AUS DEM LEHRSTUHL FÜR KINDER- UND JUGENDMEDIZIN
PROF. DR. MICHAEL MELTER DER FAKULTÄT FÜR MEDIZIN DER UNIVERSITÄT REGENSBURG
CHOLESTATISCHE ERKRANKUNGEN IM FRÜHEN KINDESALTER: ANALYSE DER EXPRESSION VON HEPATOBILIÄREN TRANSPORTERN UND
NUKLEÄREN REZEPTOREN
Inaugural - Dissertation zur Erlangung des Doktorgrades
der Medizin
der
Fakultät für Medizin der Universität Regensburg
vorgelegt von Victoria Cisar
2019
AUS DEM LEHRSTUHL FÜR KINDER- UND JUGENDMEDIZIN
PROF. DR. MICHAEL MELTER DER FAKULTÄT FÜR MEDIZIN DER UNIVERSITÄT REGENSBURG
CHOLESTATISCHE ERKRANKUNGEN IM FRÜHEN KINDESALTER: ANALYSE DER EXPRESSION VON HEPATOBILIÄREN TRANSPORTERN UND
NUKLEÄREN REZEPTOREN
Inaugural - Dissertation zur Erlangung des Doktorgrades
der Medizin
der
Fakultät für Medizin der Universität Regensburg
vorgelegt von Victoria Cisar
2019
Dekan: Prof. Dr. Dirk Hellwig 1. Berichterstatter: Prof. Dr. Thomas Weiß 2. Berichterstatter: Prof. Dr. Christa Büchler Tag der mündlichen Prüfung: 08.10.2019
3
Inhalt
1. Einleitung ... 7
1.1. Die Leber als größtes Stoffwechselorgan des Menschen ... 7
1.2. Gallebildung und –sekretion ... 8
1.2.1. Gallensäurebildung ... 8
1.2.2. Bilirubinstoffwechsel ... 9
1.2.3. Exportsysteme in Hepatozyten und Cholangiozyten ... 9
1.2.4. Enterohepatischer Kreislauf ... 11
1.2.5. Biotransformation in der Leber ... 12
1.2.6. Nukleäre Rezeptoren ... 12
1.3. Cholestase ... 13
1.3.1. Obstruktive Ursachen ... 14
1.3.2. Hepatozelluläre Ursachen ... 16
1.3.3. Pruritus als Symptom der Cholestase ... 17
1.4. Aktueller Stand der Forschung ... 18
2. Fragestellung der Arbeit ... 21
3. Material und Methoden ... 22
3.1. Kindergewebeproben ... 22
3.2. Datenerhebung zu den Gewebeproben ... 23
3.2.1. Daten aus SAP ... 23
3.2.2. Fragebogen an die Familien ... 25
3.2.3. Daten der klinischen Chemie ... 26
3.3. RNA-Isolation ... 26
3.4. Qualitätskontrolle der totalen RNA ... 28
3.5. Genexpressionsmessung ... 30
3.6. Statistische Auswertung ... 33
4. Ergebnisse ... 36
4.1. Charakterisierung der Gewebeproben ... 36
4.1.1. Klinische Parameter ... 36
4.1.2. Histopathologische Parameter ... 38
4.1.3. Laborchemische Parameter ... 39
4
4.1.4. Cholestatische Symptome ... 42
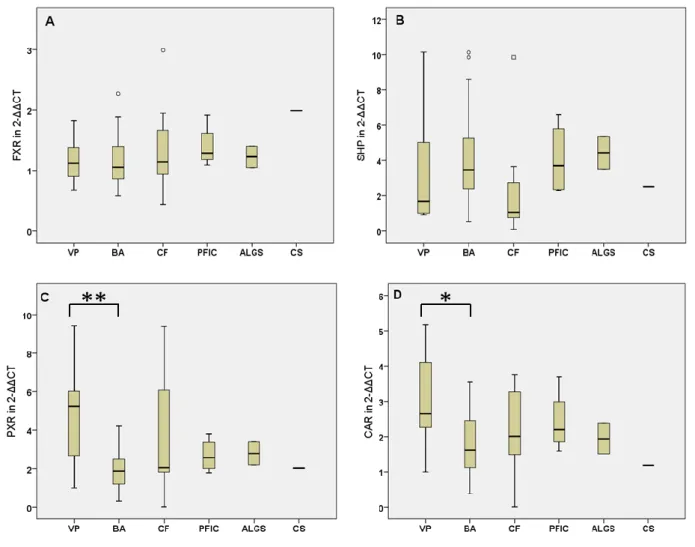
4.2. Expression hepatobiliärer Transporter und nukleärer Rezeptoren in cholestatischen Gewebeproben und Vergleichsproben ... 44
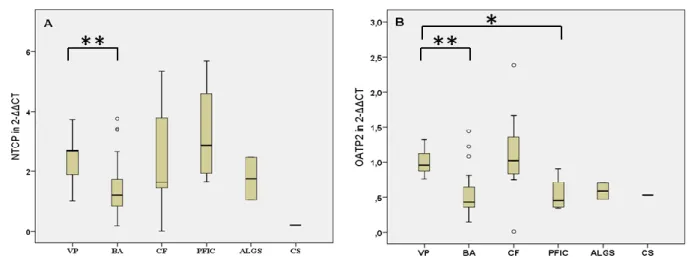
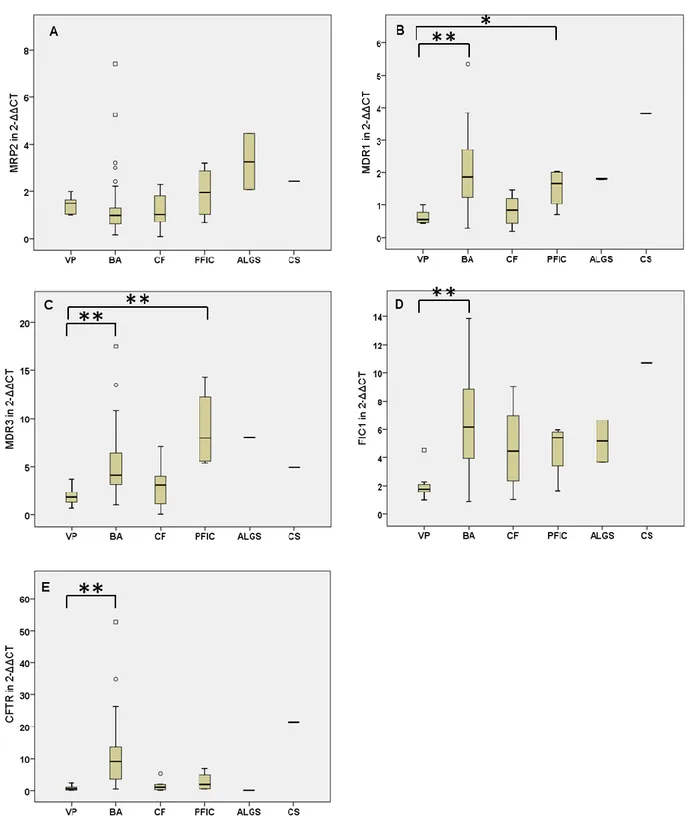
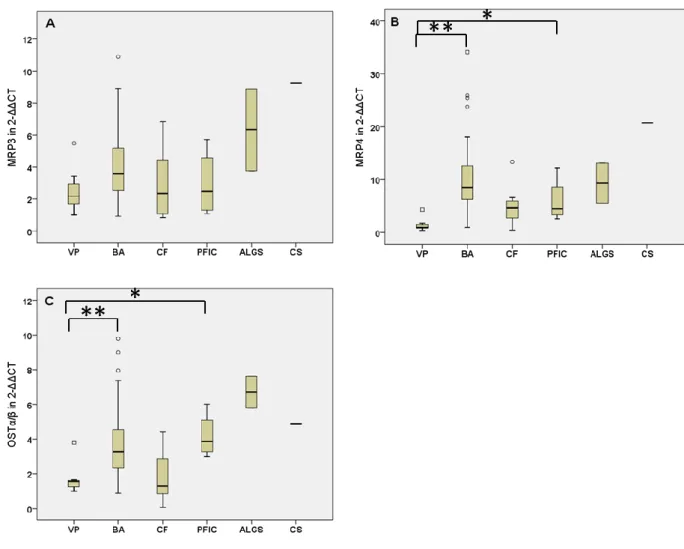
4.2.1. Basolaterale Aufnahmetransporter ... 45
4.2.2. Kanalikuläre Effluxmechanismen und Cholangiozytentransporter ... 45
4.2.3. Basolaterale Effluxmechanismen ... 46
4.2.4. Nukleäre Rezeptoren ... 48
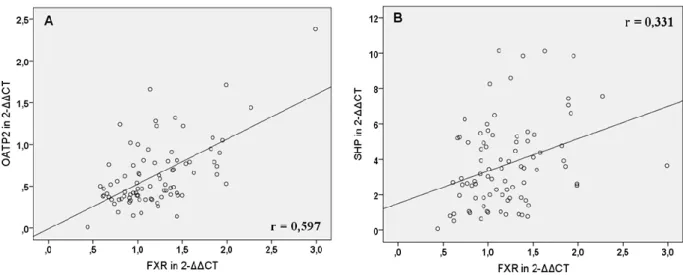
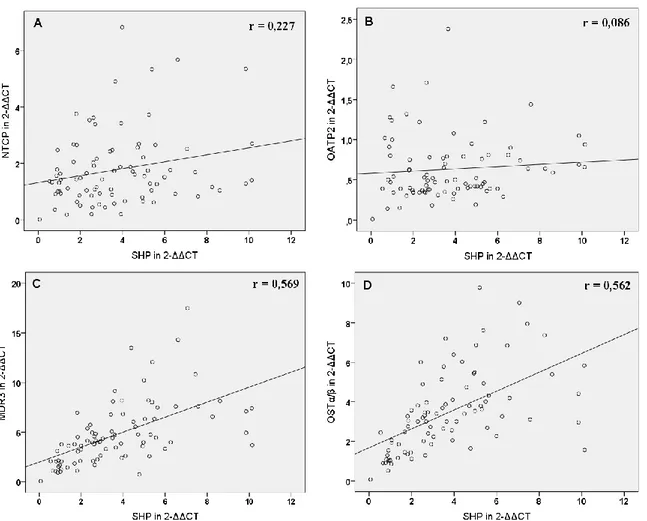
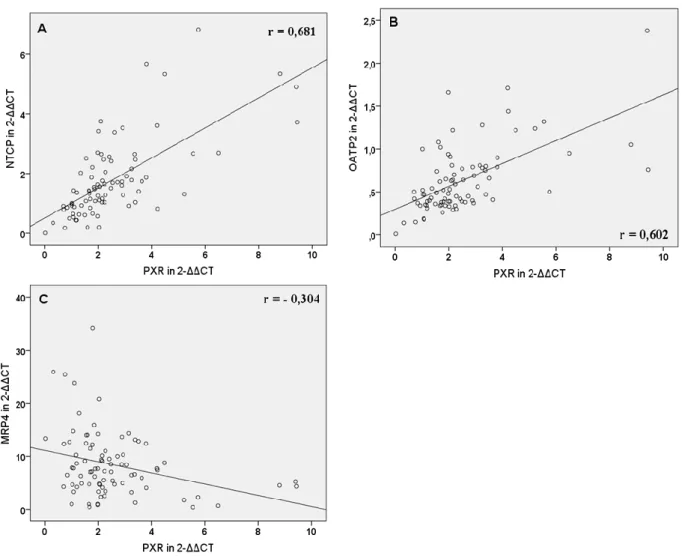
4.3. Korrelation der mRNA-Expression von nukleären Rezeptoren und hepatobiliären Transportern und mögliche regulatorische Zusammenhänge ... 49
4.4. Korrelation von laborchemischen, histopathologischen und klinischen Parametern mit der mRNA-Expression der hepatobiliären Transporter und nukleären Rezeptoren ... 53
4.4.1. Laborchemische Parameter ... 53
4.4.2. Histologische Parameter ... 56
4.4.3. Klinische Parameter ... 63
4.5. Einfluss herkömmlicher Antipruritus-Medikamente auf die mRNA-Expression der hepatobiliären Transporter und nukleären Rezeptoren... 67
5. Diskussion ... 71
5.1. Charakterisierung der Gewebeproben ... 71
5.2. Expression hepatobiliärer Transporter und nukleärer Rezeptoren in cholestatischen Gewebeproben und Vergleichsproben ... 77
5.3. Korrelation der mRNA-Expression hepatobiliärer Transporter mit laborchemischen Parametern und mit der mRNA-Expression nukleärer Rezeptoren ... 82
5.4. Korrelation von histopathologischen und klinischen Parametern mit der mRNA- Expression der hepatobiliären Transporter und nukleären Rezeptoren ... 86
5.5. Einfluss herkömmlicher Antipruritus-Medikamente auf die mRNA-Expression der hepatobiliären Transporter und nukleären Rezeptoren... 92
6. Zusammenfassung ... 95
7. Literaturverzeichnis ... 98
8. Abbildungsverzeichnis ... 102
9. Tabellenverzeichnis ... 104
10. Anhang ... 105
11. Danksagung... 119
5
Abkürzungsverzeichnis
A. Arteria/ Arterie
ABC ATP-Binding-Cassette
ALGS Alagille-Syndrom
AP Alkalische Phosphatase
ATP Adenosintriphosphat
BA Biliäre Atresie/ Extrahepatische Gallengangatresie
Bili Bilirubin
BSEP Bile-Salt-Export-Pump
CAR Constitutive-Androstane-Rezeptor
cDNA complementary-DNA
CF Cystische Fibrose
CFLD Cystic Fibrosis-associated Liver Disease
CFTR Cystic-Fibrosis-Transmembrane-Conductance-Regulator
CHE Cholinesterase
CK Cholestatisches Kollektiv
CRP C-reaktives Protein
CS Caroli-Syndrom
CT Treshold-Cycle
CYP Cytochrom-P450-Enzym
DNA Desoxyribonukleinsäure
FIC1 Familial-Intrahepatic-Cholestasis-Protein 1
FXR Farnesoid-X-Rezeptor
GLDH Glutamatdehydrogenase
GOT Glutamat-Oxalacetat-Transaminase GPT Glutamat-Pyruvat-Transaminase
GS Gallensäure(n)
HPRT Hypoxanthin-Guanin-Phosphoribosyltransferase
LDH Laktatdehydrogenase
MDR Multiple-Drug-Resistance-Protein
miRNA micro-RNA
mM Millimolar
mRNA messenger-RNA
MRP Multidrug-Resistance-Related-Protein
6
NR Nukleäre(r) Rezeptor(en)
NTCP Natrium-Taurocholate-Cotransporting-Polypeptide OATP Organic-Anion-Transporting-Polypeptide
OST Organic-Solute-Transporter
PB Phenobarbital
PBC Primär Biliäre Zirrhose/ Cholangitis
PFIC Progressive Familiäre Intrahepatische Cholestase
PXR Pregnane-X-Rezeptor
qRT-PCR quantitative Real-Time Polymerasekettenreaktion
RIN RNA Integrity Number
RMP Rifampicin
RNA Ribonukleinsäure
SD Standardabweichung
SHP Small-Heterodimer-Partner SULT Sulfotransferase
TPN Totale Parenterale Nutrition/ Ernährung
UDCA Ursodeoxycholsäure
UDP Uridindiphosphat
UGT UDP-Glucuronyltransferase
V. Vena/ Vene
VLDL Very-Low-Density-Lipoprotein
VP Vergleichsproben/ Kontrollproben/ Normalgewebeproben γGT Gamma-Glutamyl-Transferase
7
1. Einleitung
1.1. Die Leber als größtes Stoffwechselorgan des Menschen
Die Leber hat als größtes inneres Organ des Menschen ihren Sitz im rechten Oberbauch und ist beim gesunden Menschen fast vollständig vom Brustkorb verdeckt (1). Als zentrales Stoff- wechselorgan des Körpers kommen ihr lebenswichtige Synthese-, Abbau- und Entgiftungs- funktionen zu (2). Aufgrund ihrer daraus resultierenden, außergewöhnlichen Stoffwechsel- intensität fließt rund ¼ des Herzminutenvolumens durch die Leber (3). Sie erhält eine doppelte Blutversorgung aus zwei Gefäßsystemen: 25% des Blutes gelangt aus dem systemischen Kreislauf über die Arteria (A.) hepatica zur Leber und ist daher sauerstoffreich, während die restlichen 75% des Blutes der Leber über die Vena (V.) portae zugeführt werden.
Diese drainiert venöses, nährstoffreiches, jedoch sauerstoffarmes Blut aus dem Gastrointestinaltrakt, einschließlich Pankreas, Milz und dem abdominellen Fettgewebe zur Leber (3, 4). Auf diese Weise unterliegen fast alle aus der Nahrung resorbierten Bestandteile einem First-Pass-Mechanismus durch die Leber, bevor sie dem Systemkreislauf zugeführt werden. Verantwortlich für nahezu alle Stoffwechselprozesse sind die Hepatozyten, die in der Leber ein Trabekelwerk bilden. Mit ihrer basolateralen Seite grenzen sie an die Sinusoide, in die das Blut aus der A. hepatica und der V. portae mündet, während ihre apikale Seite die blind endenden Gallekanalikuli umgibt (4).
Als wichtigstes Stoffwechselorgan erfüllt die Leber zahlreiche Aufgaben im Intermediär- stoffwechsel aller Makronährstoffe (4). So hält sie einen konstanten Blutglukosespiegel aufrecht, indem sie überschüssige Glukose als Glykogen speichert. Aus diesem Notfalldepot und durch Gluconeogenese kann zwischen den Mahlzeiten bei Bedarf rasch Glukose ins Blut abgegeben werden (3, 4). Die Leber verwertet außerdem die Triglyceride aus der Nahrung oder synthetisiert sie aus Fettsäuren und gibt diese anschließend in Form von Very-Low- Density-Lipoprotein (VLDL)-Triglyceriden an die Peripherie ab. Übersteigt die Triglyceridproduktion der Leber die Kapazität der VLDL-Produktion, werden Triglyceride in den Hepatozyten eingelagert und eine Fettleber kann entstehen (4). Weiterhin synthetisiert die Leber Cholesterin und baut Überschüsse beider Gruppen ab (1). Der aus dem bakteriellen Stoffwechsel und Aminosäurestoffwechsel entstehende Ammoniak wird im Harnstoffzyklus fixiert und so entgiftet und zahlreiche Plasmaproteine werden von der Leber produziert. So synthetisiert sie Albumin, ein universales Transportprotein und das wichtigste Serumprotein zur Aufrechterhaltung des kolloidosmotischen Drucks. Außerdem synthetisiert sie weitere
8
Transportproteine, Protease-Inhibitoren sowie zahlreiche Komponenten des Blutgerinnungs- und des Komplementsystems (3, 4).
Die Leber ist zudem das wichtigste Entgiftungsorgan des Körpers (1). Viele Fremdstoffe, aber auch körpereigene Stoffe, wie Steroidhormone, Schilddrüsen- und Pankreashormone, werden in der Leber einer Biotransformation unterzogen. Diese besteht aus mehreren enzymatisch katalysierten Reaktionen, die die Funktion haben, ihre Substrate in besser ausscheidbare, meist wasserlösliche Formen umzuwandeln (3, 4).
Zuletzt ist die Leber als exokrine Drüse tätig, indem sie Galle bildet, die sich hauptsächlich aus von ihr synthetisierten Gallensäuren, Phospholipiden, Cholesterin und Bilirubin- Diglucuroniden zusammensetzt. Gallensäuren und Phospholipide sind als Detergenzien wichtig für die Fettverdauung, zudem halten Phospholipide als amphiphile Verbindungen das Cholesterin der Galle in Lösung. Bilirubin wird als Abbauprodukt des Häms in der Leber konjugiert und so für die Ausscheidung wasserlöslich gemacht. Es ist als Gallefarbstoff für die charakteristische gelbe Färbung der Galle verantwortlich (3, 4).
1.2. Gallebildung und –sekretion
Pro Tag werden von der Leber circa 600 ml Primärgalle produziert, auch Lebergalle genannt.
Diese setzt sich zusammen aus 75 Millimolar (mM) Gallensäuren, 25mM Phospholipiden, 10mM Cholesterin, 5mM Bilirubin-Diglucuroniden und anderen Produkten der Biotrans- formation. Durch Rückresorption von Natriumchlorid und darauffolgenden Entzug von Wasser wird die Galle in der Gallenblase auf 1⁄5bis1⁄10ihres Volumens eingedickt, woraufhin sie auch Blasengalle genannt wird (3).
1.2.1. Gallensäurebildung
Gallensäuren werden aus Sterol-Vorstufen gebildet (4). Verschiedene Hydroxylasen wandeln Cholesterol auf dem klassischen Weg zu Cholsäure oder auf dem alternativen Weg zu Chenodesoxycholsäure um, wobei beim Menschen der klassische Weg dominiert (5). Das erste geschwindigkeitsbestimmende Enzym des klassischen Weges ist die Cholesterol-7α- Hydroxylase (Cytochrom-P450-Enzym 7A1 = CYP7A1) (6). Es folgt eine komplexe Abfolge aus 16 verschiedenen Reaktionen, unter anderem Hydroxylierungen am C12 und C27 durch die Enzyme CYP8B1 und CYP27A1, an deren Ende durch eine Konjugationsreaktion mit
9
H2O oder den Aminosäuren Glycin oder Taurin je nach Reaktion Cholsäure oder die konjugierten Gallensäuren Glyko- oder Taurocholsäure entstehen (4, 5, 6).
1.2.2. Bilirubinstoffwechsel
Bilirubin entsteht als Abbauprodukt Häm-haltiger Proteine wie dem Hämoglobin der Erythrozyten. Diese leben im Durchschnitt 120 Tage, bevor sie in Milz, Leber und Knochenmark durch Makrophagen abgebaut werden (3). Täglich entstehen circa 250mg Häm- Gruppen, die nicht weiter verwertet werden können (3, 4). Sie werden durch die Hämoxygenase zu Biliverdin abgebaut, aus welchem durch Reduktion Bilirubin entsteht.
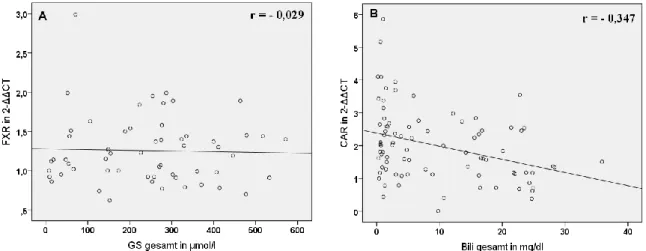
Bilirubin, das durch seine hydrophoben Gruppen an der Außenseite wasserunlöslich ist, wird an Albumin gebunden zur Leber transportiert. Dieses an Albumin gebundene Bilirubin wird auch indirektes Bilirubin genannt (3). Über Transporter für organische Anionen (OATPs) wird Bilirubin in die Hepatozyten aufgenommen und dort durch die Uridindiphosphat (UDP)- Glucuronyltransferase (UGT) zu Bilirubinmono- oder -diglucuronid konjugiert. Das konjugierte Bilirubindiglucuronid wird auch als direktes oder konjugiertes Bilirubin bezeichnet und durch aktiven Transport über den Multidrug-Resistance-Related-Protein 2 (MRP2)-Transporter in die Galle und in geringerem Umfang über MRP3-Transporter ins Blut sezerniert und renal eliminiert (4). Unter physiologischen Bedingungen liegt die Bilirubinkonzentration im Plasma unter 1mg/dl. Ab einer Plasmakonzentration von > 2mg/dl diffundiert Bilirubin ins Interstitium, wo es sich ablagert und die charakteristische gelbe Färbung von Haut und Skleren verursacht (Ikterus). Im Kolon wird das konjugierte Bilirubin durch bakterielle Enzyme deglucuronisiert und in mehreren Reduktionen schließlich zu Stercobilin und Urobilin abgebaut, die für die charakteristische dunkle Farbe des Stuhls verantwortlich sind (3).
1.2.3. Exportsysteme in Hepatozyten und Cholangiozyten
Abbildung 1 gibt einen Überblick über die Transportsysteme in Hepato-, Cholangio- und Enterozyten. Gallensäuren werden, konjugiert an Glycin oder Taurin, über Adenosin- triphosphat (ATP)-abhängige Exportpumpen an der apikalen Membran der Hepatozyten ausgeschieden. Wichtige Transportsysteme sind hierbei der Bile-Salt-Export-Pump (BSEP)- Transporter, der zur Familie der ATP-Binding-Cassette (ABC)-Transporter gehört und eine hohe Affinität für konjugierte Gallensalze besitzt und der MRP2-Transporter. Dieser befördert sowohl glucuronidierte und sulfatierte Gallensäuren als auch organische Anionen wie konjugiertes Bilirubin in die Kanalikuli. Außerdem vermittelt er den Export
10
Abb. 1: Transportsysteme in Hepato-, Cholangio- und Enterozyten des Menschen
NTCP = Natrium-Taurocholate-Cotransporting-Polypeptide, OATP = Organic-Anion-Transporting-Polypeptide, MRP = Multidrug-Resistance-Related-Protein, BSEP = Bile-Salt-Export-Pump, MDR = Multiple-Drug- Resistance-Protein, FIC1 = Familial-Intrahepatic-Cholestasis-Protein 1, CFTR = Cystic-Fibrosis- Transmembrane-Conductance-Regulator. (5)
vieler Medikamente, Toxine und Schwermetalle (5, 7). Phospholipide, vor allem Phosphatidylcholin, werden durch die ATP-abhängige Flippase Multiple-Drug-Resistance- Protein 3 (MDR3) auf die Außenseite der kanalikulären Membran transloziert. Dort bilden sie mit Gallensäuren und Cholesterol Lipidmizellen und schützen die Gallengänge so vor Schäden durch nicht in Mizellen gebundene Gallensäuren (6, 7, 8). In der kanalikulären Hepatozytenmembran befindet sich außerdem das Familial-Intrahepatic-Cholestasis-Protein 1 (FIC1)-Genprodukt, eine ATPase, die vermutlich eine Rolle im Gallensäure- und
11
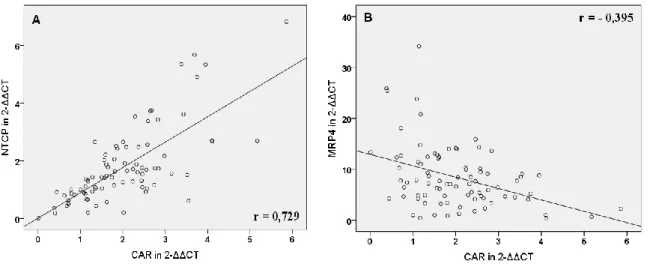
Phospholipidtransport spielt, sowie der MDR1-Transporter. Dieser gehört zur Familie der P- Glykoproteine und sorgt sowohl in den Hepatozyten als auch in der Oberfläche von Enterozyten für den Export zytotoxischer Kationen wie Digoxin oder Cyclosporin in die Gallekanalikuli sowie ins Darmlumen (5, 7, 9). An der basolateralen Membran der Hepatozyten sorgt der Transporter MRP3 für den Efflux organischer Anionen wie Bilirubindiglucuronid, während der MRP4-Transporter und der Organic-Solute-Transporter α/β (OSTα/β) vor allem konjugierte Gallensäuren retrograd ins Blut der Sinusoide sezernieren (5, 8). MRP3, MRP4 und OSTα/β sind unter Normalbedingungen nur schwach exprimiert und stellen bei Cholestase einen alternativen Sekretionsweg bereits wasserlöslicher Komponenten zurück ins Blut dar, woraus diese dann renal eliminiert werden können (6). Über den Cystic- Fibrosis-Transmembrane-Conductance-Regulator (CFTR)-Transporter in der apikalen Cholangiozytenmembran wird Chlorid dem elektrochemischen Gradienten folgend in die Kanalikuli abgegeben und schließlich im Antiport gegen Bicarbonat ausgetauscht. Natrium und Wasser folgen den Anionen (Gallensäuren und Bicarbonat) passiv nach, wodurch das Sekret aus den Kanalikuli ausgetrieben wird (4, 7).
1.2.4. Enterohepatischer Kreislauf
Gallensäuren haben eine Schlüsselfunktion im Signalmechanismus des enterohepatischen Kreislaufs. Durch ausgeprägte Feedback-Autoregulation, die die Effektivität des Kreislaufs zwischen Leber und Darm gewährleistet, gehen täglich nur circa 0,5g Gallensäuren über den Stuhl verloren, was über Neusynthese in der Leber kompensiert wird (5). Die Gallensäuren halten Cholesterol in der Galle in Lösung und bilden im Darm gemischte Mizellen im Rahmen der Fettresorption (4, 5). Im distalen Dünndarm und in den Cholangiozyten werden konjugierte und unkonjugierte Gallensäuren über den Apical-Sodium-dependent-Bile-Acid- Transporter (ASBT) in die Enterozyten aufgenommen (5). Ein Großteil gelangt daraufhin auf der basolateralen Seite entweder passiv über die Anionenaustauschsysteme OSTα/β in die V.
portae oder über den MRP3-Transporter, der sowohl Gallensäuren als auch Bilirubin- Glucuronide aus den Enterozyten und Cholangiozyten ins Pfortaderblut abgibt (4, 5).
Gallensäuren werden im Blut an Albumin und Lipoproteine gebunden transportiert und an der basolateralen Hepatozytenmembran über den Natrium-Taurocholate-Cotransporting- Polypeptide (NTCP)-Transporter oder über verschiedene OATPs aufgenommen (5). Der NTCP stellt im Hepatozyten den wichtigsten Gallensäureaufnahmemechanismus dar, da mehr als 80% der hepatischen Aufnahme konjugierter Gallensäuren über ihn erfolgt (4, 7).
Unkonjugierte Gallensäuren und Bilirubin werden über Transporter für organische Anionen
12
(OATPs) aufgenommen (8). Menschliche Hepatozyten exprimieren hauptsächlich OATP2 (5, 6). Durch die Regulation all dieser Transportsysteme wird ein Gallensäurepool von 3-5g im Körper aufrechterhalten. Unterbrechungen des enterohepatischen Kreislaufs, z.B. durch Funktionsstörung des Ileums, resultieren in Störungen der Fettresorption und in Fettstühlen (4).
1.2.5. Biotransformation in der Leber
Die Leber ist als wichtigstes Entgiftungsorgan Ort der Biotransformation. Sowohl lipophile, reaktionsträge Verbindungen, die im Körper gebildet wurden, z.B. Steroidhormone, als auch viele Fremdstoffe (Xenobiotika) können in ihrer Form nur schlecht oder gar nicht ausgeschieden werden. Die Aufgabe der Leber ist es deshalb, diese in besser ausscheidbare, wasserlösliche Formen zu überführen, was auch Biotransformation genannt wird (1, 4). Diese besteht aus mehreren enzymatisch katalysierten Reaktionen, die die Sekretion der Stoffe ermöglichen sollen (3). In der Phase I der Biotransformation werden durch Oxidationen, Reduktionen und Hydrolysen reaktive Gruppen in die abzubauenden Stoffe eingefügt. Der erste Schritt ist hierbei meist die Oxidation durch Monooxygenasen der CYP450-Familie, deren häufigster Vertreter CYP3A4 ist. Es wird vermutet, dass 60% aller therapeutisch eingesetzten Wirkstoffe CYP3A4-Substrate sind (3, 4). In den Phase-II-Reaktionen der Biotransformation werden Konjugate zwischen den in der Phase I eingefügten reaktiven Gruppen und hydrophilen Resten hergestellt, sodass Produkte entstehen, die in der Regel wasserlöslicher sind als ihre Ausgangssubstrate. Quantitativ am wichtigsten ist hierbei die Übertragung von Glucuronsäure auf OH-, COOH-, NH2- und SH-Gruppen durch die UGT.
Weitere häufig beteiligte Enzyme der Phase II sind Sulfotransferasen (SULTs) und die Glutathion-S-Transferase, die Sulfatgruppen oder Glutathion übertragen (3, 4). In der Phase III der Biotransformation finden die bereits erwähnten Transportprozesse zur Ausscheidung der gebildeten Konjugate wie des Bilirubindiglucuronids statt (4).
1.2.6. Nukleäre Rezeptoren
Die Gallensäure-Transportsysteme unterliegen einer strengen Regulation, vor allem auf Ebene der Gentranskription (5). So wirken Gallensäuren und auch Xenobiotika bei intrazellulärer Akkumulation als Liganden für nukleäre Rezeptoren (NR). Diese Liganden-regulierten Transkriptionsfaktoren binden im Folgenden an Promotoren der unterschiedlichen Gene des Gallensäuresynthese- und -transportsystems und des Fremdstoffmetabolismus und beeinflussen so deren Transkription (4, 7). Der Farnesoid-X-Rezeptor (FXR) ist vor allem in
13
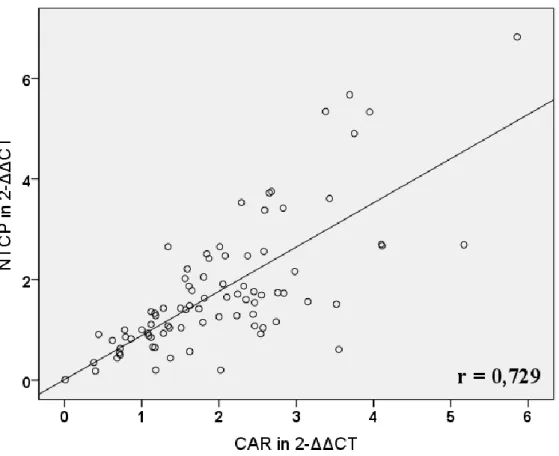
der Leber und im Darm vorhanden und wird durch Gallensäuren und nur schwach durch Ursodeoxycholsäure (UDCA) induziert (5, 6, 7). In Folge dessen wird der Gallefluss durch Induktion der Transportsysteme BSEP, MRP2, MDR3 und OSTα/β stimuliert und die Detoxifikation durch CYP3A4, UGT2B7 und SULT2A1 gefördert. FXR induziert jedoch auch den nukleären Rezeptor Small-Heterodimer-Partner (SHP), der die Expression von CYP7A1, CYP8B1, NTCP und OATP2, die für Gallensäuresynthese und -aufnahme verantwortlich sind, reduziert (6, 8). Der Pregnane-X-Rezeptor (PXR) hat als Liganden hauptsächlich Xenobiotika, darunter Rifampicin (RMP), Phenobarbital (PB), UDCA und Johanniskraut und aktiviert die Bildung von CYP3A4, OATP2, MDR1 und MRP2, die allesamt für gesteigerte Verstoffwechslung oder Efflux der Xenobiotika verantwortlich sind (9). Auch der Constitutive-Androstane-Rezeptor (CAR) hat wie PXR als Liganden vor allem Xenobiotika wie PB und bewirkt eine gesteigerte Transkription der Exportpumpen MRP3, MRP4 und MRP2 sowie der Detoxifikationsenzyme CYP3A4 und SULT2A1 (6, 8). Bilirubin ist ebenfalls ein CAR-Ligand und bewirkt dessen Translokation in den Nukleus (5, 6).
1.3. Cholestase
Cholestase bezeichnet einen gestörten Abfluss von Galle oder ihrer Bestandteile und somit eine Retention sämtlicher gallepflichtiger Substanzen im Körper (10, 11). Leitsymptome der Cholestase sind Juckreiz und Gelbsucht (Ikterus) (10). Beim Ikterus kommt es in Folge einer erhöhten Bilirubinkonzentration im Blut zu Ablagerungen in Haut und Skleren und so zu deren Gelbfärbung, während Juckreiz (Pruritus) vermutlich durch eine Einlagerung von Gallensäuren in die Haut und eine daraus resultierende Irritation peripherer Nervenendigungen entsteht (11, 12).
Cholestase kann eine Folge gestörter intrahepatischer Galleproduktion, gestörten transmembranären Galletransports oder mechanischer Gangobstruktion sein (13). Hyperbili- rubinämie und Ikterus können jedoch auch prähepatisch durch gesteigerte Hämolyse und somit durch erhöhtes indirektes Bilirubin verursacht sein, wie dies beim Neugeborenenikterus der Fall ist (3, 11). Hier liegt eine physiologische Erhöhung des unkonjugierten Bilirubins auf bis zu 15 mg/dl durch eine unreife UGT und somit eine harmlose Ursache des Ikterus vor (3).
Bei 2,4 - 15% der Neugeborenen kommt ein solcher Ikterus in den ersten zwei Lebenswochen vor und verschwindet meist spontan ohne Intervention (14).
14
Infolge der Cholestase lagern sich Gallebestandteile in den kleinen Gallengängen und in den Hepatozyten ab und rufen deren Zerstörung sowie eine Regeneratbildung aus Hepatozyten (Zirrhose) oder Gallekapillaren hervor. Die Leberzirrhose zeichnet sich durch knotige Regeneration des zerstörten Leberparenchyms mit bindegewebigem Umbau der Läppchen- und Portalfeldstruktur aus. Im weiteren Verlauf der Erkrankung wird der Durchfluss des Portalvenenblutes durch die Leber behindert, wodurch der Pfortaderdruck steigt (portale Hypertension) und Umgehungskreisläufe gebildet werden (10). Die portale Hypertension ist die Hauptkomplikation der Leberzirrhose und hat als häufigste direkte Folge die Entwicklung gastroösophagealer Varizen und deren Blutung (15). Weitere Folgen der portalen Hypertension sind die Entwicklung von Splenomegalie und Aszites. Im Laufe der zirrhotischen Lebererkrankung kommt es auch zu Leberfunktionsstörungen mit Gerinnungsstörungen, Hypalbuminämie und Ammoniakintoxikationen, die zu einer Enzephalopathie führen können (10). Durch die fehlenden Gallebestandteile im Darm kommt es bei Cholestase außerdem zu acholischen, entfärbten Stühlen, einer Malabsorption von Fetten und fettlöslichen Vitaminen sowie zu Fettstühlen (4, 14).
Die Ursachen von Cholestase in der Kindheit können grob in obstruktive und hepatozelluläre Ursachen eingeteilt werden (2).
1.3.1. Obstruktive Ursachen
Extrahepatische Gallengangatresie
Bei der extrahepatischen Gallengangatresie (Biliäre Atresie = BA) handelt es sich um eine chronisch progrediente Cholangiopathie mit fortschreitender fibröser Obliteration der extra- und später auch der intrahepatischen Gallengänge. Die Ätiologie der Erkrankung ist bisher unbekannt. Vermutet wird ein infektiöses Agens, das, prä- oder postnatal erworben, die fortschreitende Obliteration auslöst (2). Die Schätzungen der Häufigkeit liegen bei 1:6000 - 1:18000 Geburten, wobei die Inzidenz in Afrika und in der Pazifikregion am höchsten ist und Frauen etwas häufiger betroffen sind als Männer (14, 16). Die BA ist die häufigste Indikation für eine pädiatrische Lebertransplantation weltweit (16). Sie präsentiert sich mit persistieren- dem oder nach freiem Intervall in den ersten Lebenswochen erneut auftretendem Ikterus, acholischen Stühlen, dunklem Urin und Hepatomegalie (2, 16). Nach anfangs guter Entwicklung kommt es plötzlich zu Gewichtsverlust (16). Über ein Cholestasesyndrom führt die Erkrankung zur progressiven Leberzirrhose mit starkem Juckreiz, Gerinnungsstörungen und Mangel an fettlöslichen Vitaminen. Ohne Intervention stirbt die Mehrzahl der Patienten
15
in den ersten drei Lebensjahren (2). Die Diagnose sollte optimalerweise bis zu einem Alter von 30 Tagen durch eine verkleinerte oder fehlende Gallenblase in der Sonographie, durch den Nachweis fehlenden Galleflusses in der Szintigraphie oder durch Cholangiographie gestellt werden, um eine palliative Portoenterostomie nach Kasai noch vor dem zweiten Lebensmonat zu ermöglichen. Durch diese Operation kann häufig ein verbesserter Gallefluss sowie ein Rückgang der Cholestasesymptome erzielt werden (2, 16, 17). Eine frühe Diagnosestellung ist sehr wichtig, da die Erfolgsrate stark mit dem Alter bei Eingriff assoziiert ist (13). Durch die Operation wird die Lebenserwartung verbessert, langfristiges Überleben ist jedoch in den meisten Fällen nur mit Lebertransplantation möglich (2).
Caroli-Syndrom
Choledochuszysten sind angeborene oder erworbene sackförmige Erweiterungen des Gallengangsystems unklarer Ursache. Sie sind selten und treten meist ohne andere Fehlbildungen auf. Eine Sonderform dieser obstruktiven Fehlbildung stellt das Caroli- Syndrom (CS) dar (2). Dieses ist charakterisiert durch multiple segmentale zystische Erweiterungen der intrahepatischen Gallengänge, assoziiert mit kongenitaler Leberfibrose.
Ursache ist eine Entwicklungsanomalie der Gallengänge durch eine autosomal-rezessive Vererbung von Mutationen im PKHD1-Gen, das auch eine Rolle bei polyzystischer Nierenerkrankung spielt (18). Die Klinik setzt sich zusammen aus portaler Hypertension mit Ösophagusvarizen aufgrund der kongenitalen Leberfibrose und aus Cholestase mit rekurrenter Cholangitis, Gallensteinen und Ikterus, die das klinische Bild dominiert. Diagnostisch ist der Beweis der Kommunikation von Sacculi und Gallengängen durch Sonographie, Computertomographie, MRCP oder ERCP (Magnetresonanz-/ Endoskopisch retrograde Cholangiopankreatikographie) wichtig, während häufig auch ein Tumor getastet werden kann (2, 18). Therapeutisch werden die veränderten extrahepatischen Gallengänge mitsamt Gallenblase komplett entfernt und eine Anastomose zum Dünndarm gebildet. Verbleibende dysplastische Anteile erhöhen das Risiko der Entstehung eines Cholangiokarzinoms (2).
Aussagen über die Prognose und den weiteren Verlauf der Erkrankung können aufgrund mangelnder Daten über das Krankheitsbild nicht getroffen werden (17).
Alagille-Syndrom
Beim Alagille-Syndrom (ALGS) handelt es sich um eine syndromale Gallengangshypoplasie mit einem Mangel an intralobulären Gallengängen (2, 10, 14). Sie tritt bei einem von circa 70.000 Lebendgeborenen auf und wird autosomal-dominant durch eine Mutation im Jagged- 1-Gen vererbt. Klinisch sieht man eine chronische Cholestase mit schwerem Pruritus, die
16
typische dysmorphe Gesichtsform sowie weitere häufige syndromale Fehlbildungen wie z.B.
Pulmonalarterienstenosen, Schmetterlingswirbel und okuläres Embryotoxon (2, 14). Die Therapie erfolgt meist symptomatisch durch UDCA und durch Behandlung des schweren Juckreizes mit Antipruritusmedikamenten wie Opioidantagonisten, PB, RMP oder Cholestyramin (10). Das Outcome hängt stark von der individuellen Klinik ab (14). Nur in Einzelfällen wird eine Lebertransplantation erforderlich (2).
1.3.2. Hepatozelluläre Ursachen
Progressive familiäre intrahepatische Cholestasen
Bei den progressiven familiären intrahepatischen Cholestasen (PFICs) handelt es sich um drei verschiedene, autosomal-rezessiv vererbte Störungen verschiedener Gene, die für die Galle- bildung verantwortlich sind (14). Bei der PFIC1, auch Byler´s Disease genannt, liegt eine Mutation von FIC1 vor, das für den Transport von Phospholipiden verantwortlich ist. Es kommt zu schweren, wässrigen Diarrhöen und Cholestase (5, 14). Bei PFIC2, dem Byler´s Syndrome, ist die Ursache eine Mutation des BSEP, die zu einer Akkumulation von Gallensäuren in den Hepatozyten führt (5). Sowohl bei PFIC1 als auch bei PFIC2 entwickelt sich eine Cholestase noch vor dem ersten Lebensjahr bei hohen Serumkonzentrationen von Gallensäuren und normalen Gamma-Glutamyl-Transferase (γGT)- und Cholesterolwerten.
Viele sprechen auf eine partielle biliäre Diversion oder eine ileale Exklusion an, während die anderen meist vor dem 10. Lebensjahr eine Lebertransplantation erhalten (14). Die Prognose von M. Byler ist ohne Transplantation meist infaust (10). Bei der PFIC3 liegt eine homozygote Mutation im MDR3-Gen vor, einer Phospholipid-Exportpumpe, in deren Folge es aufgrund der fehlenden Phospholipide zu einer toxischen Schädigung des Gallengang- epithels kommt (5). Hier liegen klinisch hohe γGT-Werte und Entzündungsinfiltrate vor, die schon frühzeitig zu biliärer Zirrhose führen (5, 14). Der Juckreiz ist bei PFIC3 oft milder und spricht auf UDCA an (14). Die Mutationen der jeweiligen PFIC-Gruppen können mit molekulargenetischen Tests nachgewiesen werden (13).
Cystische Fibrose
Die Cystische Fibrose (CF) ist der häufigste genetische Defekt in der kaukasischen Bevölkerung (19). Ursächlich für diese Stoffwechselkrankheit ist eine Mutation im CFTR- Gen auf Chromosom 7, die autosomal-rezessiv vererbt wird (10, 19, 20, 21). Das Genprodukt ist ein Chloridkanal in der apikalen Membran vieler Epithelzellen, darunter Zellen der Lunge und des Pankreas sowie Cholangiozyten der Leber, weshalb die CF eine Systemkrankheit
17
darstellt (19, 21). Morbidität und Mortalität werden zwar durch den Grad der Beteiligung des respiratorischen Systems bestimmt, trotzdem zählt die Cystic Fibrosis-associated Liver Disease (CFLD), die etwa 30% der CF-Patienten betrifft, als dritthäufigste Todesursache bei CF. Es sind mehr als 1900 unterschiedliche Mutationen im CFTR-Gen bekannt, die häufigste Mutation ist jedoch eine Deletion von Phenylalanin an Position 508, die in Europa und Nordamerika bei 60-70% der Patienten vorliegt und in einem funktionsbeeinträchtigten CFTR-Kanal resultiert (20, 21). Durch den Austausch von Chlorid und Bicarbonat nimmt der CFTR-Kanal eine entscheidende Funktion in der Regulation des Wasser- und Elektrolytgehalts der Galle sowie in deren Alkalisierung ein und sorgt so für den Gallefluss (4, 7, 21). Liegt ein abnormes CFTR-Protein vor, resultiert ein Stau verdickter, visköser Galle, die weniger alkalisch ist und das Lumen der Gallekanäle verlegt. Es kommt zu einer intrahepatischen Gallengangsobstruktion und in deren Folge durch toxische Komponenten zur Schädigung der Hepatozyten mit Entwicklung einer langsam progredienten Leberzirrhose mit portaler Hypertension. Die Leberdysfunktion zeigt sich klinisch meist in den Jahren vor dem Teenageralter mit Bauchschmerzen, Übelkeit, Müdigkeit, Ikterus und Juckreiz (19, 21).
Therapeutisch werden fettlösliche Vitamine substituiert und UDCA für alle CF-Patienten empfohlen, da diese einen positiven Effekt auf Galledrainage und Histologie der Leber haben soll (21). Bei stabiler Leberfunktion kann das portale Gefäßsystem durch einen operativen Shunt entlastet werden, während bei fortgeschrittenen Stadien eine Lebertransplantation das Leben verlängern und die Lebensqualität verbessern kann (19, 21).
1.3.3. Pruritus als Symptom der Cholestase
Wie zu Beginn des Kap. 1.3. bereits erwähnt, stellt Pruritus eines der Leitsymptome der Cholestase dar (10). Die Entstehung des Juckreizes bei cholestatischen Erkrankungen ist noch nicht ausreichend geklärt, wobei jedoch eine Stimulation peripherer Nervenfasern der Haut durch ein enterohepatisch zirkulierendes Pruritogen wie beispielsweise Gallensäuren angenommen wird. Gestützt wird diese Hypothese dadurch, dass das Anionenaustauscherharz Cholestyramin, das den enterohepatischen Kreislauf der Gallensäuren durch vermehrte Bindung im Darm unterbricht, bei vielen cholestatischen Patienten eine deutliche Minderung des Juckreizes bewirkt (22). Eines der gängigsten Antipruritus-Medikamente bei Cholestase ist UDCA. Als hydrophile, nicht-toxische Gallensäure ersetzt sie bei oraler Gabe den Pool an hydrophoben, toxischen Gallensäuren (23). Zudem induziert UDCA, wie in Kap. 1.2.6.
erklärt, den nukleären Rezeptor FXR und sorgt so durch Reduktion der Aufnahme- und Synthesemechanismen und durch Aktivierung apikaler und basolateraler
18
Sekretionsmechanismen für einen Schutz der Hepatozyten (6, 8, 24). Auch dem Antibiotikum RMP wird ein antipruritogener Effekt nachgesagt. Durch Induktion von PXR werden, wie in Kap. 1.2.6. dargestellt, viele metabolisierende Enzyme und Transporter wie CYP3A4 und UGT aktiviert und so die Verstoffwechslung cholestatischer Pruritogene wie Gallensäuren und Bilirubin gefördert (23, 25). PB ist ein vor allem bei Kindern genutztes Medikament zur Antipruritusbehandlung (25). Als CAR-Ligand sorgt auch PB durch die Aktivierung verschiedener Detoxifikations- und Sekretionsmechanismen für die Sekretion der Pruritogene Bilirubin und Gallensäuren (6, 8, 25).
1.4. Aktueller Stand der Forschung
Wie in den Kap. 1.2.2., 1.2.3. und 1.2.4. dargestellt, ist bereits sehr viel über die genaue Lokalisation und Funktionsweise sowie die Substrate der hepatobiliären Transportsysteme bekannt. Durch die Fertigstellung des Humangenomprojektes wurde es möglich, diese Transporter zu klonen und es wurde festgestellt, dass bei einigen familiären, cholestatischen Störungen eine Mutation in einem bestimmten Gen, welches für einen solchen Transporter codiert, ursächlich ist, wie beispielsweise bei der PFIC oder bei der CF (8, 21, 26). Zahlreiche Autoren, darunter Kullak-Ublick et al., Zollner et al. sowie Boyer beschäftigten sich außerdem, wie im Kap. 1.2.6. erläutert, bereits mit der Regulation der Genexpression durch die nukleären Rezeptoren FXR, SHP, PXR und CAR. Sie zeigten, dass verschiedene Liganden, darunter Gallensäuren, ihren eigenen Metabolismus durch nukleäre Rezeptoren steuern und dass auch viele bereits genutzte Medikamente wie UDCA, PB und RMP modulierend auf nukleäre Rezeptoren Einfluss nehmen können (5, 6, 7, 8, 9, 27). Wie von Boyer und Zollner et al. zusammengefasst, wurde gezeigt, dass bei Cholestase über diese nukleären Rezeptoren verschiedene adaptive Mechanismen in Gang gesetzt werden, die die Leber vor weiterem Schaden bewahren sollen (6, 8). Es wurde beobachtet, dass Gallensäuresynthese- und -aufnahmesysteme reduziert und Detoxifikations- sowie Effluxmechanismen aktiviert werden (6, 7, 8, 27). Viele dieser Erkenntnisse wurden anhand von Tiermodellen der Cholestase, vor allem bei Ratten, gewonnen, bei denen durch Gallengangsligatur eine künstliche Cholestase induziert wurde (8, 26, 27, 28). Zollner et al.
untersuchten jeweils die adaptiven Mechanismen einer Auswahl an hepatobiliären Transportsystemen bei cholestatischen Erkrankungen bei Erwachsenen wie der Primären Biliären Zirrhose/ Cholangitis (PBC), der entzündungsinduzierten Cholestase und der
19
chronischen Hepatitis C im Vergleich zu Kontrollproben (29, 30). Keitel et al. erforschten die Genexpressionsunterschiede mehrerer hepatobiliärer Transporter bei zehn Kindern mit PFIC2 oder PFIC3, während Chen et al. die Expression einer großen Auswahl an Transportern im Lebergewebe von jeweils acht Kindern mit frühem und spätem Stadium einer BA betrachteten (31, 32). Als frühes Stadium galt bei Letzteren Gewebe, das während der Kasai- OP entnommen wurde, als spätes Stadium galt Lebergewebe, das bei Lebertransplantation gewonnen wurde. Bei beiden Studien wurde nicht-cholestatisches Lebergewebe als Kontrollgewebe verwendet (31, 32). Da Ergebnisse aus Tierstudien nur unzureichend auf den Menschen übertragen werden können und die meisten Genexpressionsuntersuchungen der hepatobiliären Transporter bei cholestatischen Erkrankungen des Erwachsenen stattfanden, herrscht somit ein Mangel an Arbeiten, die die adaptiven Mechanismen bei Cholestase sowie deren Wechselwirkungen mit dem Organismus in ihrer Gesamtheit gegenüberstellend bei cholestatischen Kinderkrankheiten untersuchen (31). Dies gilt vor allem für seltenere cholestatische Erkrankungen wie die PFIC, das ALGS oder das CS. Auch die häufigste Krankheit der cholestatischen Kinderkrankheiten, die BA, zählt mit einer Häufigkeit von 1:10.000 – 1:19.000 zu den seltenen Krankheiten, was patientenbasierte Forschung und den Erwerb neuer Behandlungsmethoden erschwert (17).
Die Auswirkung etablierter Antipruritus-Medikamente auf die Genexpression hepatobiliärer Proteine war bereits Gegenstand einiger Forschungsarbeiten. Marschall et al. untersuchten die Veränderung der Genexpression mehrerer hepatobiliärer Transport- und Detoxifikations- systeme bei ansonsten gesunden Patienten mit Gallensteinen, die vor der Operation RMP, UDCA oder keine derartigen Medikamente erhielten (33). Hagenbuch et al. werteten die Genexpression einiger Transporter für Gallensäuren und organische Anionen bei Ratten aus, nachdem ihnen für fünf Tage PB oder NaCl intraperitoneal verabreicht wurde (34). Benson et al. inkubierten primäre humane Hepatozyten 24 Stunden mit RMP und maßen die Expression von 410 Transporter-Genen der Leber, während Fickert et al. Mäuse für sieben Tage mit UDCA oder Cholsäure fütterten und anschließend die Expression mehrerer Transportsysteme untersuchten (35, 36). Auch hier gibt es zwar bereits einige Forschungsarbeiten, es herrscht jedoch ebenfalls ein Mangel an Daten zur Auswirkung von Antipruritus-Medikamenten auf die Genexpression bei cholestatischen Kinderkrankheiten.
Um bessere Kenntnisse zu Ätiologie, Pathologie und Behandlung cholestatischer Kinder- krankheiten zu gewinnen, liegt deshalb die Notwendigkeit vor, an weiteren Patienten-
20
kollektiven die adaptiven Mechanismen bei kindlicher Cholestase sowie die Wirksamkeit von Antipruritus-Medikamenten zu untersuchen.
21
2. Fragestellung der Arbeit
In der vorliegenden Arbeit werden die verschiedenen cholestatischen Kinderkrankheiten und nicht-cholestatischen Vergleichsproben hinsichtlich klinischer, laborchemischer und histopa- thologischer Gesichtspunkte charakterisiert. Es werden mRNA-Expressionsunterschiede der nukleären Rezeptoren und hepatobiliären Transporter untersucht und die adaptiven Mechanismen bei Cholestase sowie deren Wechselwirkungen mit klinischen, histopatho- logischen und laborchemischen Aspekten bei verschiedenen Krankheitsbildern dargestellt.
Weiterhin wird die Auswirkung herkömmlicher Antipruritus-Medikamente auf die Genexpression analysiert. Das Hauptaugenmerk der Arbeit liegt auf den cholestatischen Kinderkrankheiten BA, CF, PFIC sowie dem ALGS und dem CS, jeweils im Vergleich zu nicht-cholestatischen Kontrollproben. Die restlichen cholestatischen Erkrankungen werden als cholestatisches Kollektiv in die Analyse der adaptiven Mechanismen mit einbezogen. Bei den zu untersuchenden Transportsystemen handelt es sich um die Aufnahmetransporter NTCP und OATP2, die kanalikulären Effluxmechanismen MRP2, MDR1, MDR3 und FIC1, den Cholangiozytentransporter CFTR sowie die basolateralen Effluxmechanismen MRP3, MRP4 und OSTα/β. Zusätzlich sollen die nukleären Rezeptoren FXR, SHP, PXR und CAR betrachtet werden. Im Einzelnen beschäftigt sich die vorliegende Arbeit mit folgenden Fragestellungen:
- Wie sind die cholestatischen Kinderkrankheiten und Vergleichsproben hinsichtlich klinischer, histologischer und laborchemischer Befunde charakterisiert?
- Wie unterscheidet sich die Expression der hepatobiliären Transporter und nukleären Rezeptoren zwischen verschiedenen cholestatischen Erkrankungen und Vergleichsproben?
- Inwieweit korreliert die mRNA-Expression nukleärer Rezeptoren mit der mRNA- Expression hepatobiliärer Transporter? Gibt es mögliche regulatorische Zusammenhänge?
- Inwieweit korrelieren klinische, histopathologische und laborchemische Parameter mit der mRNA-Expression nukleärer Rezeptoren und hepatobiliärer Transporter?
- Inwieweit beeinflussen Antipruritus-Medikamente die mRNA-Expression hepato- biliärer Transporter und nukleärer Rezeptoren?
22
3. Material und Methoden
3.1. Kindergewebeproben
Die Lebergewebeproben der Kinder wurden über einen Zeitraum von sieben Jahren, von Januar 2008 bis Dezember 2014, im Rahmen von Leberresektionen und Leberbiopsien (Tab.
1), die im Uniklinikum Regensburg stattfanden, gewonnen. Das Lebergewebe wurde jeweils auf Eis direkt vom Operationssaal in die Pathologie des Uniklinikums gebracht, wo ein kleiner Teil für spätere Untersuchungen abgeschnitten wurde. Diese Proben wurden zur Stabilisierung sofort in RNAlater gegeben, mit flüssigem Stickstoff schockgefroren und zur weiteren Verwendung bei -80°C aufbewahrt. Von den insgesamt 84 Lebergewebeproben wurden 77 bei Leberresektionen aufgrund verschiedener cholestatischer Erkrankungen gewonnen (Tab. 1). Sieben Proben von Kindern mit Lebererkrankungen nicht-cholestatischer Ursache dienten als Vergleichsproben (VP). Bei drei dieser Vergleichsproben wurde eine Leberresektion bei Hepatoblastom durchgeführt, während die anderen vier Vergleichsproben durch Leberbiopsien bei Morbus Meulengracht, unklarer Transaminasenerhöhung, heterozygotem α1-Antitrypsinmangel und bei Leberfibrose Grad 1 gewonnen wurden. Bei dem entnommenen Gewebe handelte es sich folglich um, hinsichtlich Cholestase, nicht- pathologisch verändertes Lebergewebe. Die cholestatischen Grunderkrankungen bei Leberresektion waren vielfältig. Bei 56 Kindern wurde eine BA diagnostiziert, bei sieben
Tab. 1: Krankheitsbilder bei cholestatischen Lebergewebeproben und Vergleichsproben Cholestatische Lebergewebeproben
(n = 77)
Vergleichsproben (n = 7)
Extrahepatische Gallengangatresie (n = 56)
Cystische Fibrose (n = 7)
PFIC (n =4) o PFIC 1 (n = 1) o PFIC 2 (n = 1) o PFIC 3 (n = 2)
Alagille-Syndrom (n = 2)
Cholangiodysplasie (n = 2)
Sekundär sklerosierende Cholangitis (n = 2)
Caroli-Syndrom (n = 1)
Kongenitale Leberfibrose (n =1)
Duktalplattenmalformation (n = 1)
Kurzdarmsyndrom mit TPN (n = 1)
Hepatoblastom (n = 3)
Morbus Meulengracht* ( n = 1)
Unklare Transaminasenerhöhung*
(n = 1)
Heterozygoter α1-Antitrypsin- Mangel* (n = 1)
Leberfibrose Grad 1* (n = 1)
* = mittels Leberbiopsie entnommen.
PFIC = Progressive Familiäre Intrahepatische Cholestase, TPN = Totale Parenterale Nutrition/ Ernährung.
23
Kindern eine CF. Vier Kinder litten unter PFIC (1x Typ1, 1x Typ2, 2x Typ3). Bei jeweils zwei Kindern wurde ein ALGS, eine Cholangiodysplasie oder eine sekundär sklerosierende Cholangitis diagnostiziert. Jeweils ein Kind litt an CS, kongenitaler Leberfibrose, Duktal- plattenmalformation oder Kurzdarmsyndrom mit langjähriger totaler parenteraler Ernährung (TPN). Vor Eingriff oder mittels Aufklärungsschreiben im Rahmen des Fragebogens (Anhang A und B) wurden die Eltern/ Angehörigen über eine mögliche Nutzung der Gewebeproben sowie krankheitsrelevanter Daten im Rahmen wissenschaftlicher Forschungsprojekte aufge- klärt und eine Einwilligung diesbezüglich eingeholt.
3.2. Datenerhebung zu den Gewebeproben
3.2.1. Daten aus SAP
Die klinischen Daten zu den vorliegenden Lebergewebeproben wurden mittels des SAP- Programms der Universität Regensburg erhoben und als Tabelle in den Programmen Excel®
2010 und SPSS® Statistics 23 gesammelt. Jedem Kind wurde aus Datenschutzgründen eine Transplantations- oder eine Vergleichsprobennummer zugeordnet. Fehlende Werte oder Informationen wurden bei SPSS als solche gekennzeichnet. Tabelle 2 gibt an, welche Daten zur Charakterisierung der Gewebeproben erfasst wurden.
Zunächst wurde die Krankheitsgeschichte der Kinder, soweit in den Arztbriefen und im Operationsbericht erwähnt, in Form von Geburtsdatum, Geschlecht, Alter bei Symptombeginn und Datum des Eingriffs vermerkt (Tab. 2). Aus dem Geburtsdatum und dem Datum des Eingriffs wurde das Alter bei Eingriff errechnet.
Die klinischen Parameter Aszites und Milzlänge wurden, soweit vorhanden, aus Befunden der Abdomensonographie entnommen. Aus der Milzlänge wurde mithilfe einer Normtabelle (Anhang C) für jedes Kind anhand des Alters bestimmt, ob eine Splenomegalie vorlag. Falls in den Sonographiebefunden keine Angaben zu Milzlänge oder Aszites gemacht wurden, wurde das Vorhandensein von Splenomegalie oder Aszites aus den Arztbriefen entnommen.
Die restlichen Symptome einer Cholestase (Hautikterus, Sklerenikterus, Juckreiz, dunkler Urin, acholischer Stuhl und Ansprechen auf Antipruritus-Medikamente) wurden nicht zuverlässig in den Arztbriefen erwähnt, weshalb diese Daten aus Fragebögen entnommen wurden, die im Rahmen der Datenerhebung an die Familien geschickt wurden (Anhang B).
24
Tab. 2: Daten zur Charakterisierung der Gewebeproben Krankheitsgeschichte
Geburtsdatum, Geschlecht, Alter bei Symptombeginn (Jahre), Datum des Eingriffs, Alter bei Eingriff (Jahre)
Klinische Daten der Erkrankung
Aszites (j/ n), Splenomegalie (j/ n), Milzlänge (cm)
Aus Fragebögen: Haut-/ Sklerenikterus (j/ n), Juckreiz (j/ n), dunkler Urin (j/ n), acholischer Stuhl (j/ n), Ansprechen auf Antipruritusmedikation (j/ n/ kein Juckreiz vorhanden)
Klinische Chemie
Cholestaseparameter: AP (U/l), Bilirubin direkt/ gesamt (mg/dl),
Gallensäuren Glyko-/ Tauro-/ gesamt (µmol/l), γGT (U/l) Leberzellschädigung: GOT (U/l), GPT (U/l), GLDH (U/l), LDH (U/l)
Entzündungsmarker: CRP (mg/l)
Synthesefunktion: CHE (U/l), Gesamteiweiß (g/l), Quick-Wert (%) Medikamente
Antipruritus-Medikamente: Ursodeoxycholsäure, Rifampicin, Phenobarbital Histopathologische Befunde
Gewebetyp: Leberresektion/ Leberbiopsie Cholestase: keine/ leicht/ mittel/ schwer
Inflammation: keine/ leicht/ mittel/ schwer/ vorhanden o. genaue Bezeichnung
Fibrose: j/ n
Zirrhose: j/ n
Gallengangsproliferate: j/ n
j = ja, n = nein, AP = Alkalische Phosphatase, GOT = Glutamat-Oxalacetat-Transaminase, GPT = Glutamat- Pyruvat-Transaminase, GLDH = Glutamatdehydrogenase , LDH = Laktatdehydrogenase, CRP = C-reaktives Protein, CHE = Cholinesterase.
Die Angaben zur Medikamenteneinnahme vor Eingriff wurden aus dem Arztbrief entnommen, der am nächsten zum Eingriff datiert war. Es wurde die Einnahme der Antipruritusmedikamente UDCA, PB, RMP sowie deren Kombinationen jeweils mit Dosisangabe notiert.
Die Laborwerte der Kinder wurden, wie in Kap. 3.2.3. beschrieben und in Tab. 2 dargestellt, von der klinischen Chemie angefordert.
Auch die Pathologiebefunde zum entnommenen Lebergewebe wurden konsultiert und die Entnahmeart des Gewebes (Resektion/ Biopsie) sowie typische histologische Gesichtspunkte
25
einer cholestatischen Erkrankung und deren Ausprägung erfasst. So wurde besonders auf das Vorliegen von Inflammation, Zirrhose, Cholestase und von Gallengangsproliferaten geachtet (Tab. 2).
3.2.2. Fragebogen an die Familien
In den Arztbriefen wurden vor allem die klinischen Parameter und Symptome der cholesta- tischen Erkrankungen wie Ikterus, Verfärbung von Urin und Stuhl sowie Juckreiz, die die betroffenen Kinder stark quälen, nur unzuverlässig und nicht immer vollständig erwähnt.
Auch zum Ansprechen der Kinder auf Antipruritus-Medikamente waren zu wenig sichere Daten in den Arztbriefen vorhanden. All diese Gesichtspunkte spielten jedoch für die Fragestellung dieser Arbeit und auch für das cholestatische Krankheitsbild eine große Rolle.
Da niemand die Kinder und ihre Krankheitsgeschichte besser kennt als ihre Eltern, wurde ein spezieller Fragebogen entwickelt, der die oben genannten Daten abfragt (Anhang B). Soweit noch nicht vorhanden, wurde für die entsprechenden Familien außerdem ein Aufklärungsschreiben über die Verwendung der Lebergewebeproben und des von den Kindern gewonnenen Blutes sowie eine Einverständniserklärung über die Nutzung des Gewebes erstellt (Anhang A). Es wurde darauf hingewiesen, dass Kinder ab einem Alter von 14 Jahren, die in der Lage sind, die Informationen zu verstehen und sich selbst ein Urteil zu bilden, eigenständig die Einwilligungserklärung unterschreiben oder ablehnen dürfen. Die Fragebögen wurden anschließend an die Familien der Kinder mit cholestatischer Erkrankung verschickt.
Der Fragebogen fragte ab, ob die Eltern bei ihrem Kind im Zeitraum vor der Lebertransplantation eine Gelbfärbung von Haut und/ oder Augen, eine intensive Gelb- oder Braunfärbung des Urins und/ oder eine Aufhellung (gelbliche Färbung) des Stuhls festgestellt haben. Zur Klärung der Stuhlfarbe wurde dem Fragebogen eine Kuno-Kids-Farbkarte beigelegt, auf der verdächtige Stuhlfarben abgebildet sind (Anhang D). Die Eltern wurden aufgefordert, anzugeben, ob ein verdächtig gefärbter Stuhl bei ihrem Kind aufgetreten ist und falls ja, welche Nummer der Farbkarte zutraf. Weiterhin wurde gefragt, ob bei dem jeweiligen Kind Juckreiz vorhanden war und falls ja, ob dieser auf eine Körperregion beschränkt oder am ganzen Körper präsent war und ob Hautveränderungen wie Ausschlag, Kratzspuren oder aufgekratzte Hautstellen bei den jeweiligen Kindern bemerkt wurden. Zum Schluss wurde die Frage gestellt, ob die Kinder auf das jeweils erhaltene Medikament zur Juckreizbehandlung und -prophylaxe angesprochen haben. Es wurde für jedes Kind ein individueller Brief
26
gedruckt, sodass jeweils nur die erhaltenen Medikamente abgefragt wurden. Im Anhang A sind Aufklärungsschreiben und Einverständniserklärung abgebildet, während Anhang B den Fragebogen zeigt.
3.2.3. Daten der klinischen Chemie
Die Laborwerte der 84 Kinder wurden von der Klinischen Chemie des Universitätsklinikums Regensburg angefordert und auch hier Befunde festgehalten, die möglichst nah vor erfolgtem Eingriff lagen. Die einzelnen Werte sind in Tab. 2 dargestellt. Ein besonderes Augenmerk wurde, auch im Hinblick auf die Auswertung, auf den Entzündungsmarker C-reaktives Protein (CRP) und die Cholestasemarker Alkalische Phosphatase (AP), Bilirubin (direkt und gesamt), Gallensäuren (Glyko-, Tauro-, gesamt), sowie γGT gelegt. Die Transaminasen Glutamat-Oxalacetat-Transaminase (GOT) und Glutamat-Pyruvat-Transaminase (GPT) sowie die Laktatdehydrogenase (LDH) und die Glutamatdehydrogenase (GLDH) wurden als Marker für den Leberzellschaden notiert. Die Parameter Cholinesterase (CHE), Gesamteiweiß und Quick-Wert dienten zur Prüfung der Lebersynthesefunktion.
3.3. RNA-Isolation
Die Isolation der totalen Ribonukleinsäure (RNA) aus den Lebergewebeproben der Kinder erfolgte unter Verwendung des mirVana™ miRNA Isolation Kits und des dazugehörigen mirVana™ miRNA Isolation Kit Protokolls (Ambion®, Life Technologies) (37).
Gemäß des Protokolls wurden vor Beginn der RNA-Isolation die Waschlösungen mit der jeweiligen Menge an 100% Ethanol angemischt und der Arbeitsplatz sowie die verwendete Pinzette mit RNase-Dekontaminationsspray (RNaseZAP™, R2020 SIGMA) gereinigt. Das in RNAlater (RNAlater®, R0901 SIGMA) oder pur eingefrorene Lebergewebe wurde in flüssigem Stickstoff aus dem -80°C-Kühlschrank geholt und in kleinen Waagschälchen (2149.2, Roth) abgewogen.
Das verwendete Gewebe, das meist unter 100mg und nur in vereinzelten Fällen etwas mehr wog, wurde mittels eines Einwegskalpells (Feather, No.22) zerkleinert und zusammen mit 1ml Lysepuffer in M-Tubes (gentleMACS™ Tubes) auf Eis gegeben. Im gentleMACS™
Dissociator (MACS, Miltenyi Biotec) wurde das Gewebe anschließend 60 - 84 Sekunden mit dem Programm RNA02.01 geschreddert, bis es vollständig in Lösung gegangen und keine Gewebeteilchen mehr sichtbar waren. Waren nach einem Durchlauf noch Gewebestückchen
27
zu sehen, wurde das Programm nach einer Kühlphase auf Eis wiederholt. Das Homogenisat wurde fünf Minuten bei 4°C bei 4,000 x g zentrifugiert.
Zur organischen Extraktion wurde anschließend so viel Lysat wie möglich aus dem Plastiktube in 2ml Tubes überführt und 1⁄10 des gewonnenen Volumens an microRNA (miRNA) Homogenate Additive zugegeben. Die Mischung wurde kurz gevortext und zehn Minuten auf Eis gegeben. Es folgte die Zugabe der gleichen Menge Chloroform wie an Lysat vor Zugabe des Homogenate Additive vorhanden war. Es wurde 30 Sekunden gevortext und anschließen bei Raumtemperatur fünf Minuten auf Maximalgeschwindigkeit (10,000 x g) zentrifugiert, um die wässrige von der organischen Phase zu trennen. Nach der Zentrifugation folgten die Entnahme der obersten, wässrigen Phase sowie die Überführung in 1,5ml Tubes.
Das entnommene Volumen wurde notiert und zur finalen RNA-Extraktion die 1,25 fache Menge an 100% Ethanol zugegeben und wiederum kurz gevortext. Die gewonnene Ethanol/
Lysat-Mischung wurde in 700µl-Schritten auf spezielle Filtereinsätze im Collection Tube gegeben und jeweils für 15 Sekunden bei 10,000 x g zentrifugiert, bis die komplette Mischung durch den Filter gelaufen war. Der Durchlauf wurde jeweils verworfen.
Anschließend wurde die im Filter verbliebene RNA einmal mit 700µl RNA Waschlösung 1 und zweimal mit 500µl Waschlösung 2/ 3 gereinigt und jeweils der Durchfluss verworfen.
Zum Schluss folgte die erneute Zentrifugation für eine Minute, um die übriggebliebene Flüssigkeit zu entfernen.
Der Filter mit der RNA wurde auf ein frisches Collection Tube aufgebracht und die RNA mit 100µl RNase-freiem Wasser und anschließender 30-sekündiger Zentrifugation eluiert. Der Durchlauf, der nach diesem Schritt die RNA beinhaltete, wurde in ein kleineres Tube überführt und die Konzentration der enthaltenen RNA mittels des NanoDrop 2000 UV-Vis Spectrophotometers (Thermo Scientific) im RNA-Programm bestimmt. Bei den vier vorhandenen Leberbiopsien war so wenig Gewebe verfügbar, dass am Ende zweimal hintereinander mit 30µl statt 100µl RNase-freiem Wasser eluiert wurde, um die Konzentration der enthaltenen RNA zu erhöhen. Die gewonnenen RNA-Isolationen wurden zur weiteren Verwendung bei -80°C aufbewahrt (37).