Charakterisierung des biologischen Abbaus von Azofarbstoffen am Beispiel
von DRIMARO sowie des sonochemischen Abbaus von
ausgewählten perfluorierten Tensiden
Inaugural‐Dissertation
zur Erlangung des Doktorgrades
der Mathematisch‐Naturwissenschaftlichen Fakultät der Universität zu Köln
vorgelegt von Jochen Ohrem
aus Frechen
Berichterstatter:
Prof. Dr. Axel G. Griesbeck (Universität zu Köln) Prof. Dr. Astrid Rehorek (Fachhochschule Köln)
Tag der letzten mündlichen Prüfung: 16.01.2013
interessanten Themen, die intensive Betreuung, die rege Diskussionsbereitschaft, die hilfreichen Ratschläge, die konstruktiven Korrekturen dieser Arbeit sowie für die Übernahme der Berichterstattung und den Beisitz. Insbesondere gilt mein Dank ihrer sehr engagierten Art, sowie dem mir entgegengebrachten Vertrauen.
Prof. A. Griesbeck danke ich ganz herzlich für die Betreuung, die Übernahme des kooperativen Promotionsverfahrens, die freundlichen Ratschläge, Korrekturen, Motivationen sowie die Übernahme der Berichterstattung und des Beisitzes.
Prof. Ruschewitz danke ich für die Übernahme der Berichterstattung.
Herrn Dr. von der Gönna danke ich für die Übernahme des Beisitzes.
Meiner Familie danke ich besonders herzlich für jedwede Unterstützung, mein Studium und für die vielen netten, unterstützenden Worte.
Maria danke ich besonders herzlich für famose Unterstützung, fortwährendes Ver‐
ständnis, bewundernswerte Geduld, ausdauernde Kraft, liebevolle Zuwendung und Zuneigung und ganz besonders dafür, dass sie mir stets zur Seite steht.
Kai und Marc und vielen anderen Freunden danke ich für ihr aktives Zuhören, die vielen Tipps und für schöne Zeiten.
Bei der Arbeitsgruppe von Prof. Rehorek bedanke ich mich für die Anregungen und kollegialen Unterstützungen, das angenehme Arbeitsklima und insbesondere für die schöne und kreative Zeit.
Allen Mitarbeitern möchte ich danken, insbesondere Herrn Müller und Herrn Gusakov für die schnelle und unkomplizierte Lösung vieler technischer Probleme.
Nestor danke ich für die gute Zusammenarbeit und die freundliche Aufnahme an der Universität zu Köln sowie insbesondere für die Hilfe bei den NMR Messungen.
Bei Prof. Metwally bedanke ich mich für die Diskussionen der Struktur‐Vorschläge und Abbaumechanismen.
Der AG Prof. Waldvogel, Universität Mainz, und insbesondere Robert Francke, danke ich für den Aufenthalt und die Einweisung in die Messung der Cyclovoltamogramme.
1. EINLEITUNG 1
2. ZIELSTELLUNG 4
2.1. E
NTWICKLUNG EINESA
BBAUVERFAHRENS FÜR PERFLUORIERTET
ENSIDE5
2.1.1. Z
IELSETZUNG FÜR DIESEU
NTERSUCHUNGEN5
2.2. E
NTWICKLUNG EINESA
BBAUVERFAHRENS FÜR DENA
ZOFARBSTOFFDRIMARO 6
2.2.1. Z
IELSETZUNG FÜR DIESEU
NTERSUCHUNGEN6
3. THEORIE 8
3.1. P
ERSISTENTE,
PERFLUORIERTEV
ERBINDUNGEN8
3.1.1. Ü
BERSICHT PERFLUORIERTERV
ERBINDUNGEN8
3.1.2. A
NWENDUNGSGEBIETE9
3.1.3. R
ICHTLINIEN FÜRPFOS
[28,
29] 10
3.1.4. G
ESUNDHEITSGEFÄHRDUNG DURCH PERFLUORIERTEV
ERBINDUNGEN12
3.1.5. A
NREICHERUNG IN DERU
MWELT13
3.1.6. H
ERSTELLUNGSVERFAHREN VONPFT 14
3.1.7. Ü
BERSICHT DER ABBAUENDENB
EHANDLUNGSMETHODEN16
3.2. U
LTRASCHALL19
3.2.1. E
RZEUGUNG VONU
LTRASCHALL19
3.2.2. A
KUSTISCHEK
AVITATION20
3.3. C
HARAKTERISIERUNG DERA
QUASONOLYSE23
3.3.1. P
HYSIKALISCHEE
FFEKTE23
3.3.2. C
HEMISCHEE
FFEKTE26
3.4. A
NALYTIK PERFLUORIERTERV
ERBINDUNGEN27
3.5. F
ARBSTOFFE29
3.5.1. T
EXTILIENFÄRBUNG30
3.5.2. A
UFZIEHGRADE VONF
ARBSTOFFEN31
3.5.3. R
EAKTIV‐
UNDA
ZOFARBSTOFFE32
3.5.4. H
ERSTELLUNGSVERFAHREN33
3.6. DRIMARO 34
3.7. I
NDUSTRIELLEA
BWÄSSER DERT
EXTILINDUSTRIE35
3.7.1. G
ESETZLICHEG
RUNDLAGEN36
3.8. R
EINIGUNGSVERFAHREN FÜRA
ZOFARBSTOFFE[153] 37
3.8.1. S
TOFFTRENNENDEV
ERFAHREN37
3.8.2. S
TOFFZERSTÖRENDEV
ERFAHREN38
3.9. B
IOLOGISCHEB
EHANDLUNG VONA
ZOFARBSTOFFEN41
3.9.3. A
NAEROBER
EAKTOREN43
3.9.4. A
EROBERA
BBAU46
3.9.5. S
TAND DESW
ISSENS ZUMA
BBAU VONR
EACTIVER
ED141
ALSDRIMARO‐A
NALOGON49
3.10. A
NALYSEVERFAHREN MITLC‐MS
N50
3.10.1. A
NALYTIK LÖSLICHERA
ZOFARBSTOFFE50
3.10.2. I
ONENPAARCHROMATOGRAPHIE(IPC) 50
3.10.3. R
EVERSED‐P
HASE‐C
HROMATOGRAPHIE(RPC) 51
3.10.4. M
ASSENSPEKTROMETRIE51
4. EXPERIMENTELLES 55
4.1. B
ESTIMMUNG DERR
ADIKALBILDUNGSRATE55
4.2. M
ESSUNG DESE
NERGIEEINTRAGS58
4.3.
19F‐NMR
M
ESSUNGEN DER PERFLUORIERTENS
UBSTANZEN59 4.4. O
NLINE‐A
NALYSENSYSTEMIPC‐DAD‐S
UPRESSOR‐ESI‐MS
N FÜR DENB
IOREAKTOR60
4.4.1. C
HROMATOGRAPHISCHET
RENNUNG[154] 63
4.4.2. M
ASSENSPEKTROMETRIE64
4.5. A
NAEROB‐
AEROBB
IOREAKTOR[154] 67
5. ERGEBNISSE UND DISKUSSION 72
5.1. B
ESTIMMUNG DERR
ADIKALBILDUNGSRATE72
5.1.1. H
INTERGRUND72
5.1.2. S
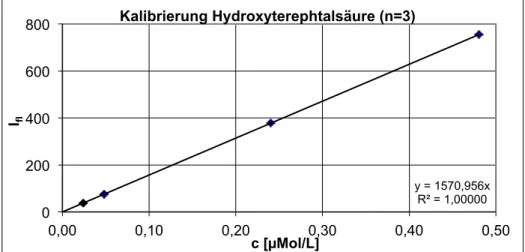
YNTHESE VONH
YDROXYTEREPHTHALSÄURE UNDR
EINHEITSBESTIMMUNG MITHPLC 72
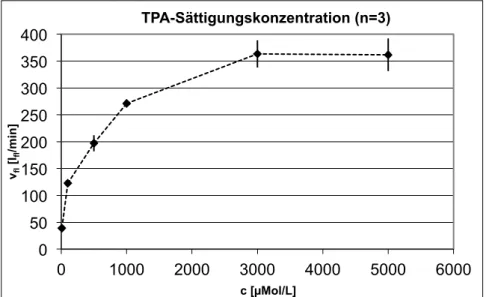
5.1.3. B
ESTIMMUNG DERS
ÄTTIGUNGSKONZENTRATION74
5.1.4. B
ESTIMMUNG DERU
MSETZUNG VONT
EREPHTHALSÄURE ZUH
YDROXYTEREPHTHALSÄURE76
5.1.5. B
ERECHNUNG DERR
ADIKALBILDUNGSRATE78
5.2. M
ESSUNG DESE
NERGIEEINTRAGS80
5.3.
19F‐NMR
M
ESSUNGEN DER SONOCHEMISCHENB
EHANDLUNG VONPFC 82 5.3.1.
19F‐NMR
M
ESSUNGEN DERB
EHANDLUNGEN BEIP
AC=64W
(
MITTLEREI
NTENSITÄT) 84 5.3.2.
19F‐NMR
M
ESSUNGEN DERB
EHANDLUNG BEIP
AC=134W
(
MAX.
I
NTENSITÄT) 86 5.3.3.
19F‐NMR
M
ESSUNGEN DERB
EHANDLUNG VONPFOA
BEIP
AC=134W
UND PH
9 88 5.3.4. B
ESTIMMUNG DERG
ESCHWINDIGKEITSKONSTANTEN K90 5.3.5. A
BBAUMECHANISMUS DER PERFLUORIERTENS
ULFONSÄUREN94 5.3.6. A
BBAUMECHANISMUS DER PERFLUORIERTENC
ARBONSÄUREN95 5.4. B
IOLOGISCHEB
EHANDLUNG DESA
ZOFARBSTOFFSDRIMARO 97
5.5. LC‐MS
NM
ETHODENENTWICKLUNG98
5.9. I
DENTIFIZIERTEI
NTERMEDIATE115
5.10. V
ORGESCHLAGENEA
BBAUMECHANISMEN119
5.10.1. A
NAEROBERA
BBAUMECHANISMUS120
5.10.2. A
EROBERA
BBAUMECHANISMUS125
5.11. B
EHANDLUNG EINESG
EMISCHS AUSDRIMARO,
NYROF,
CABVBF 132 5.12. S
ONOCHEMISCHEB
EHANDLUNG DESU
LTRAFILTRATS134 5.13. U
NTERSUCHUNGEN MIT ZYKLISCHERV
OLTAMMETRIE136 5.14. E
RMITTLUNG DES OPTIMALENR
EDOXPOTENTIALS UND PH‐W
ERTS DER ANAEROBENB
EHANDLUNGSSTUFE139
6. AUSBLICK 141
7. ZUSAMMENFASSUNG 142
8. ABSTRACT 144
9. MATERIALIEN UND METHODEN 146
9.1. S
UBSTANZEN UNDL
ÖSUNGEN146
9.2. L
ABORGERÄTE UNDM
ETHODEN148
10. QUELLEN 152
11. VERZEICHNISSE 167
11.1. A
BBILDUNGEN167
11.2. T
ABELLENVERZEICHNIS169
11.3. F
ORMELVERZEICHNIS170
11.4. A
BKÜRZUNGSVERZEICHNIS171
1. Einleitung
Wasser ist für alle Menschen lebenswichtig. Daher wird das Gesundheitsrisiko durch toxische Wasserinhaltsstoffe in der Bevölkerung mit Sorge wahrgenommen [1].
Angesichts der Bedeutung des wichtigsten Lebensmittels ist Trinkwasserhygiene besonders entscheidend [2]. Neue Messmethoden, Nachweisgrenzen und wissenschaftliche Erkenntnisse führen oft zu Anpassungen der Hygienean‐
forderungen an das Nutzwasser [3]. Neben seiner Rolle als Lebensmittel wird Wasser für die industrielle Nutzung als Energieträger, Reaktionspartner, Reinigungsmittel, Lösungsmittel, Prozessmedium, Absorbens und Adsorbens verwendet [4]. Diese Anwendungen gefährden Wasser als Bestandteil der natürlichen Umwelt. Wasser ist der einzige Rohstoff, der nicht nachgebildet, sondern im Kreislauf geführt wird.
Die Bestrebungen der EU zur Harmonisierung der gesetzlichen Anforderungen an das Trinkwasser und die Überführung der kommunalen Trinkwasserversorger in wettbewerbsorientierte Wirtschaftsunternehmen stellt die Wasserversorgungen vor neue Herausforderungen und Fragen zur hygienischen Sicherheit des Trinkwassers [1]. Die Trinkwasserverordnung von 1975, die auf dem alten nationalen Bundes‐
seuchengesetz basierte, hat sich zu einer Verordnung entwickelt, die neben anderen Zielen den Anforderungen der EG genügt [5]. Aus der staatlichen Wasserversorgung als Einrichtung der kommunalen Selbstversorgung sind Dienstleistungsunternehmen geworden, die mit dem wirtschaftlichen Wettbewerb konfrontiert werden.
Hygienische Anforderungen erscheinen unter Berücksichtigung wirtschaftlicher Betrachtung in einem ganz neuen Licht.
Seit Ende der 60er Jahre sind Befürchtungen in der Bevölkerung eine Gesundheits‐
schädigung durch toxische Wasserinhaltsstoffe zu erleiden sehr hoch [27]. Akut toxische Stoffe im Trinkwasser spielen hierbei weniger eine Rolle als chronisch toxische Stoffe, bei denen die lebenslange Aufnahme die Gesundheit schädigen kann.
Das Spektrum an Stoffen, die zu Gesundheitsgefährdungen führen können, hat kontinuierlich zugenommen.
Im Rahmen dieser Arbeit werden für zwei bedeutende, als chronisch toxisch und persistent geltende Stoffgruppen, anhand von industrierelevanten Vertretern, Behandlungsverfahren in Abbauversuchen eingesetzt: Azofarbstoffe und per‐
fluorierte Tenside.
Die Schlagwörter PFT (perfluorierte Tenside), PFC (perfluorinated compounds) und
PFOS (Perfluoroctansulfonsäure) sind 2006 spektakulär in den Medien aufgetaucht
[6‐9]. Eher zufällig stießen Forscher vom Hygiene‐Institut der Uni Bonn im Juni 2006
auf einen der bislang größten NRW‐Umweltskandale. Sie untersuchten die
Konzentration von schwer abbaubaren Schadstoffen entlang des Rheins und stellten
dabei auffällig erhöhte Werte von perfluorierten Tensiden an der Mündung der Ruhr
fest. Das Team verfolgte die Spur flussaufwärts und fand mehrere Schadstoffquellen
am Oberlauf der Möhne (Scharfenberg). Auch das Trinkwasser und die Fische in Möhne, Ruhr und Rhein weisen gesundheitsschädlich hohe PFT‐Konzentrationen auf.
Mindestens 15.400 Tonnen des als „Bodenhilfsstoff“ deklarierten Sondermülls der Firma GW Umwelt gelangten auf Feld, Wald und Wiese [6‐9]. Mehr als drei Jahre nach dem PFT‐Skandal um giftigen Müll hat die Staatsanwaltschaft Anklage gegen sieben Personen erhoben. Durch die illegale Entsorgung belgischer Industrieabfälle waren die Flüsse Ruhr und Möhne verseucht worden [6]. Die Kontamination des Wassers ist auf die Abschwemmung der PFT von landwirtschaftlich genutzten Flächen zurück‐
zuführen, die mit PFT‐belastetem Dünger (Bioabfallstoffgemischen) behandelt wurden. Seit den Vorfällen ist die Aufmerksamkeit der Bevölkerung für perfluorierte Tenside als belastende Stoffe in Oberflächenwasser besonders gestiegen.
Perfluorierte Tenside sind eine Gruppe von anthropogen erzeugten Substanzen, die aufgrund ihrer besonderen Eigenschaften (zugleich fett‐ und wasserabweisend) in vielen Industriebereichen und Produkten eingesetzt werden, z.B. Feuerlöschschaum, Teflon
®‐Herstellung (PTFE), Galvanik, Halbleiterlithographie. Diese Verbindungen sind global verteilt, persistent und bioakkumulierbar. Toxizitätsuntersuchungen stufen diese Verbindungen im Tierversuch als lebertoxisch, tumorpromovierend, cancerogen und feminisierend ein [10, 11]. Aufgrund ihrer außergewöhnlichen chemischen Stabilität sind diese Substanzen biologisch nicht abbaubar. Sie stellen kommunale Kläranlagen vor eine große Herausforderung. Durch den Einsatz von sonolytischen Verfahren, die aufgrund des hohen Energieeintrages zum Kettenabbau und zur Oxidation von PFT führen, ist der Abbau jedoch möglich [12‐15].
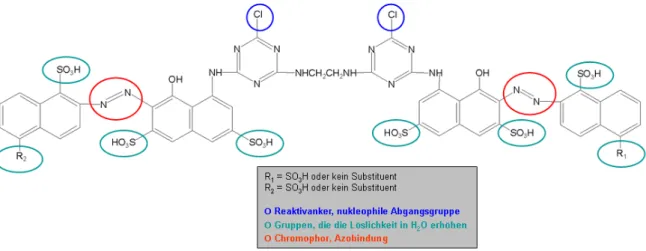
1954 wurden von der englischen Firma ICI die Reaktivfarbstoffe eingeführt und seitdem in großen Mengen für die Textilienfärbung eingesetzt [16]. Reaktivfarbstoffe bilden eine feste kovalente Bindung zwischen Farbstoff und Cellulose‐Faser. Die Beobachtung, dass bestimmte Farbstoffe, die einen Dichlortriazin‐Rest tragen, unter alkalischen Färbebedingungen überraschend waschechte Färbungen lieferten, wurde richtigerweise so gedeutet, dass über diesen Dichlortriazin‐Rest eine kovalente Bindung zur Cellulose aufgebaut wird [17]. Die Hydroxylgruppen der Glucoseeinheiten der Cellulose verdrängen am Triazin einen der Chlorreste und bauen so eine stabile Bindung auf.
Als Chromophore (griech. „Farbträger“) für Reaktivfarbstoffe kommen prinzipiell alle bekannten Chromophore in Frage. 75% der bekannten Chromophore werden von der Gruppe der Azofarbstoffe gestellt, mit denen praktisch alle Nuancen von Gelb bis Schwarz abgedeckt werden können [17].
Die Hydrolyse des Reaktivfarbstoffs während der Textilienfärbung ist eine ernst zu
nehmende Nebenreaktion. Da das Hydrolyseprodukt nur ungenügend auf die Faser
aufzieht, muss es durch Seifungsschritte komplett abgewaschen werden und gelangt
so letztlich ins Abwasser, aus dem es entfernt werden muss. Die Ökonomie und
Ökologie der Reaktivfärbung hängt also stark von der sogenannten Fixierausbeute ab
[17]. Damit ist der prozentuale Anteil der im Färbebad angebotenen Farbstoffmenge gemeint, der kovalent an die Faser gebunden wird.
Reaktivfarbstoffe mit zwei Reaktivhaken (bifunktionelle Farbstoffe) sind in den letzten Jahren aus ökologischen und ökonomischen Gründen immer wichtiger geworden, da sie zu Fixierausbeuten von 90% und mehr führen [18, 19]. Einerseits sucht die Textil‐ und Farbstoffindustrie nach Möglichkeiten zur Verbesserung der Fixierausbeute, andererseits bedarf es innovativer, effizienter Verfahren zur Reinigung des farbigen und toxischen Abwassers [20, 21]. Hier sind spezielle Verfahren notwendig, da kommunale Kläranlagen nicht in der Lage sind, das farbige Abwasser ausreichend zu reinigen [3]. Daher wird im Rahmen dieser Arbeit ein zweistufiges Verfahren zur Behandlung des roten Azofarbstoffs DRIMARO charakterisiert und weiterentwickelt.
Hinsichtlich dieser beiden Stoffgruppen stellt sich die Frage, welche Möglichkeiten der Abwasserreinigung effektiv und sicher sind.
2. Zielstellung
Ziel dieser Arbeit sind Untersuchungen sowie Bewertungen zum Einsatz von Verfahren zur Behandlung von industrierelevanten, aktuell problematischen, höhermolekularen organischen, teils stark persistenten und bioakkumulierbaren Stoffen in Wasser. Im Fokus stehen organische Verbindungen, die mit etablierten Verfahren der kommunalen Abwasserreinigung nicht befriedigend zu beseitigen sind, sondern bei denen es besonderer Behandlungsverfahren bedarf.
Im Vordergrund stehen die Entwicklung von geeigneten Behandlungsverfahren und die Entwicklung von analytischen Methoden zur Beurteilung der chemischen Veränderungen während des Abbaus. Die folgenden Verfahren werden als spezielle Behandlungsmethoden getestet, die als Einzelmethode oder in Kombination mit etablierten Verfahren zum Einsatz kommen sollen:
• Biologischer Abbau (anaerob und aerob)
• Ultraschallbehandlung.
Alle Verfahren kommen ohne den Einsatz zusätzlicher, belastender Chemikalien aus.
Gezeigt werden soll, dass sich diese Verfahren für die konkreten Stoffe zeichnen sich durch Umweltverträglichkeit, Effizienz sowie eine geeignete Raum/Zeit‐Ausbeute auszeichnen. Wünschenswert ist die Mineralisierung der problematischen Chemikalien, zumindest aber eine molekulare Degradierung der Substanzen.
Bei der Verfahrensentwicklung wird sich auf zwei verschiedene Stoffgruppen konzentriert, die industrielle Relevanz haben. Zum einen werden perfluorierte Substanzen (Perfluorinated compounds = PFC), zum anderen wird ein industrierelevanter Azofarbstoff untersucht. Die Stoffe stellen beide Problemfälle in der industriellen sowie in der kommunalen Abwassertechnologie dar. Werden die Stoffe durch die Behandlung nicht mineralisiert, müssen die entstehenden Intermediate aufgeklärt und auf ihre Toxizität untersucht werden.
Ein Schwerpunkt der Untersuchungen ist die Aufklärung der Abbaumechanismen bzw. des Metabolismus durch quantitative Online‐Analytik der entstehenden Produkte sowie die qualitative Strukturanalyse dieser Abbauprodukte.
Zusammenfassend sind die wichtigsten Ziele die Aufklärung der Reaktions‐
mechanismen, die Optimierung der Prozessparameter sowie die Bewertung der jeweiligen Behandlungsverfahren.
Grundlage für diese Promotionsarbeit sind die seit 1998 zahlreichen Diplomarbeiten, Masterarbeiten und Dissertationen [21, 98, 292, 320, 323] in der Arbeitgruppe von Frau Prof. Dr. A. Rehorek sowie Erfahrungen der Arbeitsgruppe.
2.1. Entwicklung eines Abbauverfahrens für perfluorierte Tenside
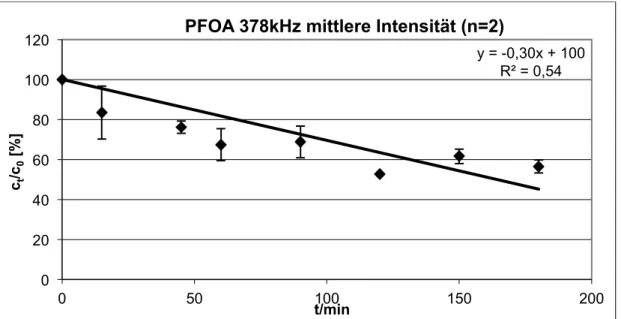
Untersucht werden soll das Abbauverhalten von aliphatischen perfluorierten Tensiden unterschiedlicher Kettenlänge und einer perfluorierten Sulfonsäure durch sonochemischen Energieeintrag.
Bei einem Reaktionsvolumen von 250mL sollen die sonochemischen Behandlungen erfolgen. Die Behandlungsart kann variiert werden, und zwar durch vier zur Verfügung stehende Frequenzen sowie durch unterschiedlichen Leistungseintrag durch die Veränderungen der Intensität der Amplitude.
Durch quantitative
19F‐NMR Messungen sollen die Abbaukinetik der Edukte ermittelt und die Produkte identifiziert und quantifiziert werden. Zur Betrachtung der Wirtschaftlichkeit sollen Energieeintrag und Radikalausbeute bestimmt werden.
2.1.1. Zielsetzung für diese Untersuchungen
1. Entwicklung eines geeigneten Monitoring‐Verfahrens für die Bestimmung von perfluorierten Tensiden.
2. Entwicklung eines neuen sonochemischen Verfahrens zur Behandlung von verschiedenen perfluorierten Tensiden: Frequenz, Behandlungsdauer sowie Leistungseintrag sollen für verschiedene Substanzen optimiert werden.
3. Beurteilung der Ultraschallbehandlungen hinsichtlich einer Einschätzung des Reaktionsmechanismus als eher pyrolytischen oder überwiegend radikalischen Mechanismus.
4. Untersuchung des Einflusses des pH‐Werts auf die Behandlung.
5. Ermittlung von Unterschieden des Abbaus von perfluorierten Carbon‐ und Sulfonsäuren sowie der Kohlenstoffkettenlänge.
2.2. Entwicklung eines Abbauverfahrens für den Azofarbstoff DRIMARO
Ziel der Untersuchungen ist die Entwicklung eines anaerob‐aeroben Abbauverfahrens für den Azofarbstoff DRIMARO, wodurch der Eintrag farbiger und toxischer Stoffe in den Vorfluter erheblich vermindert werden soll.
Im Vordergrund für die Auslegung eines Abbauverfahrens soll die Aufklärung von Abbaumechanismen bzw. Metabolismen durch quantitative Online‐Analytik der ent‐
stehenden Produkte sowie qualitativer Strukturanalyse mit Hilfe von LC‐DAD‐IC‐ESI‐
MS
nstehen.
Durch quantitative Messungen kann die Kinetik ermittelt werden und durch qualitative Messungen können Metabolite identifiziert werden.
Die analytischen Bestimmungen sollen die Abbau‐Kinetik zur Bestimmung der Reaktionsordnung und Geschwindigkeitskonstanten liefern. Ziel ist es die Prozessparameter zu optimieren. Mit Hilfe der bekannten Reaktorgeometrie können Reaktorverweilzeiten berechnet werden und zu einem Scale Up auf ähnliche Reaktortypen angewandt werden.
2.2.1. Zielsetzung für diese Untersuchungen
1. Entwicklung eines geeigneten Online‐Analytik‐Monitoring Verfahrens: Eingesetzt werden eine getauchte Inline‐Sonde, ein Pumpensystem, ein Bypass sowie die Kombination aus Ionenpaarchromatographie (HPLC), UV‐Vis‐Detektor, Supressor und ein Triple‐Quadrupol‐Massenspektrometer mit Elektrosprayionisierung und Ionenfalle.
2. Charakterisierung des anaeroben bzw. anaeroben‐aeroben Verfahrens zur Ent‐
färbung anhand eines akut problematischen, industriellen Azofarbstoffes: Biologische Verfahren konkurrieren auf dem Gebiet der Entfärbung von Farbstoffen mit physiko‐
chemischen Verfahren. Biologische Verfahren verbrauchen weniger Energie; sie sind kostengünstiger und nachhaltiger.
3. Ermittlung der Abbau‐Kinetik zur Bestimmung und Optimierung der Entfärbe‐
kapazität des Bioreaktors unter verschiedenen Betriebsbedingungen: Temperatur, pH‐Wert, Redox‐Potential, mechanische Belastung und Cosubstrate/Induktoren sind Parameter, die den Stoffwechsel von Biozönosen beeinflussen. Durch Variation dieser Betriebsparameter soll ein optimaler Reaktorbetrieb mit minimalen Verweilzeiten erzielt werden.
4. Spezielle, mechanistische Untersuchungen zur Öffnung der Azobindung in Abhängigkeit vom Redox‐Potential: Die anaerobe Biozönose liefert Redox‐
Äquivalente für die reduktive Spaltung der Bindung. Untersucht wird die Öffnung der
Bindung in Abhängigkeit vom Redox‐Wert sowie dem Zustand der eingesetzten Schlammart.
5. Testung einer sonochemischen Nachbehandlung der persistenten Metabolite: Die Azofarbstoffe können biologisch nicht vollständig metabolisiert und mineralisiert werden. Die Behandlung mit Ultraschall eignet sich hierfür. Frequenz, Be‐
handlungsdauer und Leistungseintrag müssen optimiert werden.
3. Theorie
3.1. Persistente, perfluorierte Verbindungen
3.1.1. Übersicht perfluorierter Verbindungen
Perfluorierte Verbindungen sind Substanzen mit der allgemeinen Formel F(CF
2)
n‐R, bestehend aus einer hydrophoben Alkylkette unterschiedlicher Länge (typischer‐
weise C
4bis C
16) und einer hydrophilen Endgruppe. Die teilweise fluorierten Substanzen beinhalten −CH
2CH
2– zwischen dem hydrophilen Teil und der perfluorierten Kette mit der allgemeinen Formel (F(CF
2)
n‐CH
2CH
2‐X). Diese Substanzen werden als Telomere bezeichnet. Telomere sind Precursor einiger in der Umwelt auffindbarer perfluorierter Verbindungen [22].
Name Formel Acronym CAS Nr.
aSulfonsäuren
Perfluorbutansulfonsäure F(CF
2)
4SO
3‐PFBS 375‐73‐5 Perfluoroctansulfonsäure F(CF
2)
8SO
3‐PFOS 1763‐23‐1
Carbonsäuren
Perfluorbutanat F(CF
2)
3CO
2‐PFB 375‐22‐4
Perfluoroctanat F(CF
2)
7CO
2‐PFOA 335‐67‐1
Perfluornonansäure F(CF
2)
8CO
2‐PFN 375‐95‐1
Alkohole
Perfluorhexylethanol F(CF
2)
6CH
2CH
2OH 8‐2 FTOH 678‐39‐7
Andere
1H,1H,2H,2H‐Perfluor‐
octansulfonsäure
F(CF
2)
6CH
2CH
2SO
3‐6‐2 FTS 27619‐97‐2 Perfluoroctylsulfonamid F(CF
2)
8SO
3NH
2PFOSA 754‐91‐6
Tab. 1: Beispiele für verschiedene perfluorierte Verbindungen [22]
aFür Säuren ist die CAS Nr. die protonierte Form
In Tab. 2 sind Strukturen von perfluorierten Verbindungen dargestellt, die im
Rahmen dieser Arbeit behandelt werden.
Substanz‐
klasse
Vertreter Struktur
Perfluorierte Alkylsulfonate
(PFAS)
Perfluoroktan‐
sulfonsäure
(PFOS)
Perfluorbutan‐
sulfonsäure
(PFBS)
Perfluorierte Carbonsäuren
(PFCA)
Perfluoroktan‐
säure
(PFOA)
Fluortelomere Telomersulfonat
Tab. 2: Strukturen von speziellen perfluorierten Verbindungen
3.1.2. Anwendungsgebiete
Perfluorierte Verbindungen sind amphiphil und oberflächenaktiv, weshalb sie industriell breit genutzt werden. Das Spektrum verschiedener Industriebranchen spricht für sich: die Textilindustrie in Form von Scotchgard
®und Goretex
®als Imprägnierungsmittel für atmungsaktive Jacken [23], die Papierindustrie für schmutz‐, fett‐ und wasserabweisende Papiere, die Haushaltsindustrie für Pfannen‐
und Topfbeschichtung (z.B. PTFE/ Teflon
®, Reinigungsmittel, Kleber, Farben, Lacke, Polituren), für fett‐ und wasserabweisende Lebensmittelverpackungen (Pizza–
kartons, Fastfoodverpackungen, Backpapier) [24, 25], die Feuerwehr für Feuerlösch–
schäume, die Landwirtschaft zur Verbesserung der Sprüheigenschaften von Pestiziden, die Möbel‐, Teppichindustrie, die Glas‐, Chip‐, Metallindustrie (Galvanik) sowie Luft‐ und Raumfahrt (vergleiche Tab. 3) [26, 27].
Industriezweig Verwendung
Textilindustrie Imprägnierungsmittel: atmungsaktive Jacken (z.B. Scotchgard, Goretex)
Papierindustrie schmutz‐, fett‐ und wasserabweisende Papiere Möbel‐,
Teppichindustrie Imprägnierung, Polituren, Reinigungsmittel Glasindustrie Antifoggingmittel
Chipindustrie Antistatika
Haushaltsindustrie Pfannen‐ und Topfbeschichtung (z.B. Teflon, Reinigungsmittel, Kleber, Farben, Lacke, Polituren
Lebensmittel‐
Verpackungen
fett‐ und wasserabweisende Verpackungen, Pizzakarton, Fastfood‐
verpackungen Feuerwehr Feuerlöschschäume
Landwirtschaft Pestizide (zur Verbesserung der Sprüheigenschaft)
Metallindustrie Chrombäder (um Aerosolbildung im Sinne des Arbeitsschutzes zu verhindern)
Sport Zusatz für Skiwachs, Outdoorbekleidung, Regenbekleidung Luft‐ und
Raumfahrt Hydraulikflüssigkeiten
Tab. 3: Verwendung von perfluorierten Verbindungen [27]
3.1.3. Richtlinien für PFOS [28, 29]
Mit der Richtlinie 2006/122/EG erfolgte eine Änderung der Richtlinie 76/769/EWG.
Damit wird ab dem 27.06.2008 das Inverkehrbringen und Verwenden von PFOS und PFOS‐haltigen Zubereitungen und Erzeugnissen weitestgehend verboten.
Folgende Regeln sind in Kraft getreten:
1. PFOS darf nicht als Stoff oder Bestandteil von Zubereitungen in einer
Konzentration von 0,005 Massenprozent oder mehr in Verkehr gebracht oder
verwendet werden.
2. PFOS darf nicht in Halbfertigerzeugnissen oder Erzeugnissen oder Bestandteilen davon in Verkehr gebracht werden, wenn die Massenkonzentration 0,1% oder mehr beträgt, berechnet im Verhältnis zur Masse der strukturell oder mikrostrukturell verschiedenartigen Bestandteile, die PFOS enthalten, oder bei Textilien oder anderen beschichteten Werkstoffen mit einem PFOS‐Anteil von 1μg/m² oder mehr des beschichteten Materials.
3. Abweichend hiervon gelten die Nummern 1 und 2 nicht für folgende Produkte und die für deren Erzeugung erforderlichen Stoffe und Zubereitungen:
• Fotoresistlacke und Antireflexbeschichtungen für fotolithografische Prozesse,
• Fotografische Beschichtungen von Filmen, Papieren und Druckplatten,
• Antischleiermittel für nicht‐dekoratives Hartverchromen (Chrom VI) und Netzmittel für überwachte Galvanotechniksysteme, bei denen die Menge der PFOS‐
Emissionen in die Umwelt durch vollständigen Einsatz der einschlägigen besten verfügbaren Technologien, die im Rahmen der Richtlinie 96/61/EG des Rates vom 24. September 1996 über die integrierte Vermeidung und Verminderung der Umweltverschmutzung entwickelt worden sind, auf ein Mindestmaß reduziert wird,
• Hydraulikflüssigkeiten für die Luft‐ und Raumfahrt.
Das Ministerium für Klimaschutz, Umwelt, Landwirtschaft, Natur‐ und Verbraucher‐
schutz des Landes NRW schreibt, dass PFT „hauptsächlich durch gewerbliche Ab‐
wassereinleitungen in die Gewässer gelangen [30].“ Das hatte zur Folge, dass das Umweltministerium am 7. August 2007 mit den entsprechenden Industrieverbänden eine freiwillige Vereinbarung getroffen hat mit dem Ziel der Reduzierung des Eintrags von PFT ins Abwasser. Unterzeichnet wurde diese vom Zentralverband Oberflächen‐
technik e.V., vom Verband Deutscher Papierfabriken, vom Verband der Chemischen Industrie – VCI (Landesverband Nordrhein‐Westfalen), vom Verband der Nord‐
deutschen Textil‐ und Bekleidungsindustrie, vom Verband TEGEWA e.V., vom Chemieverband Imaging und Photo e.V., vom Gesamtverband Textil und Mode sowie vom Verband Deutscher Maschinen‐ und Anlagenbau. Man einigte sich über folgende Agenda [30]:
• Verwendung anderer PFT‐freier Einsatzstoffe,
• Umstellung auf digitale Medien im Bereich des Einsatzes von Röntgenfilmen,
• Schließung von Wasserkreisläufen in der Produktion,
• Abtrennung und getrennte Entsorgung der mit PFT belasteten Abwasserteil‐
ströme,
• Behandlung des Abwassers mit Aktivkohle.
In der Galvanikindustrie, die ca. 80 Prozent der PFOS‐haltigen Abwassereinleitungen
verursacht, sind die Umstellungsprozesse sehr viel aufwändiger [30]. Die Veredler
von Oberflächen schreiben, sie „sind sich ihrer Verantwortung bewusst und haben
Antworten zur Minimierung des Eintrags in die Umwelt gefunden.“ Als Ersatzstoff
wird das sogenannte H
4PFOS (C
8F
13H
4SO
3‐) vorgeschlagen [31]. H
4PFOS ist poly‐
fluoriert und nicht perfluoriert, ebenfalls schwer abbaubar, mobil im Grundwasser und es ist im Vergleich zu PFOS eine höhere Einsatzmenge erforderlich. Über die toxischen bzw. ökotoxischen Eigenschaften und das sonstige Umweltverhalten dieser Produkte ist bisher wenig bekannt [29]. Ob es sich um einen echten Ersatz mit weniger toxischen Wirkungen handelt oder ob es nur eine vom Gesetz nicht abgedeckte Nische ist, ist fraglich. Die Suche nach geeigneten Alternativen ist somit noch nicht abgeschlossen.
3.1.4. Gesundheitsgefährdung durch perfluorierte Verbindungen
Perfluorierte Verbindungen haben eine Gemeinsamkeit: Einen anthropogenen Ur‐
sprung. Dennoch kommen sie mittlerweile ubiquitär vor. Aufgrund ihrer Bio‐
akkumulierbarkeit wurden sie in Polarbären, Robben sowie in vielen Nahrungs‐
mitteln nachgewiesen [14, 32, 33].
Perfluorierte Verbindungen sind im Tierversuch als lebertoxisch und tumor‐
promovierend eingestuft. Sie sind cancerogen, im Fischversuch feminisierend, Fertilitäts‐ und Reproduktionstoxisch. Viele Vertreter dieser Substanzgruppe (z.B.
PFOS, PFOA) weisen eine sehr hohe Verweilzeit (Halbwertszeit 8‐9 Jahre) im menschlichen Körper auf. Ein Zusammenhang zwischen der Exposition von PFOS und Blasenkrebs sowie zwischen der Exposition von PFOA und Prostatakrebs konnte bei Arbeitern nachgewiesen werden [10].
PFOS und PFOA zirkulieren im Körper, indem sie an Serumproteine binden und in der Leber akkumulieren [34]. Es wird vermutet, dass diese Verbindungen vom Körper als Gallensäuren erkannt und über den enterohepatischen Kreislauf recycliert werden [35]. Ein möglicher Mechanismus, welcher mit der Karzinogenese in Verbindung gebracht wird [11], ist die Inhibierung der interzellulären Kommunikation über sogenannte gap junctions, die durch verschiedene perfluorierte Verbindungen erfolgt [35‐37].
Perfluorierte Verbindungen haben in den letzten Jahren eine große Bedeutung bekommen, was sich auch an der Anzahl der Publikationen in der wissenschaftlichen Literatur widerspiegelt:
Abb. 1: Anzahl der “peer‐reviewed” Literatur über toxikologische Effekte und Konzentrationen in der Umwelt von poly‐ und perfluorierten Substanzen, basierend auf die “keywords” in CAplus und MEDLINE für „C8, C10, PFOS, PFOA, perfluorinated, and perfluoroalkyl” [39]
3.1.5. Anreicherung in der Umwelt
Perfluorierte Verbindungen sind besonders persistent und chemisch sehr stabil.
Durch die hohe Elektronegativität des Fluors handelt es sich bei der C‐F‐Bindung um eine der stärksten in der organischen Chemie (>450 kJ/mol, Reduktions‐
potential: ‐3,6V [40]). Des Weiteren schützen drei Paar ungebundene Elektronen an jedem Fluor‐Atom die Molekülstruktur vor Reaktionen. Das zusammen bedeutet, dass ein enorm hoher Energieeintrag notwendig ist, um perfluorierte Verbindungen abzubauen. Momentan gelten PFOA und PFOS nach dem Stand der Literatur als biologisch nicht abbaubar [21, 41].
Die kritischsten Stoffeigenschaften industrieller perfluorierte Verbindungen sind: Sie haben eine sehr starke Säurefunktion, sie sind starke Tenside, haben eine unpolare perfluorierte Gruppe und eine polare Sulphonsäuregruppe. Ihre Aufnahme über die Nahrung und das Trinkwasser ist nachgewiesen [42, 43].
Der Nachweis ist in vielen Gewässern erfolgt [9, 44‐46], seit 2002 wurde ein Produktionsstop einiger PFC‐Vertreter (PFOS, PFOA) aller Chemieunternehmen in Deutschland durchgesetzt. 2009 wurde PFOS in den Anhang B der Stockholmer Konvention aufgenommen und zählt somit zum „Dirty Dozen“. Deutschland setzt sich dafür ein, dass PFOS in das POPs‐Übereinkommen (POP = persistant organic pollutants) aufgenommen wird.
Das mengenmäßig stärkste Haupteinsatzgebiet von perfluorierten Verbindungen ist Feuerlöschmittel. 2005 wurden laut einer OECD‐Schätzung 122t/a PFOS verbraucht (vergleiche Tab. 4).
Einsatzgebiet Verbrauch (t/a) Feuerlöschmittel 122
Verchromung 10
Photographie 1
Luftfahrt 0,73
Halbleiterindustrie 0,5
Tab. 4: Geschätzter PFOS‐Verbrauch in der EU pro Jahr (OECD 2005)
Aufgrund des Produktionsstops und der eingeschränkten Einsetzbarkeit wurden Stoffe zum Substituieren von PFOS entwickelt [29, 31]. Diese besitzen entweder eine kürzere C‐Kette als Grundgerüst, oder aber weniger Fluor (z.B. Fluortelomeralkohole, H
4PFOS). Perfluorierte Telomeralkohole (FTOH) werden als Precursoren bei der Herstellung fluorierter Polymere für ähnliche Anwendungen wie auf PFOS‐
basierende Sulfonamidoethanole (FOSE) eingesetzt, indem der perfluorierte Alkylrest über die Ethanolfunktion mit einem Polymerrückgrat verknüpft wird. Die weltweite Produktion von FTOH wird für die Jahre 2000–2002 auf 5000 Tonnen geschätzt [47]. Perfluortenside werden in Deutschland u.a. von der Firmen Clariant, Dyneon und ABCR verwendet bzw. vertrieben [38].
3.1.6. Herstellungsverfahren von PFT
Zur Herstellung perfluorierter Tenside werden in der industriellen Synthetisierung meist die Verfahren der elektrochemischen Fluorierung nach Simons oder der Fluortelomerisierung angewandt [48]. Jährlich werden mehrere tausend Tonnen PFT hergestellt, das Land mit der größten Produktionsmenge sind die USA.
Fluortelomerisierung [48]
Im Herstellungsprozess von perfluorierten Tensiden wie Fluortelomeralkoholen, der Fluortelomerisierung, besteht ein Prozessschritt aus einer Telomerisation. Dabei werden n Moleküle Tetrafluorethylen mit Pentafluorethyliodid umgesetzt. Weitere Derivatisierung sind aber auch möglich, wie z.B. Fluortelomeralkohole, ‐sulfonate und ‐carboxylate.
F
F F
F H
H H
H
C C
C C
C C
C C
S F F
F F
F F
F F F F
F F F F F
F F
HN
O O
R Telomerisierung +
Abb. 2: Fluortelomerisierung
Elektrochemische Fluorierung [38, 48]
Die elektrochemische Fluorierung nach Simons ist neben der Fluortelomerisierung das zweite Verfahren zur Herstellung perfluorierter Tenside. Durch eine angelegte Spannung (4,5‐7,0V) wird erreicht, dass alle Wasserstoffatome der n‐Octansäure bzw. des Octansulphonylfluorids mit wasserfreier Flusssäure im Überschuss, durch Fluoratome ersetzt werden. Die Reaktion läuft auf folgende Weise ab:
C C
C C
C C
C C
S H H
H H H H
H H H H
H H H H H
H H
O
O O
C C
C C
C C
C C
S F F
F F
F F
F F F F
F F F F F
F F
O
O O
ECF
Simons Verfahren
+ perfluorierte Nebenprodukte unterschiedlicher Kettenlänge und Verzweigungsgrades
![Abb. 30: Leistungscharakterisierung von Ultraschallsystemen [303]](https://thumb-eu.123doks.com/thumbv2/1library_info/3667446.1504126/64.892.117.606.407.759/abb-leistungscharakterisierung-von-ultraschallsystemen.webp)
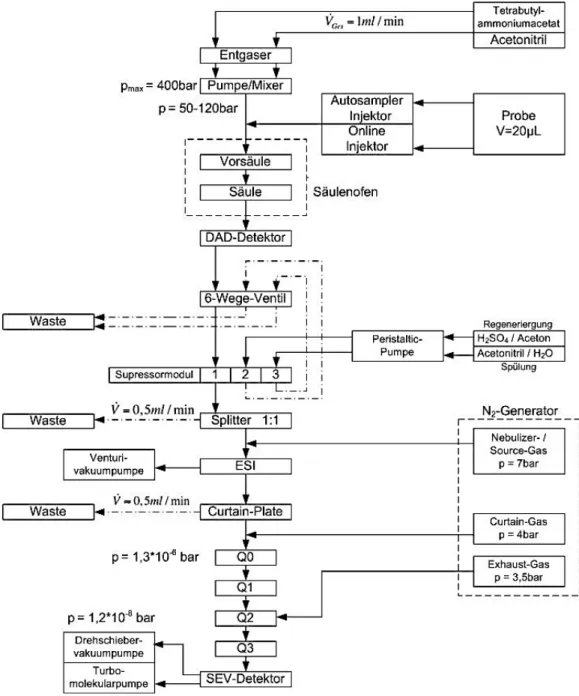
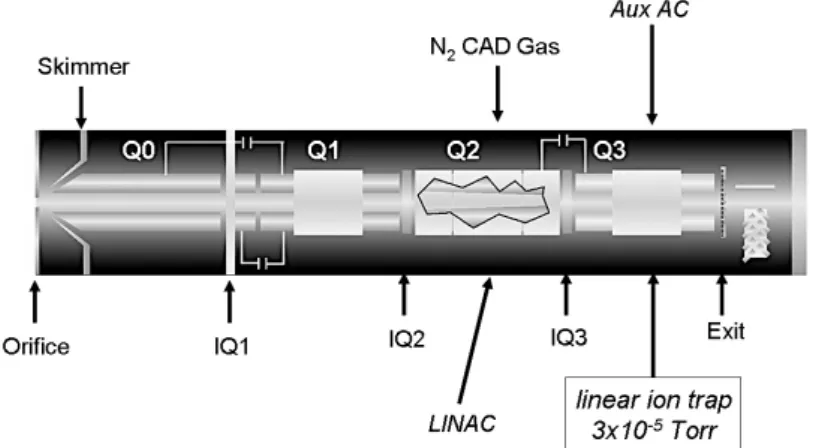
![Abb. 39: Verfahrensfließbild des anaerob‐aerob Bioreaktorsystems, Stand 2006 [154]](https://thumb-eu.123doks.com/thumbv2/1library_info/3667446.1504126/73.892.118.733.220.1057/abb-verfahrensfließbild-des-anaerob-aerob-bioreaktorsystems-stand.webp)

![Abb. 42: Labview Bedienungsoberfläche für den Anaerob‐aerob Bioreaktor [22]](https://thumb-eu.123doks.com/thumbv2/1library_info/3667446.1504126/77.892.120.742.109.509/abb-labview-bedienungsoberfläche-für-den-anaerob-aerob-bioreaktor.webp)