1
NMR- SPEKTROSKOPISCHE U NTERSUCHUNGEN VON TETRAVALENTEN A CTINID -K OMPLEXEN MODIFIZIERTER
S ALENLIGANDEN
Masterarbeit
Technische Universität Dresden
Fakultät Mathematik und Naturwissenschaften Fachrichtung Chemie und Lebensmittelchemie
Professur für Radiochemie
eingereicht von Bodo Felsner geb. am 18.04.1995
Gutachter: Prof. Dr. Thorsten Stumpf Gutachter: Dr. Peter Kaden
2
Inhaltsverzeichnis
1 Motivation und Zielstellung ... 4
2 Grundlagen und Stand der Literatur ... 6
2.1 Actinide ... 6
2.2 Iminliganden /Salenderivate ... 7
2.3 NMR-Spektroskopie... 9
3 Ergebnisse und Diskussion ... 16
3.1 Hydroxykomplexe ... 16
3.1.1 SC-XRD ... 16
3.1.2 IR-Spektroskopie ... 18
3.1.3 NMR-Spektroskopie ... 18
3.2 Fluorokomplexe - SC-XRD ... 20
3.2.1 [Th(fsalen)2] ... 20
3.2.2 [U(fsalen)2] ... 22
3.2.3 [Th(fsalpn)2] und [U(fsalpn)2] ... 24
3.3 Fluorokomplexe – IR-Spektroskopie ... 27
3.4 Fluorokomplexe – NMR-Spektroskopie... 29
3.4.1 Freie Liganden H2fsalen und H2fsalpn ... 29
3.4.2 Diamagnetische Komplexe ... 33
3.4.3 Paramagnetische Komplexe ... 39
4 Zusammenfassung und Ausblick ... 49
5 Experimentalteil ... 51
5.1 Geräte und Verfahren ... 51
5.2 Chemikalien ... 52
5.3 Experimentelle Daten ... 53
5.3.1 [Th(Hysalen)2] ... 53
5.3.2 [U(Hysalen)2]... 54
5.3.3 [Th(Hysalpn)2] ... 54
3
5.3.4 [U(Hysalpn)2] ... 55
5.3.5 [Th(Hysalcn)2] ... 56
5.3.6 [U(Hysalcn)2] ... 56
5.3.7 H2fsalen... 57
5.3.8 [Zr(fsalen)2] ... 58
5.3.9 [Hf(fsalen)2] ... 59
5.3.10 [Ce(fsalen)2] ... 60
5.3.11 [Th(fsalen)2] ... 61
5.3.12 [U(fsalen)2] ... 62
5.3.13 [Np(fsalen)2] ... 63
5.3.14 H2fsalpn ... 64
5.3.15 [Zr(fsalpn)2] ... 65
5.3.16 [Hf(fsalpn)2] ... 66
5.3.17 [Ce(fsalpn)2] ... 67
5.3.18 [Th(fsalpn)2] ... 69
5.3.19 [U(fsalpn)2] ... 70
6 Literaturverzeichnis ... 72
7 Selbständigkeitserklärung ... 74
8 Anhang... 75
4
1 Motivation und Zielstellung
Die Kernspinresonanzspektroskopie (nuclear magnetic resoncance – NMR) ist eine besonders in der organischen Chemie gängige Methode zur Analyse und Charakterisierung synthetisierter Verbindungen, ihren Ausgangsstoffen sowie Naturprodukten.[1] Neben der Anwendung in der Organik durch Aufnahme von hauptsächlich 1H NMR- und 13C NMR-Spektren, werden auch viele andere NMR-aktive Kerne in anderen Bereichen der Chemie zur Analyse genutzt. So wird beispielsweise NMR-Spektroskopie mit 129Xe dafür genutzt die Beladung von mikroporösen Feststoffen zu bestimmen.[2-3]
Zur Aufnahme von NMR-Spektren wird aufgrund geringer Sensitivität häufig eine relativ große Menge einer Probe benötigt. Die NMR-Spektroskopie bietet dafür allerdings die Möglichkeit der elementspezifischen Untersuchung von Proben und die Möglichkeit quantitative Informationen über die Verteilung eines Elements in einem Material zu erhalten. Außerdem handelt es sich um eine nichtdestruktive und nichtinvasive Methode, was eine Wiederverwendung der Probe und auch die Untersuchung radioaktiver Proben ohne Kontamination des Gerätes zulässt.[4] Für die Untersuchung von Radionukliden gibt es darüber hinaus viele sicherheitstechnische Einschränkungen, um Schutz vor Kontamination und Proliferation zu gewährleisten. Aufgrund dessen gibt es weltweit nur wenige Einrichtungen, die die Chemie der Radionuklide erforschen können und somit sind nur wenige tiefgreifende Grundlagen der Actinidchemie bekannt.
Die Actinide weisen, wie viele andere Metalle der d- und f-Elemente ungepaarte Elektronen auf, welche zu paramagnetischem Verhalten führen. Seit einigen Jahrzehnten sind die paramagnetischen Effekte vieler Metallionen auf ihre Liganden bekannt.[5] Die Einflüsse werden seitdem mit NMR-Spektroskopie untersucht und quantifiziert. Sie können in Pseudo- und Fermikontaktanteil der chemischen Verschiebung unterteilt werden und ergeben additiv mit der diamagnetischen Verschiebung die beobachtete Gesamtverschiebung. Beide Anteile bieten unterschiedliche Informationen über den Aufbau und die Struktur der Komplexe paramagnetischer Metalle.[5] Mögliche Auswertemethoden zur Auftrennung der paramagnetischen Anteile, bspw. nach Bleaney, wurden für Lanthanide entwickelt und sind seit Jahrzehnten etabliert. Seitdem werden allerdings zunehmend viele abweichende Beispiele innerhalb der Elementgruppen gefunden für die die Methoden entwickelt wurden, sodass eine kritische Betrachtung notwendig wird.[6] Für Actinide steht eine Untersuchung der Anwendbarkeit und Grenzen der Methoden, sowie eine Bestimmung vieler verwendeter Parameter noch aus.[4] Die Untersuchung von Actinidkomplexen kann somit zu einem verbesserten Verständnis der paramagnetischen Verschiebung in der NMR-Spektroskopie führen und die Basis für neue und erweiterte Modelle und Auswertemethoden für diese bilden.
5 Diese Arbeit befasst sich mit der Synthese vierwertiger Actinidkomplexe (Th, U und Np) mit salenähnlichen N,O-Donorliganden und der Synthese einiger diamagnetischer Referenzkomplexe, die nicht aus der Reihe der Actinide stammen (Zr, Hf und Ce). Als Schwerpunkt wird auf die NMR-spektroskopische Untersuchung dieser Komplexe bezüglich ihrer paramagnetischen Anteile und daraus gewinnbaren Informationen eingegangen und die Anwendbarkeit der Bleaney-Methode diskutiert. Außerdem werden die Kristallstrukturen einiger synthetisierter Actinidkomplexe verglichen.
6
2 Grundlagen und Stand der Literatur 2.1 Actinide
Die Actinide gehören wie die Lanthanide zu den f-Elementen und werden dementsprechend häufig als ihre Analoge bezeichnet, obwohl sie auch klare Unterschiede aufweisen, auf welche in den folgenden Abschnitten eingegangen wird.
Bei der Gruppe der Actinide handelt es sich um Actinium und die 14 darauffolgenden Elemente mit den Ordnungszahlen 89 bis 103.[7-8] Alle Actinide sind radioaktiv. Nur Actinium, Thorium, Uran und in Spuren Protactinium und Plutonium sind primordial. Thorium und Uran können aufgrund der hohen Halbwertszeiten ihrer stabilsten Isotope (232Th: 1,405 ∙ 1010 a, 238U: 4,468 ∙ 109 a) in größeren Mengen gefunden werden. Protactinium ist das seltenste primordiale Element der Erde und wird nur in geringen Mengen in Uranerzen als α-Zerfallsprodukt von 235U gefunden.[8-9] Neptunium ist das erste von Menschenhand erzeugte Element im Periodensystem und wurde in den 40er Jahren von McMillan und Abelson entdeckt.[8] Es entsteht hauptsächlich in Kernreaktoren durch Neutroneneinfang und kommt in beträchtlichen Mengen in high-level nuclear waste vor.[8-9] Eine Tonne der Schwermetalle aus abgebrannten Brennelementen kann beispielsweise rund 0,79 kg Np-237 enthalten.[10]
In Abbildung 1 sind die stabilen Oxidationsstufen der Actinide dargestellt. Die Vielfalt der Oxidationsstufen ist im Gegensatz zu den Lanthaniden sehr groß. Die Lanthanide weisen aufgrund ihrer energetisch niedrig liegenden f-Orbitale durchgehend +III als stabilste Oxidationsstufe auf. Auch bei den meisten Actiniden wird diese Oxidationsstufe stabilisiert. Jedoch wirken zusätzlich relativistische Effekte, wie zum Beispiel die Spin-Bahn-Kopplung, welche insbesondere die Masse der kernnahen s- und p-Elektronen erhöht. Aus diesem Grund wird die Kernladung für die d- und f-Orbitale stärker abgeschirmt, sodass diese expandieren. Dieser Effekt nimmt mit der Kernmasse und -ladung der Elemente zu. Die 5f-Orbitale werden bis in die Valenzregion der Actinide gestreckt und können somit im Vergleich zu den 4f-Orbitalen der Lanthanide nicht nur besser mit Ligandorbitalen überlappen, sondern auch leichter Elektronen abgeben, um höhere Oxidationsstufen zu erreichen.[8, 11] Bei, beispielsweise in Endlagern vorherrschenden, anoxischen Bedingungen erweisen sich dann vierwertige Actinide als besonders stabil.[7] Für die NMR-spektroskopie Untersuchung sind vierwertige Actinide außerdem interessant, da sie mit Thorium(IV) eine diamagnetische Referenz in ihrer Reihe aufweisen. Bei Untersuchung der dreiwertigen Actinide trifft dies auf Actinium(III) zu. Dieses ist jedoch aufgrund der geringen Halbwertszeit (21,77 a) und geringen Häufigkeit sehr schwierig zugänglich[8], sodass sich für die Untersuchung paramagnetischer Effekte eher die Reihe vierwertiger Actinide anbietet.
7
Abbildung 1: Stabile Oxidationsstufen der Actinide unter unterschiedlichen Bedingungen.[7]
2.2 Iminliganden /Salenderivate
Die vierwertigen Actinide Thorium und Uran wurden bereits in den siebziger Jahren mit dem Iminliganden 2,2‘-[Ethan-1,2-diylbis(azanylylidenmethanylyliden)]diphenol[12], welcher den Trivialnamen Salen trägt, chelatisiert (Abbildung 2).[13] Imine sind die stickstoffanalogen Verbindungen zu Carbonylen. Sie werden im einfachsten Fall durch Eliminierung von Wasser aus der Reaktion eines primären Amins oder Ammoniak mit einer Carbonylverbindung gewonnen. Im ersten Fall entsteht ein am Stickstoff substituiertes Imin, welches allgemein als Schiff’sche Base bezeichnet wird.[1, 14]
Der Ligand H2Salen, welcher aus Salicylaldehyd und Ethylendiamin gewonnen wird, ist ein häufig verwendeter Ligand in der Übergangsmetallchemie. Aufgrund seiner zweifach negativen Ladung in deprotonierter Form eignet er sich als tetradentater Ligand hervorragend zur Chelatisierung
Abbildung 2: Strukturformeln für die allgemeine Form eines Imins (links) und H2Salen (rechts).
8 zweiwertiger Übergangsmetalle. Außerdem weist der Ligand gleichzeitig je zwei weiche und harte Donoratome auf (Stickstoff bzw. Sauerstoff). Er bietet sich, auch aufgrund seiner leichten Zugänglichkeit, als sehr gutes Modellsystem an, um übliche, in biologischen Systemen vorkommende Donorgruppen zu untersuchen. Im Fall vierwertiger Metalle, wie die erwähnten Actinide, wird sogar eine Chelatisierung durch zwei Ligandmoleküle mit gleichzeitigem Ladungsausgleich möglich. Die so erhaltenen 2:1-Komplexe erweisen sich als sehr stabil, insbesondere gegen Luft und Feuchtigkeit.[13, 15-
16]
Die in dieser Arbeit untersuchten Liganden sind in der Aromatenregion substituierte Salenderivate mit variierenden Diaminoalkanbrücken (Abbildung 3). In der Literatur wurde bisher nur über Übergangsmetallkomplexe dieser Liganden berichtet. Diese Komplexe liegen alle im Ligand zu Metall- Verhältnis 1:1 vor. Komplexe der Hydroxyliganden wurden beispielsweise mit V, Mn, Co, Ni und Pd synthetisiert, wobei Komplexe des H2Hysalpn-Liganden nur mit Co und V und Komplexe des H2Hysalcn-Liganden nur mit Mn berichtet wurden.[17-23] Komplexe der Fluoroliganden sind ausschließlich mit Mn und Zn berichtet.[24-27] 2:1-Komplexe sind bisher mit keinem der Liganden bekannt und auch eine Komplexierung von Lanthaniden oder Actiniden wurde nicht publiziert.
Sowohl die Hydroxysubstitution, als auch die Fluorsubstitution wirken durch ihren negativen induktiven Effekt elektronenziehend. Die Hydroxygruppe weist dabei zusätzlich einen positiven mesomeren Effekt auf, der elektronenschiebend wirkt, aber den induktiven Effekt nicht ausgleicht. Der –I-Effekt sollte somit unabhängig von der Substitutionsposition die Acidität des Protons der koordinierenden Hydroxygruppe erhöhen und somit eine Deprotonierung des Liganden erleichtern, während der +M-Effekt abhängig von der Substitutionsposition unterschiedlich stark entgegenwirkt.
Die veränderte Elektronendichte am koordinierenden Sauerstoff und am koordinierenden Stickstoff
Abbildung 3: In der Arbeit verwendete Liganden. Die linken und mittigen Liganden werden in dieser Arbeit allgemein zusammengefasst als Hydroxyliganden bezeichnet, die rechten Liganden als Fluoroliganden.
9 kann auch die Bindung zu Metallionen abschwächen oder stärken.[1, 14, 28] Die elektronischen Änderungen können mit Hilfe von IR-Spektroskopie, Einkristallröntgendiffraktometrie (SC-XRD) und NMR-Spektroskopie untersucht werden.
Außerdem kann die Komplexierung durch die unterschiedlichen Diaminoalkanbrücken der Liganden sowohl sterisch, als auch elektronisch beeinflusst werden. Das Einfügen zusätzlicher Methylengruppen in die Brücke erzeugt einen positiven induktiven Effekt, was die Elektronendichte insbesondere am Stickstoff leicht erhöhen soll. Zusätzlich wird dadurch die Flexibilität des Liganden erhöht, was eine Komplexierung erleichtert. Durch die Einführung von Substituenten in der Brücke, z.B. in Form des Cyclohexanrings im H2Hysalcn-Liganden wird die Flexibilität hingegen reduziert.
2.3 NMR-Spektroskopie
Die Voraussetzung zur Untersuchung eines Kerns mit NMR-Spektroskopie ist, dass dieser ein magnetisches Moment aufweist. Dies ist der Fall für alle Kerne, die einen Spin besitzen und dementsprechend nicht gleichzeitig eine gerade Anzahl Neutronen und eine gerade Anzahl Protonen aufweisen, da dies einen Spin von 0 erzeugen würde. Das magnetische Moment µ ergibt sich somit nach (1) aus dem Plank’schen Wirkungsquantum h und dem Kernspin I.[29-30]
𝜇 = 𝛾ℎ
2𝜋∙ √𝐼(𝐼 + 1) (1)
Der dabei auftretende Proportionalitätsfaktor γ wird als gyromagnetisches Verhältnis bezeichnet, welches für jeden NMR-aktiven Kern einen spezifischen Wert annimmt.[29-30] Die gyromagnetischen Verhältnisse vieler Kerne, so auch die einiger Actinidnuklide, wie z.B. 235U, 237Np und 239Pu[3], wurden bestimmt. Bei Anlegen eines äußeren Magnetfeldes mit der magnetischen Flussdichte B0 kommt es zur Ausrichtung der magnetischen Momente. Die unterschiedlichen Spins der beobachteten Kerne verteilen sich dann auf 2I + 1 energetische Zustände. Bei der Betrachtung von Protonen mit I = ½ sind dies beispielsweise zwei Zustände. Diese energetischen Zustände werden Zeeman-Niveaus genannt.
Die Energiedifferenz dieser ergibt sich dann nach (2).[29-31]
𝛥𝐸 =𝛾ℎ
2𝜋∙ 𝐵0 (2)
10 Einer der beiden Energiezustände, die für Protonen im äußeren Magnetfeld entstehen ist energetisch günstiger. Dieser Zustand wird als Grundzustand N0 bezeichnet. Der energetisch höhere Zustand N1
weist aufgrund des höheren Energiebedarfs eine geringere Besetzung auf, was durch die Boltzmannverteilung (3), mit kB als Boltzmannkonstante und T als Temperatur, beschrieben werden kann.[29-31]
𝑁1 𝑁0= 𝑒
−𝛥𝐸 𝑘𝐵𝑇 = 𝑒
−𝛾ℎ𝐵0
2𝜋𝑘𝐵𝑇 (3)
Der Besetzungsunterschied ist die Grundvoraussetzung für die Aufnahme von NMR-Spektren und kann durch Verstärkung des B0-Feldes, Verringerung der Temperatur oder Verwendung eines Nuklids mit höherem gyromagnetischem Verhältnis erhöht werden. Durch das Einstrahlen einer Resonanzfrequenz ν0 werden Spins aus dem Grundzustand in den angeregten Zustand angehoben und auch Spins aus dem angeregten Zustand in den Grundzustand abgesenkt. Aufgrund der stärkeren Besetzung des Grundzustandes überwiegt die Absorption.[31] Am Ende des Anregungspulses kommt es so zur Sättigung, also Gleichbesetzung beider Energiezustände. Dies spiegelt sich in einer Quermagnetisierung in der x,y-Ebene wieder, welche um die z-Achse, entlang welcher das angelegte Magnetfeld wirkt, präzediert. Die Frequenz der Präzession wird dabei detektiert und führt nach einer Fourier-Transformation zu einem NMR Spektrum. Beim Relaxieren der Spinzustände löst sich die Quermagnetisierung langsam wieder auf und das vor der Anregung vorliegende Gleichgewicht mit Besetzungsunterschied wird wiederhergestellt. Die zur Anregung nötige Resonanzfrequenz ist dabei vom gyromagnetischen Verhältnis des beobachteten Kerns abhängig (4).[29, 31]
𝜈0= 𝛾
2𝜋𝐵0 (4)
Bei NMR-Spektrometern wird üblicherweise die Resonanzfrequenz der Protonen, an Stelle der magnetischen Flussdichte, angegeben. So kommt es, dass häufig zum Beispiel von einem 400 MHz Spektrometer gesprochen wird und nicht von einem 9,4 T Spektrometer. Andere Kerne können dann mit dem selben Gerät bei einer anderen Resonanzfrequenz gemessen werden, bspw. 13C bei ca.
100 MHz. Eine niedrigere Resonanzfrequenz verschlechtert dabei die Auflösung und die Sensitivität, da der Besetzungsunterschied geringer wird. Zusätzlich wird die Sensitivität durch die natürliche Häufigkeit des beobachteten Nuklides beeinflusst. Protonen weisen zum Beispiel eine natürliche
11 Häufigkeit von 99,98 % auf, während 13C-Atome nur eine von 1,11 % aufweisen.[30-32] Aus diesem Grund werden wesentlich größere Probenmengen benötigt, um ein gutes Signal-Rausch-Verhältnis bei 13C NMR-Spektren zu erhalten.
Die NMR-Spektroskopie kann jedoch keine Kerne ohne elektronische Umgebung beobachten. Da auch Elektronen einen Spin aufweisen induzieren diese ein Magnetfeld, welches dem äußeren Magnetfeld meist entgegenwirkt und dieses abschirmt, sodass sich Signale der beobachteten Kerne eines Moleküls aufgrund ihrer chemischen Umgebung unterscheiden. Der betrachtete Kern i wird dann nur durch das effektive Magnetfeld mit der magnetischen Flussdichte Bi beeinflusst. Zur Beschreibung der Abschirmung erhält jeder Kern i eine Abschirmungskonstante σi (5).[29, 31]
𝐵𝑖 = 𝐵0− 𝜎𝑖𝐵0= 𝐵0(1 − 𝜎𝑖) (5)
Die Resonanzfrequenz jedes beobachteten Kerns wird dadurch leicht verändert (6).[29, 31]
𝜈𝑖 = 𝛾
2𝜋𝐵0(1 − 𝜎𝑖) (6)
Da unterschiedliche NMR-Spektrometer häufig unterschiedliche magnetische Flussdichten aufweisen, ergeben sich dann je nach Gerät unterschiedliche Resonanzfrequenzen, trotz gleicher elektronischer Umgebung des beobachteten Kerns. Um dieser Problematik entgegenzuwirken wurde die chemische Verschiebung δ eingeführt, welche eine messfrequenzunabhängige Darstellung der NMR-Spektren ermöglicht und somit eine Vergleichbarkeit gewährleistet. Sie ergibt sich nach Formel (7).[29, 31]
𝛿𝑖 =𝜈𝑖− 𝜈𝑟𝑒𝑓 𝜈0
(7)
Dabei entspricht νref der Resonanzfrequenz der Kerne einer gewählten Referenzsubstanz, wie z.B.
Tetramethylsilan (TMS) für 1H, 13C und 29Si. Der Einfluss der Elektronen auf das Magnetfeld ist sehr gering, sodass die Differenz des betrachteten Kerns und des Referenzkerns im Hz-Bereich liegt. Da das genutzte äußere Magnetfeld im MHz-Bereich liegt, ergeben sich für die chemische Verschiebung Werte von einigen ppm.[29, 31]
12 Für diamagnetische Strukturen sind die chemischen Verschiebungen vieler bekannter Strukturgruppen tabelliert und Zuordnungen dementsprechend trivial.[29-31] Sobald allerdings Metallkomplexe betrachtet werden, verkompliziert sich die Auswertung häufig, da viele Metalle ungepaarte Elektronen und somit Paramagnetismus einbringen. Da Elektronen einen Spin besitzen, der im Falle ungepaarter Elektronen nicht durch ein weiteres Elektron mit entgegengesetztem Spin ausgemittelt wird, kann dieser eine Wechselwirkung mit dem in der NMR-Spektroskopie beobachtbaren Kernspin eingehen.
Der Elektronenspin kann sich wesentlich schneller reorientieren, als der Kernspin, weshalb in der NMR-Spektroskopie, statt eines Dubletts, ein gemittelter Elektronenspin beobachtet wird.[33] Die Besetzungsunterschiede der Zeeman-Niveaus sind für Kernspins mehrere Größenordnungen kleiner, als die der Elektronenspins, weshalb die des Kernspins vernachlässigt werden können. Aufgrund des Besetzungsunterschiedes der Elektronenspins wird allerdings das erhaltene, gemittelte Signal relativ zum Mittelpunkt des Dubletts verschoben. Dies ist die paramagnetische Verschiebung δpara.[33] Die beobachtete Gesamtverschiebung setzt sich dabei aus der paramagnetischen Verschiebung und der diamagnetischen Verschiebung δdia, welche aufgrund der Abschirmung des äußeren Magnetfeldes durch die Elektronen in den umliegenden Orbitalen entsteht, zusammen (8).[4-5]
𝛿 = 𝛿𝑑𝑖𝑎+ 𝛿𝑝𝑎𝑟𝑎 (8)
Soll der Anteil der paramagnetischen Verschiebung an der Gesamtverschiebung eines paramagnetischen Metallkomplexes berechnet werden, so wird ein diamagnetischer isostruktureller Komplex mit möglichst ähnlichem Metall gewählt und dessen Verschiebung als diamagnetischer Anteil des paramagnetischen Komplexes angenommen. Die paramagnetische Verschiebung kann dabei in zwei Anteile unterteilt werden: Den Fermikontaktanteil δFCS und den Pseudokontaktanteil δPCS (9).[5]
𝛿𝑝𝑎𝑟𝑎 ≅ 𝛿𝐹𝐶𝑆+ 𝛿𝑃𝐶𝑆 (9)
Der Fermikontaktanteil hängt dabei von der Delokalisierung der Elektronenspins ab, welche durch skalare Wechselwirkungen über die dativen Bindungen des Komplexes und anschließend durch Fermi-Kontakt über Orbitale mit großem s-Anteil von Elektronen auf Kerne übertragen werden.
Dadurch kann ein Einblick in die Bindungsverhältnisse zwischen beobachtetem Kern und paramagnetischem Metall gewährt werden. Diese Delokalisierung des Elektronenspins erzeugt ein
13 zusätzliches Magnetfeld mit der magnetischen Feldstärke ΔH am Kern und somit die Verschiebung δFCS
(10).[5]
𝛿𝐹𝐶𝑆=𝛥𝐻
𝐻0 =𝐴𝑔𝑒𝛽𝑆(𝑆 + 1) 3ℏ𝛾𝑘𝐵𝑇
(10)
Dabei entspricht H0 der magnetischen Feldstärke des angelegten Magnetfelds, A der Hyperfeinkopplungskonstante, welche die Kopplung von Elektronenspin mit beobachtetem Kernspin beschreibt, ge dem Landé-Faktor eines Elektrons, β dem Bohr‘schen Magneton, S dem Gesamtspin der Elektronen des Komplexes, welcher meist dem des Metallions entspricht und ℏ dem reduzierten Plank‘schen Wirkungsquantum. Dabei zeigt sich eine T-1 Abhängigkeit des Fermikontaktanteils, sowie die starke Abhängigkeit vom Gesamtspin, welche große Fermikontaktanteile für stark paramagnetische Komplexe vorhersagt. Bei Lanthaniden wird der Fermikontaktanteil stark durch die geringe Kovalenz der Ligand-Metallbindungen eingeschränkt, wodurch dieser nur über wenige Bindungen Entfernung messbare Hyperfeinkopplungskonstanten aufweist.[5, 34-35] Die 5f-Orbitale der Actinide hingegen überlappen stärker mit Ligandorbitalen und weisen so größere kovalente Bindungsanteile auf, was eine Beobachtung größerer und weitreichenderer Fermikontaktanteile ermöglichen kann.[11]
Der Pseudokontaktanteil beschreibt den Anteil der Spindichte, welcher nicht über Bindungen delokalisiert wird, sondern in den Orbitalen des Metalls verweilt und so einen elektromagnetischen Dipol erzeugt, der mit den magnetischen Kernspinmomenten wechselwirkt.[5] In einem System mit magnetischer Prinzipalachse ergibt sich für die Pseudokontaktverschiebung der folgende Zusammenhang:[36-38]
𝛿𝑃𝐶𝑆= 1
2𝑁𝐴𝑟3((𝜒𝑧𝑧−1
3Spur(𝜒)) (3cos2𝜃 − 1) + (𝜒𝑥𝑥− 𝜒𝑦𝑦)(sin2𝜃 cos 𝜑)) (11)
Der betrachtete Komplex wird dabei in einem Polarkoordinatensystem mit dem Metallatom als Mittelpunkt und der Prinzipalachse z betrachtet. Der Winkel θ gibt dementsprechend die Neigung des Vektors r, welcher den Abstand zwischen Metallatom und betrachteten Kern definiert, und der Prinzipalachse an. Der auf die xy-Ebene projizierte Vektor r spannt mit der x-Achse den Winkel φ auf.
NA entspricht der Avogadro-Konstante und χ der Matrix der magnetischen Suszeptibilität. Dabei sind
14 χxx, χyy und χzz die Diagonalelemente der Matrix. Für den Fall einer mindestens dreiwertigen Symmetrieachse, welche bei den 2:1-Komplexen der Salenderivate nicht vorliegt, nehmen die Elemente χxx und χyy den selben Wert an, wodurch sich (11) zu (12) vereinfacht.[39]
𝛿𝑃𝐶𝑆= 1
3𝑁𝐴𝑟3((𝜒𝑧𝑧− 𝜒𝑥𝑥)(3cos2𝜃 − 1)) (12)
Da die magnetische Suszeptibilität eine Temperaturabhängigkeit aufweist, gilt dies auch für den Pseudokontaktanteil. Bleaney gibt dabei an, dass für den Fall gelöster Lanthanidverbindungen, bei denen keine strukturelle Änderung durch eine Variation der Temperatur oder der Anzahl der ungepaarten Elektronen erfolgt, eine Abhängigkeit des Pseudokontaktanteils von T-2 gilt.[38]
Anhand der unterschiedlichen Temperaturabhängigkeiten können dementsprechend die beiden paramagnetischen Anteile getrennt werden, um die beschriebenen Strukturinformationen der beiden Anteile zu extrahieren. Dafür wird die lineare Gleichung (13) mit Anstieg a und Achsenabschnitt b genutzt, bei welcher a und b, Pseudo- bzw. Fermikontaktanteil (δPCS bzw. δFCS) nach Gleichungen (14) und (15) ergeben.
𝑇𝛿𝑝𝑎𝑟𝑎= 𝑎1
𝑇+ 𝑏 (13)
𝛿𝑃𝐶𝑆= 𝑎𝑇−2 (14)
𝛿𝐹𝐶𝑆= 𝑏𝑇−1 (15)
Diese Methode findet bereits seit mehreren Dekaden Anwendung, wurde für die Lanthanide entwickelt und basiert dabei auf einigen kritisch zu betrachtenden Annahmen. So wird angenommen, dass die Ligandenfeldaufspaltung viel kleiner als kBT ist.[6] Dies trifft bei Lanthaniden in vielen Fällen zu, kann aber bei Actiniden, welche im Schnitt eine um eine Größenordnung stärkere Ligandenfeldaufspaltung aufweisen, sehr grenzwertig erscheinen.[11, 40] Zusätzlich wird angenommen, dass das Elektron eine Punktladung im Koordinatenursprung ist, welches sofort relaxiert, und auch, dass die Position der Prinzipalachse sich innerhalb einer isostrukturellen Lanthanidkomplexserie nicht ändert.[6] Das Elektron relaxiert tatsächlich schneller als die beobachteten Kerne, sodass die sofortige Relaxation als plausible Vereinfachung gelten kann.[33] Die Betrachtung der f-Elektronendichte als Aufenthaltswahrscheinlichkeit impliziert allerdings eher eine Verteilung der Elektronendichte, als eine
15 Punktladung.[6] Zusätzlich wurde von Sessoli at al. gezeigt, dass sich die Prinzipalachse in einem C4-symmetrischen Komplex innerhalb einer Lanthanidserie ändern kann.[41-42] Es zeigen sich also bereits innerhalb der Lanthanidserie einige Probleme bei Anwendung der Bleaney-Methode, während die Anwendbarkeit für die Serie der Actinide wünschenswert wäre, aber sicher noch begrenztere Möglichkeiten aufweist und ausgiebig getestet werden muss.
Weitere Untersuchungen zur Gültigkeit der Bleaney-Methode und eine andere Variante der Auftrennung der paramagnetischen Anteile wurden von Autschbach et al. durchgeführt. Demnach gilt die einfache Temperaturabhängigkeit des Fermikontaktanteils mit T-1 und die des Pseudokontaktanteils mit T-2 nur, wenn der Gesamtspin S ≥ 1, die Nullfeldaufspaltung nennenswert und der anisotrope Anteil des g-Tensors Δg = 0 ist. Für S = 3/2 gilt die Ausnahme, dass selbst bei Δg = 0 keine saubere Aufspaltung der beiden paramagnetischen Anteile möglich ist. Dieser Sachverhalt ist in den Gleichungen (16) bis (19), in denen Fermi- und Pseudokontaktanteile für S =1 und S = 3/2 nach Autschbach et al. beschrieben sind, ersichtlich.[43]
𝑆 = 1: 𝜎𝐹𝐶𝑆= − 𝛽𝐴𝑖𝑠𝑜
3𝑔𝑁𝛽𝑁(2𝑔𝑖𝑠𝑜
𝑘𝐵𝑇 − 2𝐷∆𝑔
3𝑘𝐵2𝑇2−𝐷2𝑔𝑖𝑠𝑜
9𝑘𝐵3𝑇3) (16)
𝜎𝑃𝐶𝑆= − 𝛽∆𝐴 3𝑔𝑁𝛽𝑁(4∆𝑔
𝑘𝐵𝑇−2𝐷(𝑔𝑖𝑠𝑜+ ∆𝑔)
3𝑘𝐵2𝑇2 −2𝐷2∆𝑔
9𝑘𝐵3𝑇3) (17)
𝑆 =3
2∶ 𝜎𝐹𝐶𝑆= − 𝛽𝐴𝑖𝑠𝑜
3𝑔𝑁𝛽𝑁(5(7𝑔𝑖𝑠𝑜+ 2∆𝑔)
12𝑘𝐵𝑇 −𝐷(29∆𝑔 − 2𝑔𝑖𝑠𝑜)
9𝑘𝐵2𝑇2 −2𝐷2(𝑔𝑖𝑠𝑜− ∆𝑔)
27𝑘𝐵3𝑇3 ) (18) 𝜎𝑃𝐶𝑆= − 𝛽∆𝐴
3𝑔𝑁𝛽𝑁(5(𝑔𝑖𝑠𝑜+ 8∆𝑔)
6𝑘𝐵𝑇 −𝐷(29𝑔𝑖𝑠𝑜+ 25∆𝑔)
9𝑘𝐵2𝑇2 −2𝐷2(𝑔𝑖𝑠𝑜− ∆𝑔)
27𝑘𝐵3𝑇3 ) (19)
Dabei stellt Aiso den isotropen Teil der Hyperfeinkopplungskonstante dar und ΔA den anisotropen Teil.
giso entspricht dem isotropen Teil des Landé-Faktors des betrachteten Kerns und Δg dem anisotropen Teil. D ist der Nullfeldaufspaltungstensor, gN der Landé-Faktor des untersuchten Kerns N und βN dessen Kernmagneton. Von den in dieser Arbeit behandelten vierwertigen Metallionen besitzen außer Uran und Neptunium alle einen Gesamtspin von 0. Uran(IV) weist einen Gesamtspin von 1 und Neptunium(IV) einen von 3/2 auf.[8] Dies impliziert, dass ein Uran(IV)komplex, welcher näherungsweise keine anisotropen Anteile im g-Tensor enthält, einen Fermikontaktanteil, der etwa proportional zu T-1 ist, und einen Pseudokontaktanteil, der proportional zu T-2 ist, besitzt.[43] Die Auswertbarkeit der synthetisierten paramagnetischen Komplexe soll in dem Sinne mit Hilfe der Bleaney-Methode kritisch getestet werden. Außerdem soll getestet werden, inwiefern die Auswertbarkeit von Neptunium(IV)komplexen nach Bleaney möglich ist.
16
3 Ergebnisse und Diskussion
Im ersten Abschnitt werden die Ergebnisse der Synthesen der Hydroxyligandkomplexe diskutiert. Die erhaltenen Komplexe weisen eine geringe Löslichkeit in einer großen Anzahl gängiger Lösungsmittel auf. Die beste Löslichkeit wurde mit DMF und DMSO erreicht, weshalb diese Lösungsmittel zur Synthese bzw. Analyse mit NMR gewählt wurden. Die [M(Hysalen)2]- und [M(Hysalcn)2]-Komplexe weisen dabei eine höhere Löslichkeit auf als die [M(Hysalpn)2]-Komplexe auf. Aufgrund der Koordinationsfähigkeit von DMF und DMSO zeigen die Komplexe in Lösung teilweise geringe Stabilität auf und können durch den zweiten Hydroxysubstituenten am Aromaten polymerartige Ketten bilden.
In den darauffolgenden Abschnitten werden die Ergebnisse der Fluoroligandkomplexe diskutiert. Diese Liganden besitzen im Gegensatz zu den Hydroxyliganden keine weiteren Donorgruppen, als die koordinierenden und können somit nur unwahrscheinlich polymerartigen Ketten ausbilden. Die Polaritäten der Liganden und auch der Komplexe sind geringer und somit sind auch die Wechselwirkungen untereinander schwächer, was die Löslichkeit im Vergleich zu den Hydroxyliganden deutlich erhöht und die Nutzung einer größeren Vielfalt an Lösungsmitteln gewährt. Die Verwendung anderer Halogensubstituenten würde neben den elektronischen Änderungen auch aufgrund ihrer Größe stark in die Struktur eingreifen. Fluor und Wasserstoff weisen die geringsten Größenunterschiede in ihren Kovalenz- und Van-der-Waals-Radien auf[44] und erlauben so einen guten Vergleich, wobei Änderungen fast ausschließlich durch elektronische Eigenschaften hervorgerufen werden. Zusätzlich kann durch die Einführung von Fluoratomen ein weiterer NMR-aktiver Kern mit 100 % natürlicher Häufigkeit und protonenähnlichem gyromagnetischen Verhältnis untersucht werden.[31]
3.1 Hydroxykomplexe
Alle Hydroxykomplexe konnten über die gleiche Syntheseroute in DMF synthetisiert werden. Es wurden IR-Spektren aller Pulver und NMR-Spektren der in DMSO-d6 gelösten Pulver aufgenommen. Im Fall des [U(Hysalen)2]-Komplexes, konnten durch Zugabe von Methanol zu einer Lösung in DMF nach wenigen Tagen Kristalle gewonnen werden, welche mittels Einkristallröntgendiffraktometrie untersucht wurden.
3.1.1 SC-XRD
In Abbildung 4 ist die Struktur des [U(Hysalen)2]-Komplexes, welcher in der orthorhombischen Raumgruppe Pbca kristallisiert, zu sehen. Die Elementarzelle weist eine Größe von 7713 Å3, mit den
17 Achsen a = 11,12 Å, b = 11,16 Å und c = 62,17 Å und den Winkeln α = β = γ = 90 °, auf und enthält acht Komplexmoleküle, sowie 16 DMF-Moleküle. Das Uranatom wird von acht Donoratomen zweier tetravalenter, zweifach deprotonierter Liganden in einen Trigondodekaeder eingeschlossen. Der durchschnittliche Abstand der koordinierenden Sauerstoffatome zum Zentralatom beträgt 2,23 Å, der durchschnittliche Abstand der Stickstoffatome 2,58 Å. Die Winkel der koordinierenden Ligandatome eines Liganden mit dem Zentralatom betragen durchschnittlich ∡OUN = 70,4 °, ∡NUN = 67,4 ° und
∡OUO = 152,6 °. Alle H-Atome der unkoordinierten Hydroxygruppen des Liganden gehen Wasserstoffbrückenbindungen mit einer Carbonylgruppe je eines DMF-Moleküls ein. Jedes DMF-Molekül geht dabei je eine Wasserstoffbrückenbindung mit einer Hydroxygruppe eines Ligandmoleküls zweier unterschiedlicher Komplexmoleküle ein. Die H-Atome der Methylgruppen der DMF-Moleküle zeigen C–H-π-Wechselwirkungen mit den Phenylringen der Komplexmoleküle. Diese H-Atome befinden sich dabei teilweise in den beiden kleineren Kavitäten zwischen den Phenylringen der Liganden. In den beiden größeren Kavitäten zeigen sich C–H⋯π- und C–H⋯O-Wechselwirkungen der Wasserstoffatome eines zweiten Komplexmoleküls mit den koordinierenden Sauerstoffen sowie den Phenylringen des dargestellten Komplexmoleküls. So bilden sich abwechselnd auftretende Schichten von Komplexmolekülen und DMF-Molekülen.
Abbildung 4: Kristallstruktur von [U(Hysalen)2] ∙ 2DMF mit Wasserstoffbrückenbindungen zwischen Komplexmolekül und DMF sowie allgemeine Bezeichnungen und Lage der erwähnten Bindungswinkel. Uran - Grün, Sauerstoff - Rot, Stickstoff - Blau, Kohlenstoff - Grau, Wasserstoff - Weiß.
O1 U1
O3 O2
O4
N1 N2 N3
N4
O5
O6
N5
N6
O7
O8
O9
O10
∡NUN
∡OUN
∡OUO
18 3.1.2 IR-Spektroskopie
In Abbildung 5 sind die IR-Spektren der [M(Hysalpn)2]-Komplexe dargestellt. Diese zeigen sehr klare Ähnlichkeiten. Die Bandenlagen weichen kaum voneinander ab und auch die Intensitäten sind sehr ähnlich. Im Bereich um 3000 cm-1 kann eine breite OH-Bande der Komplexe gefunden werden. Kurz darunter liegen zwei schwache aber scharfe Banden, die durch die aliphatische Brücke der Ligandmoleküle hervorgerufen werden. Knapp unter 1600 cm-1 liegt die sehr starke Bande der substituierten Imingruppe. Bei ca. 1280 cm-1 liegt die Bande der C–O-Valenzschwingung für beide Komplexe. Kurz darunter, bei etwa 1228 cm-1, kann eine sehr intensive Bande der aromatischen OH-Gruppe gefunden werden.[31] Der einzige größere Unterschied zwischen den Spektren ist eine zusätzliche Bande unbekanntem Ursprungs im Spektrum des Urankomplexes bei 885 cm-1. Aufgrund der geringen Löslichkeit der [M(Hysalpn)2]-Komplexe scheint das Gleichgewicht durch die Fällung sehr stark auf der Seite der Komplexe zu liegen, sodass kein freier Ligand als Verunreinigung eingetragen wird. Die IR-Spektren der [M(Hysalen)2]- und [M(Hysalcn)2]-Komplexe zeigen hingegen Verunreinigungen, die eine klare Identifizierung der Komplexbanden verhindern und einen Vergleich der Spektren ausschließen.
3.1.3 NMR-Spektroskopie [M(Hysalen)2]-Komplexe
In Abbildung 6 sind die 1H NMR-Spektren der [M(Hysalen)2]-Komplexe und des freien Liganden dargestellt. Die Koordination des Liganden am diamagnetischen Thorium(IV) beeinflusst am stärksten
Abbildung 5: IR-Spektren der beiden [M(Hysalpn)2] Komplexe.
19 die Signale der Brücken- und Iminprotonen. Geringere Einflüsse sind im Aromatenbereich zu sehen.
Die koordinierende Hydroxygruppe weist im Komplex kein Signal auf, da diese deprotoniert vorliegt.
Der Vergleich des diamagnetischen Thoriumkomplexes mit dem paramagnetischen Urankomplex zeigt jedoch große paramagnetische Verschiebungen. Die Brückenprotonen werden deutlich stärker abgeschirmt und somit relativ zum Thoriumkomplex um mehr als 40 ppm hochfeldverschoben. Alle übrigen Protonen werden tieffeldverschoben. Am stärksten beeinflusst wird von diesen das Aromatenproton zwischen den beiden Hydroxygruppen. Die paramagnetischen Verschiebungen der Signale weisen Beträge auf, die sich mit einem großen winkel- und abstandsabhängigen Pseudokontaktanteil erklären lassen.
Abbildung 6: 1H NMR-Spektren von H2Hysalen und den [M(Hysalen)2]-Komplexen in DMSO-d6. Die Komplexspektren enthalten auch DMF, Methanol und Wasser.
[M(Hysalpn)2] und [M(Hysalcn)2]-Komplexe
Die Komplexe der beiden in der Brücke variierten Hydroxysalenderivate zeigen komplizierte NMR-Spektren mit mehreren schwer unterscheidbaren Signalsätzen. Bei den schwerlöslichen [M(Hysalpn)2]-Komplexen weisen diese auf mehrere Spezies, welche auf die Zersetzung oder
20 unvollständige Bildung der 2:1-Komplexe zurückzuführen sind, hin. Der [U(Hysalpn)2]-Komplex erweist sich dabei als wesentlich stabiler als der [Th(Hysalpn)2]-Komplex. Die paramagnetischen Verschiebungen des [U(Hysalpn)2]-Komplexes liegen in ähnlichen Bereichen, wie die des [U(Hysalen)2]-Komplexes können allerdings aufgrund der fehlenden diamagnetischen Referenz nicht genau quantifiziert werden.
Die [M(Hysalcn)2]-Komplexe zeigen zum Teil Aufspaltungen von Protonensignalen im 7:3-Verhältnis, was eine klare Zuordnung der Signale des [Th(Hysalcn)2]-Komplexes aufgrund der Überlagerung der Cyclohexylgruppensignale unmöglich macht. Die Aufspaltung stammt mit hoher Wahrscheinlichkeit von unterschiedlichen Konformeren der Cyclohexylbrücke des Liganden. Die stabile Sesselkonformation liegt dabei als hauptsächlich vorkommende Spezies vor. Die nächst-stabilere Twist-Konformation könnte die weniger intensiven Signale darstellen.[1] Das vorliegende Gleichgewicht der unterschiedlichen Konformationen in einer Ligandlösung kann durch die Komplexierung festgehalten worden sein und so für unterschiedlich intensive, aber sehr ähnliche Signalsätze gesorgt haben.
3.2 Fluorokomplexe - SC-XRD
Die Synthesen der Fluoroligandkomplexe wurden mit den vierwertigen Metallionen des Zirkoniums, Hafniums, Cers, Thoriums, Urans und Neptuniums in Methanol durchgeführt. Kristalle von [M(fsalen)2]-Komplexen wurden durch langsames Verdampfen von Methanol über ca. zwei Wochen aus einer methanolischen Lösung der Komplexe erhalten. Kristalle der [M(fsalpn)2]-Komplexe wurden durch Diffusion von Pentan in eine Lösung des jeweiligen Komplexes in THF nach einem Tag erhalten.
3.2.1 [Th(fsalen)2]
In Abbildung 7 ist die Struktur des [Th(fsalen)2]-Komplexes, welcher in der monoklinen Raumgruppe P21/n kristallisiert, zu sehen. Die Elementarzelle weist eine Größe von 3363 Å3, mit den Achsen a = 13,22 Å, b = 15,74 Å und c = 17,02 Å und den Winkeln α = γ = 90 ° und β = 108,29 ° auf und enthält vier Komplex- und acht Methanolmoleküle. Das Thoriumatom wird von acht Donoratomen zweier tetravalenter, zweifach deprotonierter Liganden in einen Trigondodekaeder eingeschlossen. Die Kristallstruktur zeigt zwei Methanolmoleküle pro Komplexmolekül, welche sich als parallele Ketten durch die Packung ziehen. Die Hydroxygruppen der Methanolmoleküle bilden teilweise Wasserstoffbrücken zu den koordinierenden Sauerstoffatomen aus. Die Methylgruppen der Methanolmoleküle interagieren dabei über C–H⋯π-Wechselwirkungen mit den Phenylringen der Komplexmoleküle. Die Komplexmoleküle lagern sich in diagonalen Ketten zusammen bei denen
21 Iminprotonen und eine Hälfte der Brücke mit den Phenylringen eines anderen Komplexmoleküls wechselwirken und der Phenylring dabei mit den Iminprotonen und der Brücke des anderen Komplexes wechselwirkt. Die beiden Komplexe sind dabei zueinander um 180 ° verdreht. An beiden Komplexmolekülen folgen dann in der gegenüberliegenden Kavität die Methanolmoleküle, nach denen sich jeweils wieder beschriebene Komplexpaare anlagern. In Abbildung 7 kann auch gesehen werden, dass die Phenylringe auf einer Seite des Komplexes weiter aufspannen. Dies wird hervorgerufen durch die Brücken und Iminprotonen eines benachbarten Komplexmoleküls, welche teilweise in diese Kavität eindringen und über C–H-π-Wechselwirkungen damit interagieren. Auf diese Weise werden die beschriebenen diagonalen Ketten vernetzt.
Es ergeben sich durchschnittliche Th–O-Bindungslängen von 2,30 Å und Th–N-Bindungslängen von 2,66 Å. Die durchschnittlichen Winkel zwischen Donoratomen und Zentralatom sind ∡OThN = 69,2 °,
∡NThN = 63,9 ° und ∡OThO = 157,8 °. Einzelne Bindungslängen und Winkel sind in Tabelle 1 dargestellt.
Abbildung 7: Kristallstruktur des [Th(fsalen)2]-Komplexes mit zwei MeOH-Molekülen.
Thorium - Hellblau, Sauerstoff - Rot, Stickstoff - Blau, Kohlenstoff - Grau, Wasserstoff - Weiß, Fluor - Hellgrün.
Th1
O1 O2
O3
O4 N1
N2
N3
N4 O5
O6
22
Tabelle 1: Bindungen und Winkel der Donoratome zum Zentralatom der beiden Liganden im [Th(fsalen)2]-Komplex.
Bindung/ Winkel Ligand 1 Bindung/ Winkel Ligand 2
d(Th1–O1) 2,28 Å d(Th1–O3) 2,28 Å
d(Th1–O2) 2,34 Å d(Th1–O4) 2,29 Å
d(Th1–N1) 2,70 Å d(Th1–N3) 2,69 Å
d(Th1–N2) 2,59 Å d(Th1–N4) 2,65 Å
∡O1Th1N1 69,5 ° ∡O3Th1N3 68,3 °
∡O2Th1N2 69,7 ° ∡O4Th1N4 69,2 °
∡N1Th1N2 63,4 ° ∡N3Th1N4 64,4 °
∡O1Th1O2 157,0 ° ∡O3Th1O4 158,5 °
Auffällig ist, dass Ligand 2 bei Betrachtung der Bindungslängen eher symmetrische Abstände aufweist, wohingegen Ligand 1 deutliche Unterschiede zwischen den Bindungslängen der Sauerstoffe bzw.
Stickstoffe zeigt. Grund dafür kann die Wasserstoffbrückenbindung O5–H⋯O2 des Methanolmoleküls zum koordinierenden Sauerstoff von Ligand 1 sein. Die Th1–O2-Bindung wird somit geschwächt und fällt länger aus, als die übrigen Th–O-Bindungen. Aus diesem Grund wird zum Ausgleich die benachbarte Bindung Th1–N2 verstärkt, wodurch diese kürzer ist, als die übrigen Th–N-Bindungen.
3.2.2 [U(fsalen)2]
In Abbildung 8 ist die Struktur eines [U(fsalen)2]-Komplexmoleküls, welches in der triklinen Raumgruppe P1̅ kristallisiert, zu sehen. Die Elementarzelle weist eine Größe von 4749 Å3, mit den Achsen a = 15,33 Å, b = 18,56 Å und c = 19,30 Å und den Winkeln α = 61,83 °, β = 88,61 ° und γ = 79,49 ° auf und enthält sechs Komplex- und mindestens zwei Methanolmoleküle. Das Uranatom wird von acht Donoratomen zweier tetravalenter, zweifach deprotonierter Liganden in einen Trigondodekaeder eingeschlossen. In der Struktur wird mindestens ein Methanolmolekül pro drei Komplexmoleküle eingeschlossen, weitere konnten bei der Strukturlösung nicht mit Sicherheit bestimmt werden. In der Kristallstruktur kommen drei in Bindungslängen und -winkeln leicht variierende Komplexmoleküle vor.
23 Durchschnittliche Bindungslängen und Winkel sind in Tabelle 2 im Vergleich zu denen des [Th(fsalen)2]-Komplexes dargestellt. Die Differenzen einzelnen Bindungslängen und –winkel ähneln denen des Thoriumkomplexes stark. Dementsprechend bestehen auch hier die geringen Unterschiede zwischen den beiden angelagerten Liganden.
Tabelle 2: Durchschnittliche Bindungslängen der Donoratome zu den Zentralatomen der beiden [M(fsalen)2]-Komplexe und des [U(Hysalen)2]-Komplexes.
Komplex d(M–O) d(M–N) ∡OMN ∡NMN ∡OMO
[Th(fsalen)2] 2,30 Å 2,66 Å 69,2 ° 63,9 ° 157,8 ° [U(fsalen)2] 2,23 Å 2,59 Å 70,3 ° 66,7 ° 153,2 ° [U(Hysalen)2] 2,23 Å 2,58 Å 70,4 ° 67,4 ° 152,6 °
Sowohl die M–O-, als auch die M–N-Bindungen des Urankomplexes sind durchschnittlich etwa 0,07 Å kürzer als die des Thoriumkomplexes. Die Unterschiede der Ionenradien der beiden vierwertigen Actinide betragen allerdings nur 0,05 Å, was auf eine größere Stabilität des Urankomplexes hindeutet.[45] Bei Betrachtung der Donoratombindungswinkel zeigt sich eine Vergrößerung der ∡OMN-
Abbildung 8: Ein [U(fsalpn)2]-Komplexmolekül aus der Kristallstruktur. Uran - Grün, Sauerstoff - Rot, Stickstoff - Blau, Kohlenstoff - Grau, Wasserstoff - Weiß, Fluor - Hellgrün.
U1
O1
O2 O3
O4
N1 N2
N3 N4
24 und ∡NMN-Winkel, welche durch die geringere Entfernung der Donoratome zum Zentralatom zu Stande kommt. Der ∡OMO-Winkel wird drastisch kleiner, was zeigt, dass das Uranatom deutlich weiter in die Kavität des Chelatliganden eindringt, als das Thoriumatom.
Beim Vergleich des [U(fsalen)2]-Komplexes mit dem [U(Hysalen)2]-Komplex zeigen sich fast gleiche Bindungsabstände und nur geringe Unterschiede in den Bindungswinkeln. Der größere ∡NMN-Winkel und der kleine ∡OMO-Winkel deuten bei gleichen Bindungsabständen darauf hin, dass sich das Uranatom im [U(Hysalen)2]-Komplex etwas tiefer in der Kavität befindet als im [U(fsalen)2]-Komplex.
Der Komplex des unsubstituierten Salens ([U(Salen)2]-Komplex) zeigt eine M–O-Bindungslänge von 2,23 Å und eine M–N-Bindungslänge von 2,57 Å.[46] Auch hier wird die M–O-Bindung also kaum beeinflusst. Die M–N-Bindung ist dafür noch etwas kürzer. Grund dafür sind –I-Effekte der Fluor- bzw.
Hydroxysubstituenten, welche Elektronendichte von den Donoratomen ziehen und so deren Bindung zum Metall schwächen. Da sich allerdings nur die M–N-Bindungslänge nicht aber die M–O- Bindungslänge scheint die Substitution elektronisch größeren Einfluss auf die dative Bindung des Stickstoffs zum Metall zu haben.
3.2.3 [Th(fsalpn)2] und [U(fsalpn)2]
In Abbildung 9 sind die Strukturen der [M(fsalpn)2]-Komplexe, welche beide in der triklinen Raumgruppe P1̅ kristallisieren, zu sehen. Die Elementarzelle des Thoriumkomplexes weist eine Größe von 1538 Å3 mit den Achsen a = 9,64 Å, b = 11,98 Å und c = 14,47 Å und den Winkeln α = 106,23 °, β = 105,80 ° und γ = 90,68 ° auf und enthält zwei Komplexmoleküle. Die Elementarzelle des Urankomplexes weist eine Größe von 1521 Å3 mit den Achsen a = 9,64 Å, b = 11,94 Å und c = 14,37 Å und den Winkeln α = 106,00 °, β = 105,99 ° und γ = 90,98 ° auf und enthält ebenfalls zwei Komplexmoleküle. Das Zentralatom wird bei beiden Komplexen von acht Donoratomen zweier tetravalenter, zweifach deprotonierter Liganden in einen Trigondodekaeder (siehe Abbildung 10 am Beispiel des [Th(fsalpn)2]-Komplexes) eingeschlossen. Die Packung beider Komplexe ist gleich. Die Komplexmoleküle verknüpfen sich durch C–H⋯F-Wechselwirkungen (in Abbildung 10 orange dargestellt) am Aromaten zu weiteren Komplexmolekülen um Ketten zu bilden. Die Fluoratome wechselwirken zusätzlich mit den Wasserstoffatomen der Brücken benachbarter Komplexe (nicht eingezeichnet) und prägen so Komplexmolekülschichten aus. Diese Schichten werden schließlich durch π-stacking miteinander verknüpft, sodass eine dreidimensionale Packung entsteht.
25
Abbildung 9: Kristallstrukturen der beiden [M(fsalpn)2]-Komplexe. Thorium – Hellblau, Uran - Grün, Sauerstoff - Rot, Stickstoff - Blau, Kohlenstoff - Grau, Wasserstoff - Weiß, Fluor - Hellgrün.
Abbildung 10: Elementarzelle des [Th(fsalpn)2]-Komplexes mit eingezeichneten Koordinationspolyedern in grün sowie C–H⋯F- Wechselwirkung in orange.
Ausgewählte Bindungslängen sind in Tabelle 3 und einzelne Bindungswinkel in Tabelle 4 zusammengefasst. Auch bei den [M(fsalpn)2]-Komplexen weisen die beiden Ligandmoleküle eines
Th1
O1 O2 U1 O2
O3 O1
O4 N1
N2
N3
N4
O3
N1 O4
N2 N3
N4
26 Komplexes kleine Unterschiede auf. So zeigt Ligand 1 sich um etwa 0,02 Å unterscheidende M–N- Bindungen und etwa 0,01 Å kürzere M–O-Bindungen als Ligand 2 auf. Außerdem weist Ligand 1 eine größere Differenz zwischen den beiden ∡OMN-Winkeln auf als Ligand 2. Bei Ligand 1 ist der
∡NMN-Winkel größer und der ∡OMO-Winkel kleiner als bei Ligand 2, was darauf hindeutet, dass das Zentralatom etwas tiefer in die Kavität von Ligand 1 eindringt als in die von Ligand 2.
Tabelle 3: Bindungslängen der beiden Liganden der [M(fsalpn)2]-Komplexe.
Bindung Th (Ligand 1) U (Ligand 1) Bindung Th (Ligand 2) U (Ligand 2) d(M–O1) 2,282 Å 2,228 Å d(M–O3) 2,290 Å 2,237 Å d(M–O2) 2,288 Å 2,232 Å d(M–O4) 2,293 Å 2,238 Å d(M–N1) 2,666 Å 2,624 Å d(M–N3) 2,648 Å 2,604 Å d(M–N2) 2,641 Å 2,601 Å d(M–N4) 2,642 Å 2,607 Å
Tabelle 4: Bindungswinkel der beiden Liganden der [M(fsalpn)2]-Komplexe.
Winkel Th (Ligand 1) U (Ligand 1) Winkel Th (Ligand 2) U (Ligand 2)
∡O1MN1 69,75 ° 70,45 ° ∡O3MN3 69,39 ° 70,18 °
∡O2MN2 69,04 ° 69,81 ° ∡O4MN4 69,19 ° 70,01 °
∡N1MN2 70,80 ° 70,94 ° ∡N3MN4 70,41 ° 70.49 °
∡O1MO2 152,28 ° 151,04 ° ∡O3MO4 152,77 ° 151,37 °
Bei Betrachtung der durchschnittlichen Bindungslängen und –winkel (siehe Tabelle 5) zeigt sich eine ähnliche Tendenz wie bei den [M(fsalen)2]-Komplexen. Die Unterschiede fallen jedoch etwas geringer aus. So ergeben sich etwas kleinere Differenzen in den M–O- bzw. M–N-Bindungslängen von 0,06 Å bzw. 0,04 Å. Die Unterschiede in den ∡OMN-Winkeln fallen nur etwas geringer aus, während die Differenzen der ∡NMN- und ∡OMO-Winkeln deutlich geringer sind. Grund hierfür kann die flexiblere Brücke des Liganden sein. Die Winkel müssen sich somit trotz Änderung der Bindungslängen nur wenig ändern, da die Brücke eine geringere Spannung aufweist, als im [M(fsalen)2]-Komplex. Da die Unterschiede der Bindungslängen etwa der Differenz der Ionenradien entsprechen und auch die Bindungswinkel sehr ähnlich sind, zeigt sich, dass sich die [M(fsalpn)2]-Komplexe auch in Betracht der Stabilität wesentlicher ähnlicher sind als die [M(fsalen)2]-Komplexe. Dies deutet darauf hin, dass die C3-Brücke flexibel genug ist, um eine ähnliche Stabilität der Komplexe unabhängig von der Größe des Zentralatoms zu gewährleisten.
27
Tabelle 5: Durchschnittliche Bindungslängen der Donoratome zu den Zentralatomen der [M(fsalen)2]-, [M(fsalpn)2]-Komplexe und des [U(Hysalen)2]-Komplexes.
Komplex d(M–O) d(M–N) ∡OMN ∡NMN ∡OMO
[Th(fsalen)2] 2,30 Å 2,66 Å 69,2 ° 63,9 ° 157,8 ° [U(fsalen)2] 2,23 Å 2,59 Å 70,3 ° 66,7 ° 153,2 ° [Th(fsalpn)2] 2,29 Å 2,65 Å 69,3 ° 70,6 ° 152,5 ° [U(fsalpn)2] 2,23 Å 2,61 Å 70,1 ° 70,7 ° 151,2 ° [U(Hysalen)2] 2,23 Å 2,58 Å 70,4 ° 67,4 ° 152,6 °
3.3 Fluorokomplexe – IR-Spektroskopie
Die IR-Spektren der Fluorokomplexe wurden mit den aus den Synthesen erhaltenen Pulvern aufgenommen. In Abbildung 11 sind die IR-Spektren aller [M(fsalen)2]-Komplexe im Überblick dargestellt. Es ergeben sich nur kleine Unterschiede in Bandenlage und Intensität, was auf die Isostrukturalität zurückzuführen ist.
In Abbildung 12 sind die IR-Spektren aller [M(fsalpn)2]-Komplexe im Überblick dargestellt. Auch hier sind keine wesentlichen Unterschiede in Bandenlage und –intensität erkennbar. Die Schwingungen der isostrukturellen Komplexe werden nur wenig von den koordinierten Metallatomen beeinflusst. Wie
Abbildung 11: IR-Spektren aller synthetisierten [M(fsalen)2]-Komplexe im Überblick.
28 bei den [M(fsalen)2]-Komplexen können keine Verunreinigungen, die aufgrund der Nutzung unterschiedlicher Formen der Metallsalze zu Stande kommen könnten, festgestellt werden.
In Tabelle 6 sind die charakteristischen Valenzschwingungen der substituierten Imingruppe und der aromatischen C–O-Gruppe für alle Fluoroligandkomplexe zusammengefasst. Die Schwingungen der Imingruppe weisen bei den f-Element-Komplexen eine um 6 bis 10 cm-1 für [M(fsalen)2]-Komplexe und 4 bis 7 cm-1 für [M(fsalpn)2]-Komplexe geringere Wellenzahl auf als die der jeweiligen beiden Nebengruppen-Komplexe. Durch die flexiblere Brücke zeigen sich wie auch in den Strukturdaten der Einkristalle geringere Unterschiede beim Vergleich der [M(fsalpn)2]-Komplexe als beim Vergleich der [M(fsalen)2]-Komplexe. Bei der C–O-Valenzschwingung der Fluoroligandkomplexe ergeben sich etwas größere Differenzen, allerdings unterscheiden sich auch die f-Elemente untereinander deutlich mehr, als bei den Schwingungen der Imingruppe. Die beiden in Tabelle 6 gezeigten Schwingungen unterscheiden sich nach Ligand kaum in ihrer Wellenzahl. Die einzige mittelstarke Schwingung die einen drastischen Wellenzahlunterschied aufweist ist die Gerüstschwingung der Alkylkette. Diese liegt für alle [M(fsalen)2]-Komplexe bei etwa 1040 cm-1 und für alle [M(fsalpn)2]-Komplexe bei etwa 1080 cm-1. Im Bereich von 3000 bis 3100 cm-1 sind mehrere schwache Banden der aromatischen C–H- Valenzschwingungen und im Bereich von 2850 bis 2950 cm-1 mehrere schwache aliphatische C–H- Valenzschwingungen zu finden.[31] Bei Vergleich mit dem freien Liganden fällt die Verschiebung vieler
Abbildung 12: IR-Spektren aller synthetisierten [M(fsalpn)2]-Komplexe im Überblick.
29 Gerüstschwingungen und die starke C–O-Valenzschwingung, die für die Komplexe im Bereich um 1300 cm-1 erscheint, auf.
Tabelle 6: Zuordnung ausgewählter IR-Banden der [M(fsalen)2]- und [M(fsalpn)2]-Komplexe, sowie Literaturwerte der Banden von [M(Salen)2]- und [M(Salpn)2]-Komplexen.[13]
Schwingung Zr Ce Hf Th U Np
[M(fsalen)2] ν(C=N–C) 1632 1626 1634 1624 1626 1625
ν(C–O) 1300 1286 1302 1294 1290 1291
[M(fsalpn)2] ν(C=N–C) 1631 1627 1632 1625 1626 -
ν(C–O) 1298 1286 1304 1295 1289 -
[M(Salen)2] ν(C=N–C) - - - 1632 1635 -
ν(C–O) - - - 1305 1311 -
[M(Salpn)2] ν(C=N–C) - - - 1626 1623 -
ν(C–O) - - - 1303 1306 -
Bei Vergleich der Bandenlage der [M(fsalen)2]-Komplexe mit den [M(Salen)2]-Komplexen fallen große Unterschiede in der C=N–C- und C–O-Valenzschwingung auf. Die Wellenzahlen der Schwingung sind beim Komplex des unsubstituierten Liganden knapp 10 cm-1 größer, was auf etwas stärkere Bindungen, bzw. Koordination im substituierten Liganden hinweist, welche durch den –I-Effekt des Fluoratoms hervorgerufen werden könnte. Bei Betrachtung der [M(fsalpn)2] und [M(Salpn)2]-Komplexe ist der gleiche Effekt bei der C–O-Valenzschwingung, nicht aber bei der C=N–C-Schwingung zu sehen. Dies deutet auf einen elektronenschiebenden Einfluss der zusätzlichen Methylengruppe in der Brücke des Liganden, der den elektronenziehenden des Fluorsubstituenten am Stickstoff ausgleicht, hin.
3.4 Fluorokomplexe – NMR-Spektroskopie
3.4.1 Freie Liganden H2fsalen und H2fsalpn
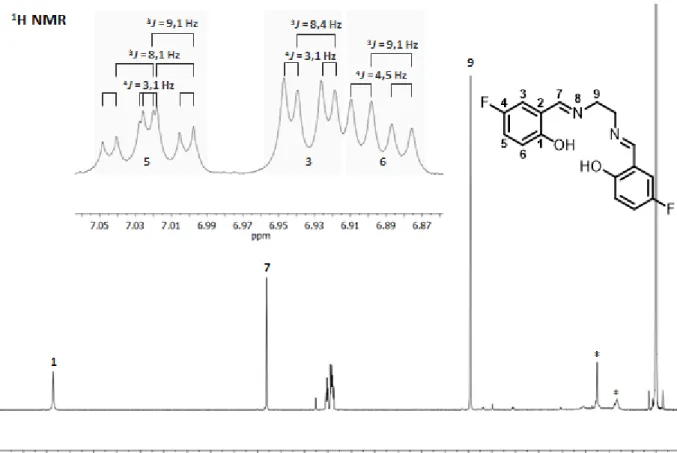
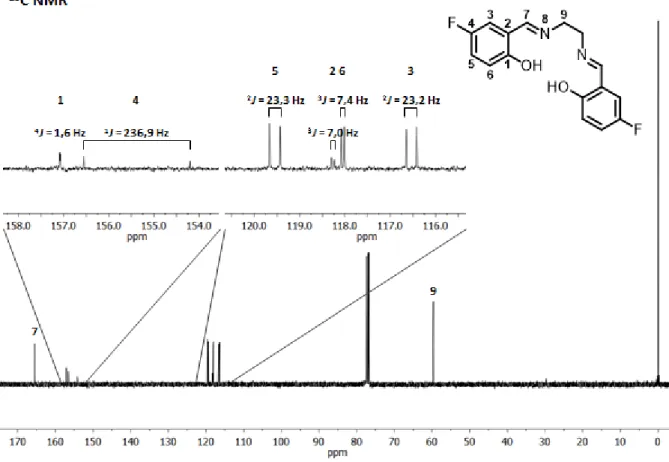
In Abbildung 13 ist das 1H NMR Spektrum des H2fsalen-Liganden zu sehen. Die aromatischen Protonen zeigen dabei durch das eingeführte Fluoratom charakteristische Kopplungsmuster, die durch Kopplungen über bis zu vier Bindungen hervorgerufen werden. Proton 3 koppelt über drei Bindungen mit Fluor und über vier Bindungen mit Proton 5. Da sich die beiden Kopplungskonstanten ausreichend unterscheiden entsteht ein Dublett-Dublett. Proton 6 koppelt mit Proton 5 und Fluor und zeigt deshalb ebenfalls ein Dublett-Dublett. Proton 5 hingegen kann mit den beiden übrigen Aromatenprotonen und
30 zusätzlich mit Fluor koppeln um ein Dublett-Dublett-Dublett zu erzeugen. Dieses Protonensignal wird in vielen der Komplexen zu einem Dublett-Triplett vereinfacht, da die 3J-Kopplungen zu Fluor und Proton 6 in vielen Fällen eine ähnliche Größe einnehmen und somit nicht mehr voneinander unterschieden werden können. Alle übrigen Protonensignale sind Singuletts. Diese Kopplungsmuster sind auch in den diamagnetischen Komplexen der Liganden sichtbar, während sie in den paramagnetischen Komplexen aufgrund von Linienverbreiterungen und kürzeren Relaxationszeiten teilweise nicht aufgelöst werden können.
Abbildung 13: 1H NMR-Spektrum des H2fsalen-Liganden in CDCl3 mit TMS. Kopplungen der Aromatenprotonen hervorgehoben.
In Tabelle 7 sind die Verschiebungen der Protonen beider verwendeter Fluoroliganden dargestellt.
Trotz Erweiterung der aliphatischen Brücke um eine Methylengruppe ergibt sich für den H2fsalpn-Liganden bei den Signalen der Imingruppe und den Aromatenprotonen keine große Verschiebung im Vergleich zum H2fsalen-Ligand. Alle vier Signale werden nur um 0,02 ppm tieffeldverschoben, also ganz leicht entschirmt. Die Brücke erhält ein zusätzliches Signal (Proton 10), welches die geringste Verschiebung und somit größte Abschirmung im Molekül aufweist, da es sich zwischen zwei Methylengruppen befindet. Die Protonen 9 sind aufgrund der zusätzlichen
![Abbildung 6: 1 H NMR-Spektren von H 2 Hysalen und den [M(Hysalen) 2 ]-Komplexen in DMSO-d 6](https://thumb-eu.123doks.com/thumbv2/1library_info/4565381.1599809/19.892.112.768.439.945/abbildung-h-nmr-spektren-hysalen-hysalen-komplexen-dmso.webp)
![Tabelle 2: Durchschnittliche Bindungslängen der Donoratome zu den Zentralatomen der beiden [M(fsalen) 2 ]-Komplexe und des [U(Hysalen) 2 ]-Komplexes](https://thumb-eu.123doks.com/thumbv2/1library_info/4565381.1599809/23.892.194.691.130.552/tabelle-durchschnittliche-bindungslängen-donoratome-zentralatomen-komplexe-hysalen-komplexes.webp)
![Abbildung 9: Kristallstrukturen der beiden [M(fsalpn) 2 ]-Komplexe. Thorium – Hellblau, Uran - Grün, Sauerstoff - Rot, Stickstoff - Blau, Kohlenstoff - Grau, Wasserstoff - Weiß, Fluor - Hellgrün](https://thumb-eu.123doks.com/thumbv2/1library_info/4565381.1599809/25.892.115.784.108.375/abbildung-kristallstrukturen-komplexe-sauerstoff-stickstoff-kohlenstoff-wasserstoff-hellgrün.webp)
![Abbildung 16: Ausschnitt aus den 1 H NMR-Spektren der diamagnetischen [M(fsalen) 2 ]-Komplexe mit allen Komplexsignalen in THF-d 8 mit TMS](https://thumb-eu.123doks.com/thumbv2/1library_info/4565381.1599809/34.892.109.789.317.779/abbildung-ausschnitt-nmr-spektren-diamagnetischen-fsalen-komplexe-komplexsignalen.webp)
![Abbildung 17: Ausschnitt aus den 13 C NMR-Spektren der diamagnetischen [M(fsalen) 2 ]-Komplexe, der die Aromatenkohlenstoffe und den Iminkohlenstoff zeigt, in THF-d 8 mit TMS](https://thumb-eu.123doks.com/thumbv2/1library_info/4565381.1599809/35.892.106.790.278.767/abbildung-ausschnitt-spektren-diamagnetischen-fsalen-komplexe-aromatenkohlenstoffe-iminkohlenstoff.webp)
![Abbildung 21: 13 C NMR-Spektren der paramagnetischen [M(fsalen) 2 ]-Komplexe gegenüber dem diamagnetischen [Th(fsalen) 2 ]-Komplex in THF-d 8 mit TMS](https://thumb-eu.123doks.com/thumbv2/1library_info/4565381.1599809/41.892.102.788.271.733/abbildung-spektren-paramagnetischen-fsalen-komplexe-gegenüber-diamagnetischen-komplex.webp)
![Abbildung 22: 1 H NMR Spektren des [U(fsalen) 2 ]-Komplexes bei Temperaturen von 243 K (unten) bis 313 K (oben) in Schritten von 5 K in THF-d 8 mit TMS](https://thumb-eu.123doks.com/thumbv2/1library_info/4565381.1599809/42.892.105.787.632.1109/abbildung-nmr-spektren-fsalen-komplexes-temperaturen-schritten-thf.webp)
![Abbildung 23: Links: Auftragung der 1 H NMR-Peaks des [U(fsalen) 2 ]-Komplexes nach Bleaney](https://thumb-eu.123doks.com/thumbv2/1library_info/4565381.1599809/43.892.116.786.810.1095/abbildung-auftragung-h-nmr-peaks-fsalen-komplexes-bleaney.webp)
