Synthese und biologische Evaluierung Sterolmodifizierter Hedgehog-Signalpeptide und Proteine
157
0
0
Volltext
(2)(3)
(4)(5)
(6)(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
(26)
(27)
(28)
(29)
(30)
(31)
(32)
(33)
(34)
(35)
(36)
(37)
(38)
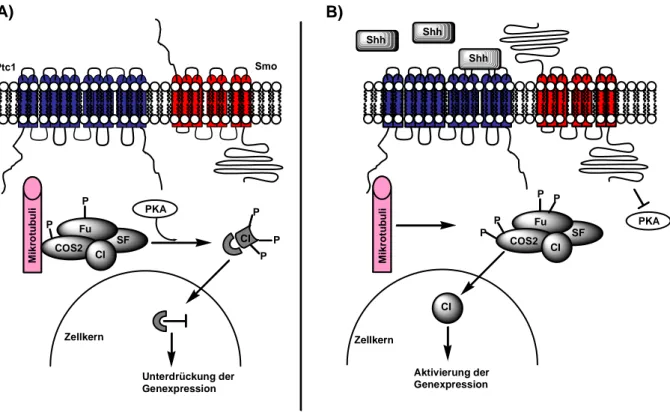
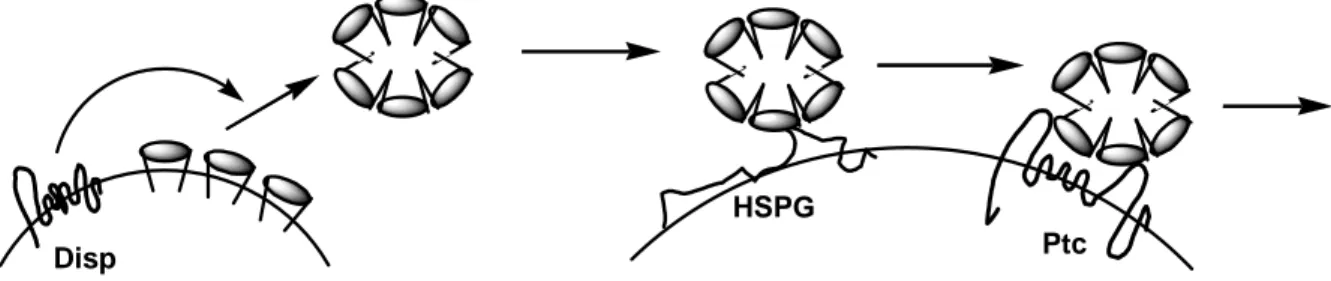
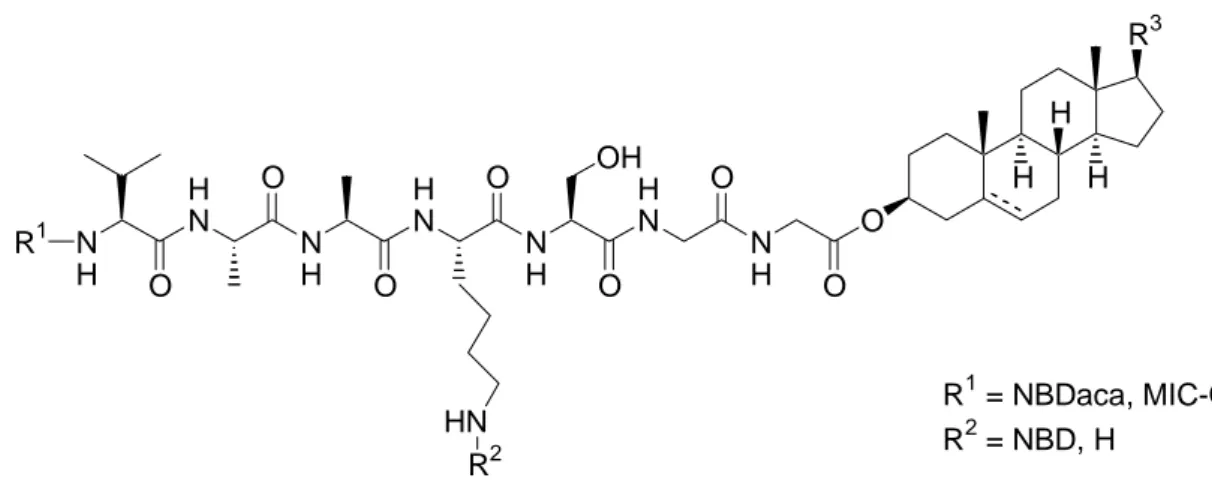
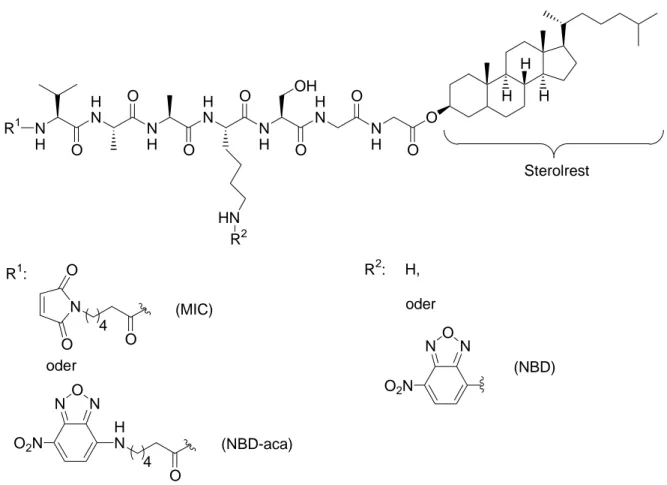
Abbildung

+7



ÄHNLICHE DOKUMENTE
Da sich im Rahmen meiner Diplomarbeit gezeigt hat, dass die exo-Methylengruppe von 33 essentiell für dessen Cytotoxizität ist, [96] das DMA-Derivat von Arglabin™
[67;77] Es konnte gezeigt werden, dass H- und N-Ras fünf bis acht Stunden nach der Translation mit dem intrazellulären Membransystem (ER- und Golgi-Apparat) assoziiert sind, was auf
Die vereinigten organischen Phasen werden dreimal mit je 10 ml gesättigter NaCl-Lösung gewaschen, über MgSO 4 getrocknet und das Lösungsmittel unter vermindertem
Das Lösungsmittel wird unter vermindertem Druck abdestilliert, 400 ml Toluol zugesetzt, die Phasen getrennt und die organische Phase dreimal mit je 300 ml Dichlormethan
Diese wurden anschließend mittels in vitro Inhibitions-Assays auf ihre biochemische Wirksamkeit gegen die Phosphatasen Cdc25A, MPTPA, PP1, PP2A, VHR und PTP1B, sowie
Die Forschung konzentrierte sich dabei vor allem auf CBI und anti-Methyl-CBI-Analoga, die mit unterschiedlichen Glykosiden und DNA-bindenden Einheiten gekuppelt wurden
65 Zur Abschätzung einer möglichen Therapie im Rahmen von ADEPT durch ein Prodrug wurden folgende Kriterien erarbeitet: 66 „Das Prodrug sollte ein adäquates Substrat für
Zudem konnte in einer Studie eine physiologische microRNA-21 Expression in der Leber von Schweinen gezeigt werden (Hinkel et al., 2020), wodurch wiederum die
