Tumorantigen-Specific CD40B Cells: Combining Enhanced Antigen-Presentation and Antibody-Secretion for Tumor Targeting
171
0
0
Volltext
(2)
(3)(4)(5)(6)(7)
(8)(9)
(10)(11)
(12)(13)
(14)
(15)
(16)
(17)
(18)(19)(20)(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
(39)
(40)
(41)(42)(43)
(44)
(45)
(46)
Abbildung
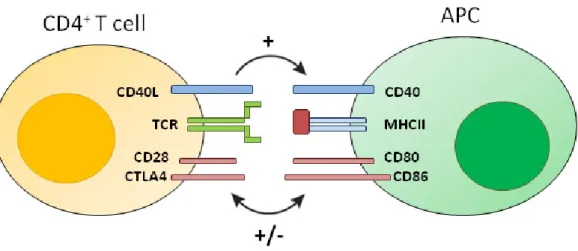
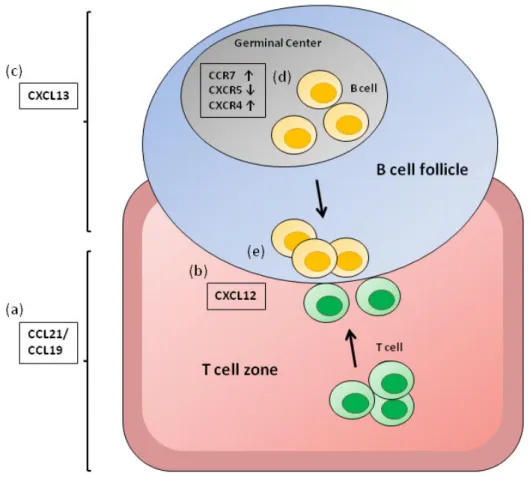
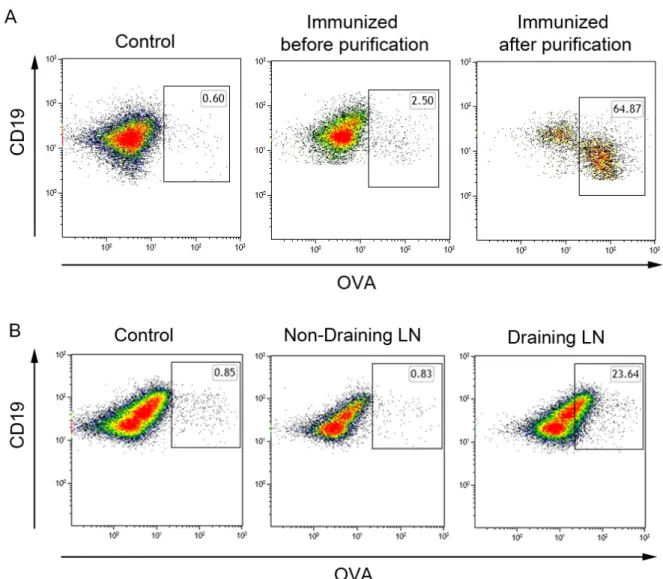
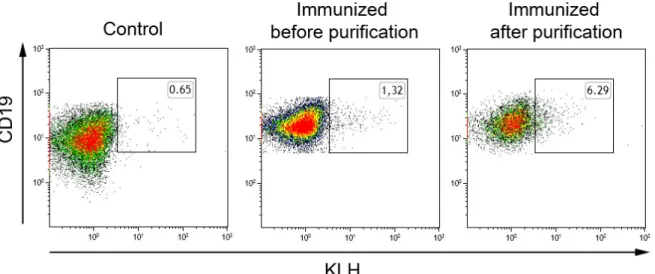
+7
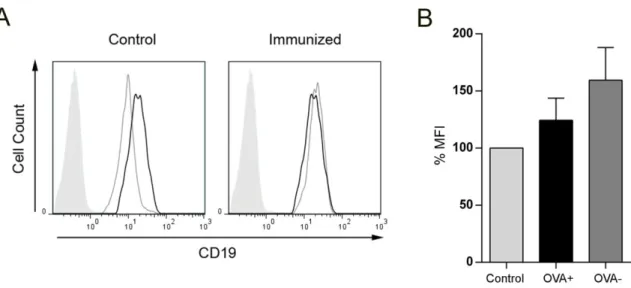
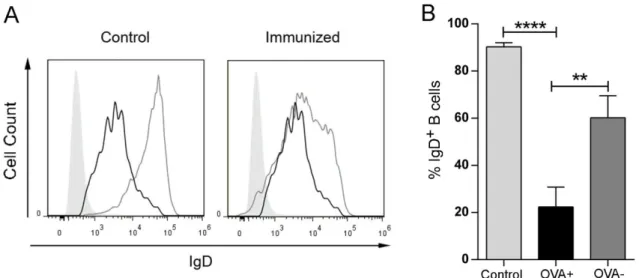
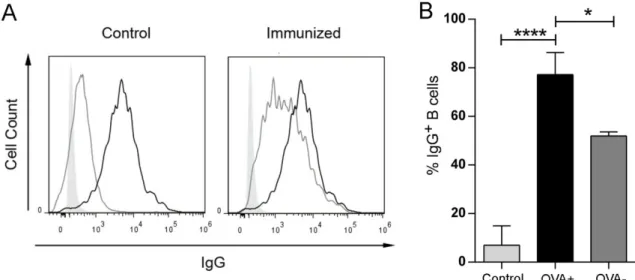
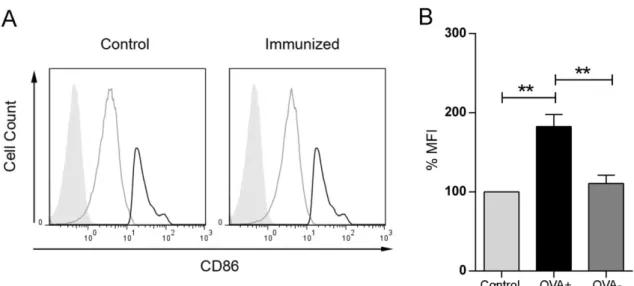
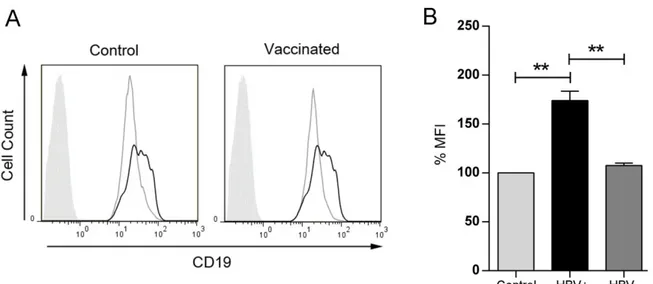
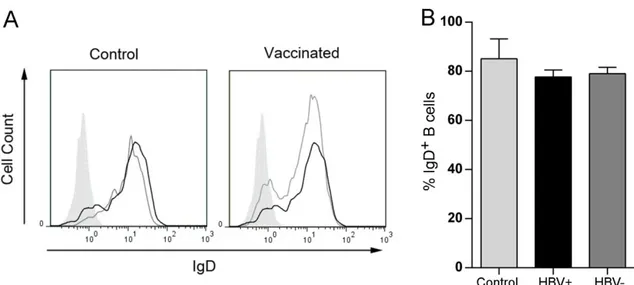
ÄHNLICHE DOKUMENTE
RESULTS: We show for the first time that EC present a quantitatively different peptide repertoire with abundance of certain peptides, compared with leukocytes. The abundance of
Among the CD1 family, CD1e is the only soluble protein, thus supporting a chaperone-like rather than an antigen-presenting function. Furthermore, CD1e is never
and the enormous sensitivity of chromium release Measuring activation of hybridomas started at 10 27 – assays are not ideal prerequisites for monitoring the 10 28 M concentrations
We did not detect secretion of suppressive cytokines (such as IL-4, IL- 10 or TGF-~) from CDS' T cells primed by LSEC that could ex- plain the Joss of specific T-cell
Minami et al. not only found BAG6 associated with chemically induced DRiPs, they
To analyze the influence of BAG6 on MHC class I surface expression, the murine fibroblast cell line B8-D b (H- 2 d + H-2D b ) and HeLa cells were subjected to BAG6
To exclude that the reduced number of virus-specific T cells in Bis VIII–treated mice at day 7 after viral infection was due to inhibition of T cell activation and reduced expansion,
and our phase I trial in cervical cancer patients [25], has demonstrated the efficacy of autologous tumor lysate pulsed DCs (TLDCs), as a next step after optimizing the rhSPAG9
