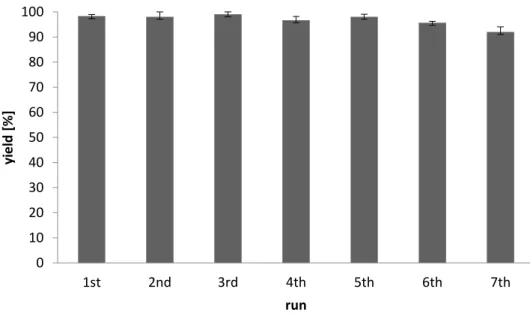
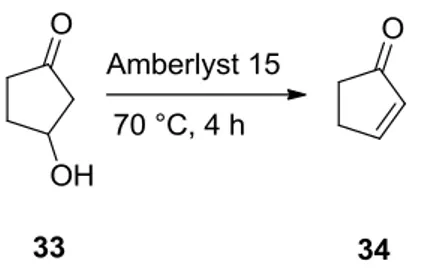
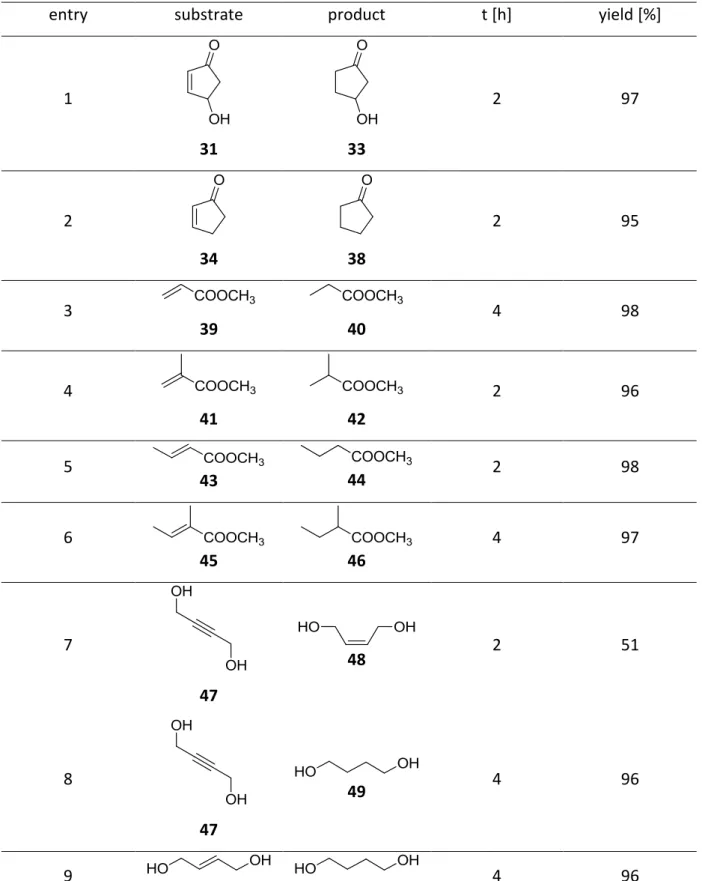
(33)
(36)
(37)
(38)
(39)
(40)
(41)
Abbildung


+7

ÄHNLICHE DOKUMENTE
This thesis describes the potential of hybrid supramolecular systems consisting of streptavidin and biotinylated piano stool complexes either as anticancer drugs or as
Take a dfRootChip containing Arabidopsis plants (see Procedure G) and place in a biosafety cabinet.. Using a magnifying glass or dissection microscope, ensure that the roots have
A Reusable Mesoporous Nickel Nanocomposite Catalyst for the Selective Hydrogenation of Nitroarenes in the Presence of Sensitive Functional Groups.. Robust Microporous Monoliths
amines. 89 Following this success, catalytic activity of manganese complexes in BH/HA reactions, particularly in N-alkylation of amines, was further explored by
1) The generation of mesoporous PD-SiCN materials utilizing a novel PS particle based one-pot synthesis route. 2) The transformation of the mesoporous SiCN material into a
We presented a method for the synthesis of colloidal stable spherical polymer brushes with a zwitterionic brush layer of pMEDSAH chains.. The extension of the shell can be influenced
In addition, mass degree of swelling (Q w,print in weight percentage) was calculated according to equation 1, where m final is the mass of the material after immersion and m
This work deals with the development of new semi-synthetic catalysts based on clay and oxides, the characterization of these catalysts obtained and finally their use in
