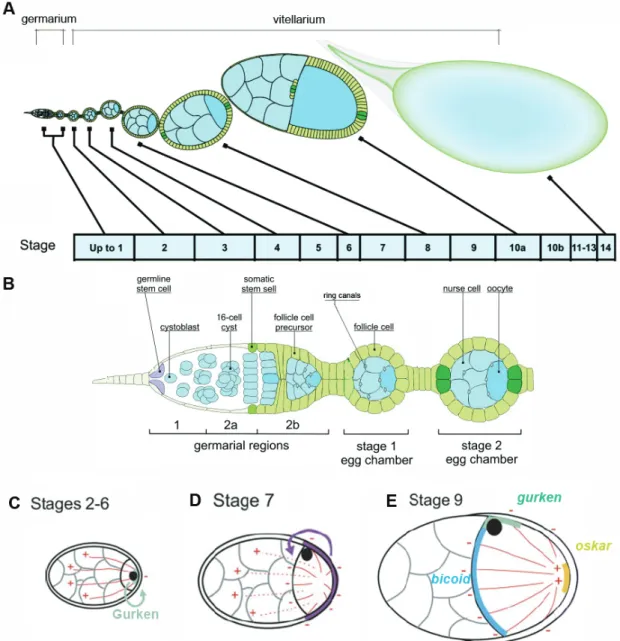
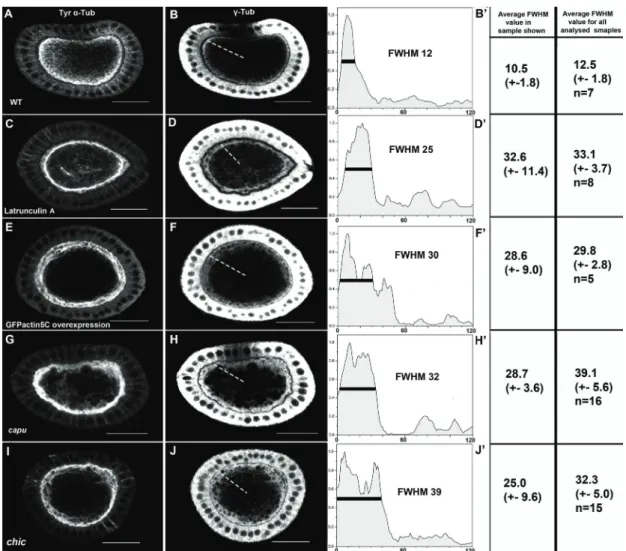
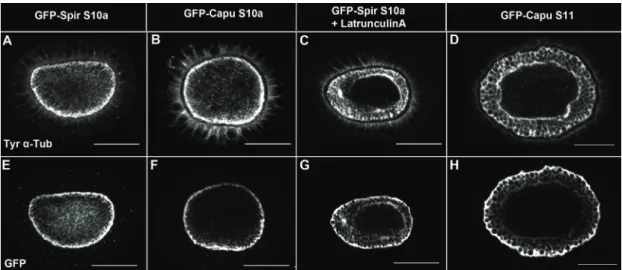
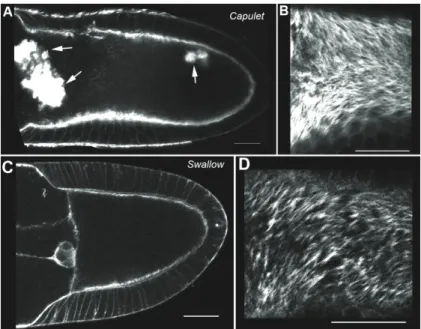
Organisation of the cytoskeleton of the Drosophila oocyte
121
0
0
Volltext
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
Abbildung



+7


ÄHNLICHE DOKUMENTE
The formation of a modern police force remained restricted to
David Evans identified the World Health Assembly (WHA) resolution on sustainable health financing which was initiated by Germany as a starting point for revisiting health
After this study, other research projects were held, about the Portuguese case (“New production models in the Portuguese industry”, and “Organization of
In the context of the Chinese vector in the activities of the Shanghai Cooperation Organisation, this trend is expressed in consistent investment in the export of products
The foundations of the “public organisation”: governance failure and the problem of external effects
Then, because we are concerned with the total effects of strategic control, we suggest that an efficient governance structure must minimise the sum of the costs of organising
A legal frame for co-operation to assure the protection of water and ecological resources and their sustainable use in the Danube River Basin. signed: 29 June
37 Малиновский Б. Антология исследований культуры.. В целом данный подход согласуется с направленностью развития материи от низших ее форм к
In the present study, we investigated the modular organisation of a standing balance task on stable and unstable ground in order to improve our understanding of the
