Isolation und Analyse der Mutation thick tassel dwarf1 aus Zea mays
91
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
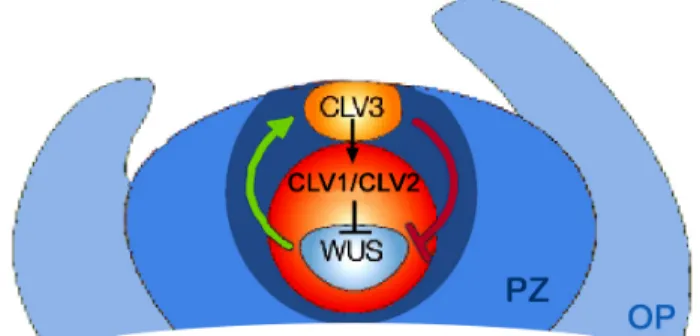
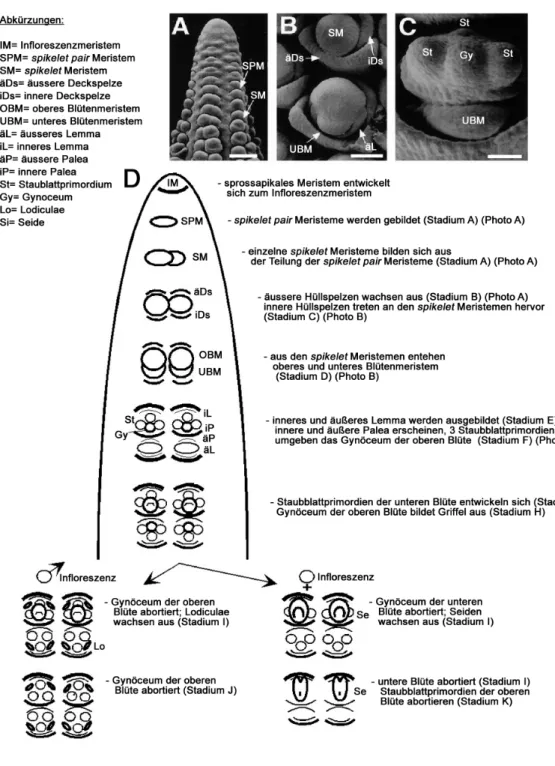
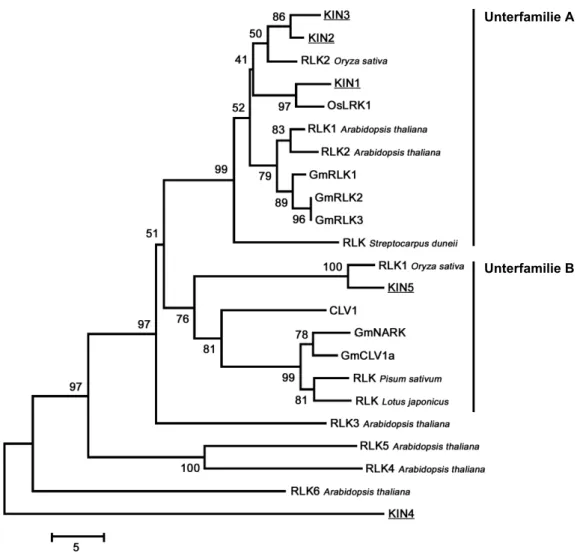
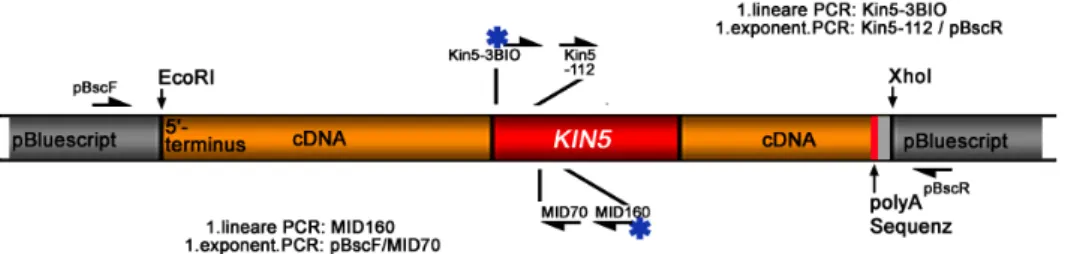
Abbildung


+7
ÄHNLICHE DOKUMENTE
Die Bestimmung der aktiven anorganischen Phosphorsäure wurde nach der Ammoniummo- lybdat-Benzidin Methode nach Tserling ausgeführt und das Kalium mit
Gleichzeitig konnte nachgewiesen werden, dass Zellen durch die verstärkte Palmitoylierung von BAX gegenüber einem Apoptose-Stimulus sensibilisiert wurden und diese mit einer
Die gefundene anti-inflammatorischen Aktivität der hergestellten Derivate zeigte, dass die Substituenten des Aglykons, sowie deren Konfiguration am jeweiligen chiralen
Darüber hinaus ließen sich die nativen HSP70-Proteine mit dem HSP70B-Antikörper in Arabidopsis- Gesamtproteinextrakt nachweisen (Abb. Da HSP70-1 und HSP70-2 zu 95 % homolog
Über das Ganze soll festgelegt werden, mit welchen zeitlichen und finanziellen Prioritäten die anstehenden Reformen angegangen werden, damit eine hohe Qualität und
Politik, Wirtschaft und Zivilgesellschaft haben die besondere Rolle von Frauen* und Mädchen* in ihren Initiativen und politischen Debatten zur Vermeidung von
Die Tatsache, daß nach der stabilen Transformation von unreifen Maisembryonen mit dem Barnase-Gen unter der Kontrolle des ZmMADS2- Promotors keine transgenen