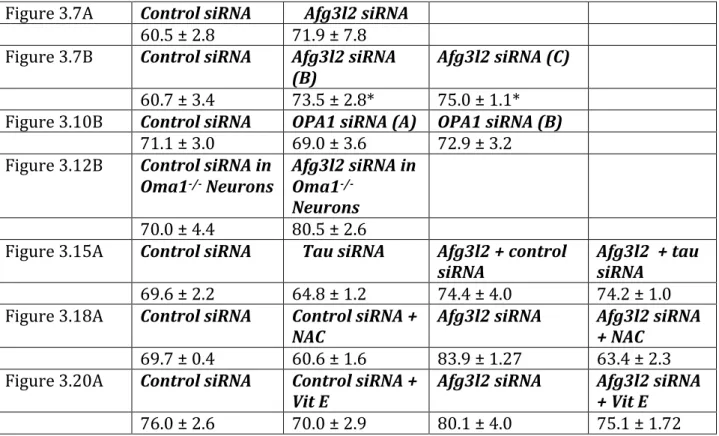
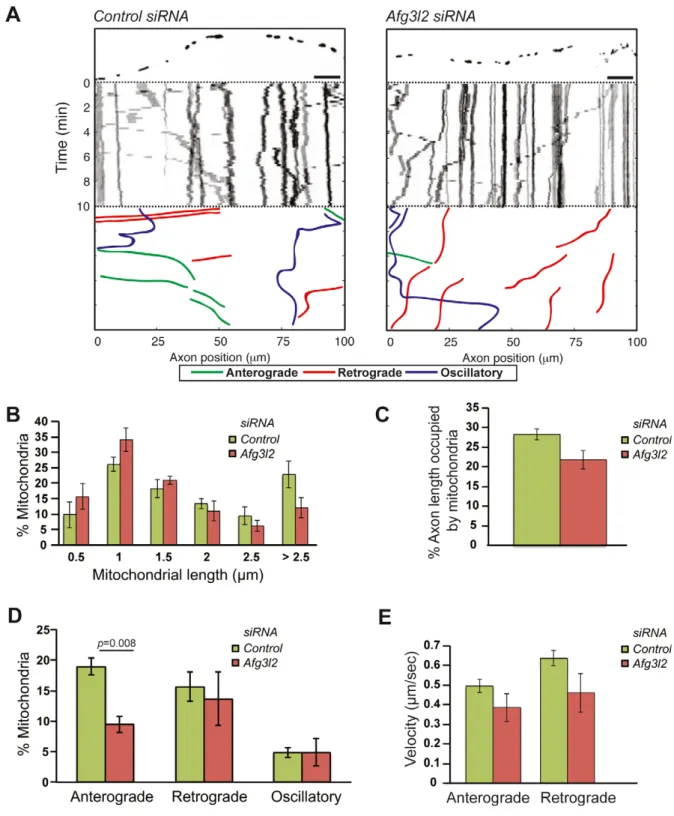
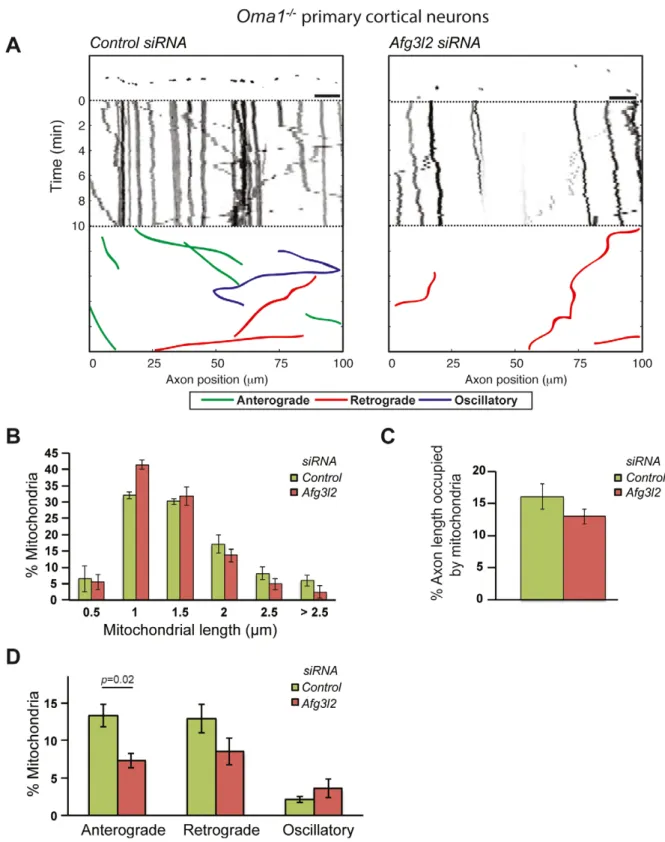
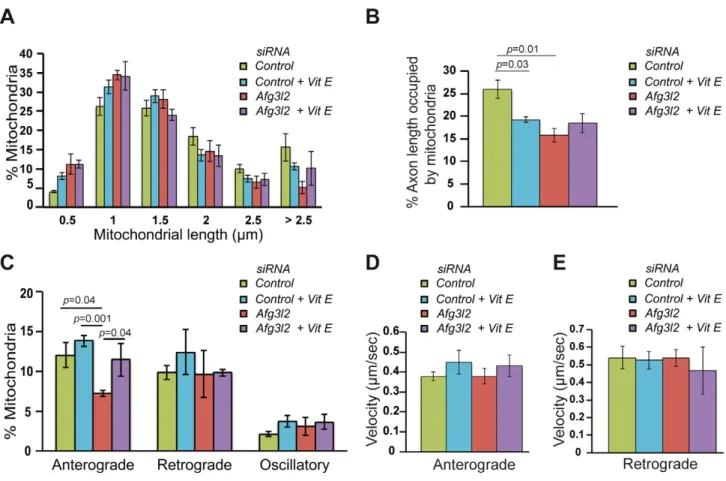
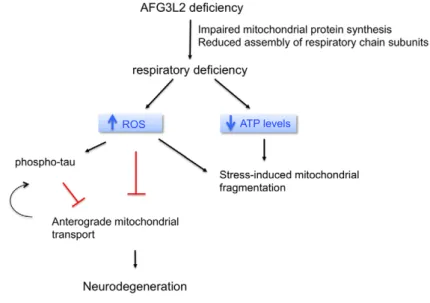
AFG3L2 deficiency impairs axonal transport of mitochondria dependent on ROS and tau levels
161
0
0
Volltext
(11)
(14)
(17)
(18)
(19)
(20)
(21)
(22)
(25)
(26)
(27)
(28)
(29)
(32)
(33)
(34)
(35)
Abbildung

+3
ÄHNLICHE DOKUMENTE
In order to evaluate whether the individual volume or the number of mitochondria caused the increase in total mitochondrial volume we estimated the number and the number-weighted
In summary, there is a great deal of data in support of localized synthesis of proteins at the mitochondrial outer membrane, yet the co-localization of cytosolic ribosomes with
“ membrane arm ” [25,26]. However, the overall shape varies in different organisms as revealed by electron microscopy. For instance, plant complex I has a second matrix-exposed
Therefore, in this thesis, the permeabilized primary cultures of cerebellar granule cells were used to characterize mitochondrial function concerning respiratory activity,
The presence of clonogenic cells in the suspension of apoptotic bodies from mela- noma cells used for pulsing of dendritic cells with tumor antigens might compromise this protocol
The cell death and survival of proliferating (clonogenic) cells were investigated in two human melanoma cell lines to assess the optimal conditions for preparation of apoptotic
Canine distemper virus encephalitis and intervertebral disk disease causing spinal cord injury represent naturally occuring translational animal models to
Figure 2: Mitochondrial complex I deficiency results in increase of branching of C. elegans sensory neurons. a) Representative confocal images (left panel) and their
